

Abstract
Purpose: Methimazole is the most convenient drug used in the management of hyperthyroid patients. However, associated with its clinical use is hepatotoxicity as a life threatening adverse effect. The exact mechanism of methimazole-induced hepatotoxicity is still far from clear and no protective agent has been developed for this toxicity.
Methods: This study attempts to evaluate the hepatotoxicity induced by methimazole at different experimental conditions in a mice model. Methimazole-induced hepatotoxicity was investigated in different situations such as enzyme induced and/or glutathione depleted animals.
Results: Methimazole (100 mg/kg, i.p) administration caused hepatotoxicity as revealed by increase in serum alanine aminotransferase (ALT) activity as well as pathological changes of the liver. Furthermore, a significant reduction in hepatic glutathione content and an elevation in lipid peroxidation were observed in methimazole-treated mice. Combined administration of L-buthionine sulfoximine (BSO), as a glutathione depletory agent, caused a dramatic change in methimazole-induced hepatotoxicity characterized by hepatic necrosis and a severe elevation of serum ALT activity. Enzyme induction using phenobarbital and/or β-naphtoflavone beforehand, deteriorated methimazole-induced hepatotoxicity in mice. N-acetyl cysteine (300 mg/kg, i.p) administration effectively alleviated hepatotoxic effects of methimazole in both glutathione-depleted and/or enzyme induced animals.
Conclusion: The severe hepatotoxic effects of methimazole in glutathione-depleted animals, reveals the crucial role of glutathione as a cellular defense mechanism against methimazole-induced hepatotoxicity. Furthermore, the more hepatotoxic properties of methimazole in enzyme-induced mice, indicates the role of reactive intermediates in the hepatotoxicity induced by this drug. The protective effects of N-acetylcysteine could be attributed to its radical/reactive metabolite scavenging, and/or antioxidant properties as well as glutathione replenishment activities.
Keywords: Enzyme induction, Glutathione, Hepatotoxicity, Methimazole, Mice, N-acetyl cysteine
Introduction
Methimazole is a worldwide used anti-hyperthyroidism drug, which its clinical use is associated with hepatotoxicity.1 Although the exact mechanism that methimazole causes hepatotoxicity through it is not clearly understood yet, but some investigations revealed that cellular glutathione reservoirs has a fundamental role in preventing methimazole-induced damage.2-5 Furthermore, the importance of glutathione in methimazole-induced cytotoxicity was shown previously in our laboratory in an in vitro model of isolated rat hepatocytes.6
Previous studies proposed the role of reactive metabolites in methimazole-induced toxicity.7,8 It has been shown that N-methylthiourea as a suspected reactive intermediate of methimazole is produced at in vitro models.8,9 N-methylthiourea is further metabolized by cytochrome (CYP) and/or flavin monooxygenase (FMO) enzymes to sulfenic acid species.8,10 Sulfenic acids are reactive electrophilic metabolites11 and could interact with different intracellular targets, which might consequently encounter toxicity.11 In a study on methimazole-induced toxicity in rat olfactory mucosa, it has been shown that the metabolic pathways and CYP enzymes play a major role in the toxicity induced by this drug.5 In addition, glyoxal as another metabolite of methimazole8, might plays a role in methimazole-induced hepatotoxicity.6
This study attempted to evaluate the role of hepatic glutathione reservoirs in methimazole-induced hepatotoxicity. In addition, the role of N-acetyl cysteine (NAC) as a protective agent in this situation was studied. To investigate the effect of metabolism and metabolic pathways, the adverse effects of methimazole was studied in enzymeinduced animals and the protective role of NAC was also evaluated in this situation.
Serum alanine amino transferase (ALT) levels, lipid peroxidation, hepatic glutathione (GSH) contents, and liver histopathological changes were assessed in different conditions after methimazole administration alone and/or in combination with NAC.
Materials and Methods
Chemicals
Methimazole was purchased from Medisca pharmaceutique (Montreal, Canada). 5,5´-dithionitrobenzoic acid (DTNB) and L-buthionine sulfoximine (BSO) were purchased from Sigma-Aldrich (St. Louis, USA). Thiobarbituric acid (TBA) was obtained from SERVIA (Heidenberg, New York). Trichloro acetic acid (TCA), β-naphtoflavone, Sodium dodecyl sulfate (SDS), Phenobarbital, and Hydroxy methyl amino methane (Tris) were purchased from Merck (Dardamstd, Germany). The kit for alanine aminotransferase (ALT) analysis was obtained from Pars Azmun Company (Tehran, Iran). All salts for preparing buffer solutions were of the highest grade commercially available.
Animals
Male Swiss albino mice, 6 weeks old (25-40 g weight), were obtained from Tabriz University of Medical Sciences (Tabriz, Iran). Mice were housed in cages on wood bedding at a temperature of 25±3 °C. Unless otherwise stated, mice had free access to food and water. The animals were handled and used according to the animal handling protocol approved by the Tabriz University's ethics committee.
Animals were randomly divided equally into eleven groups of five animals. The treatments were as follows: Group A (control): vehicle (0.9% saline solution) only. Group B: 100 mg/kg methimazole (i.p, dissolved in 0.9% saline). Group C: 100 mg/kg methimazole + BSO (1 g/kg, i.p). Group D: 100 mg/kg methimazole + BSO (1 g/kg) + NAC (300 mg/kg, i.p). Group E: β-naphtoflavone-induced animals + methimazole 100 mg/kg. Group F: β-naphtoflavone-induced animals+ methimazole 100 mg/kg + NAC 300 mg/kg. Group G: Phenobarbital-induced animals + methimazole 100 mg/kg. Group H: Phenobarbital-induced animals + methimazole 100 mg/kg + NAC 300 mg/kg. There was no significant differences between groups; I (BSO-treated animals), J (β-naphtoflavone-induced animals), and group K (Phenobarbital-induced animals), and the control (vehicle-treated) animals in the parameters assessed in this study.
Glutathionedepleted animals
Mice were treated with buthionine sulfoximine (BSO) (1 g/kg) as a model for hepatic glutathione depletion in animals.12 One hour later, BSOtreated animals were given methimazole (100 mg/kg i.p). Food was removed at 15 hours before dosing with BSO and supplied again at 2 hours after methimazole administration.3
Enzyme-induced mice
β-naphtoflavone-induced animals were treated with β-naphtoflavone (40 mg/kg, i.p) for three consecutive days.13 Phenobarbital-induced animals were treated with 80 mg/kg of phenobarbital (i.p injection for three days) before the experiments.13 At the forth day, animals were treated with methimazole (100 kg/kg, i.p).
Serum biochemical analysis and liver histopathology
Blood was collected from the abdominal vena cava under pentobarbital anesthesia, and the liver was removed. The blood was allowed to clot at 25°C, and serum was prepared by centrifugation (1000 g, for 20 minutes).14 Serum alanine transaminase (ALT) activities were measured with a commercial kit. For histo-pathological evaluation, samples of liver were fixed in formalin (10%). Paraffin-embedded sections of liver were prepared and stained with haematoxylin and eosin (H&E) before light microscope viewing.14
Liver glutathione content
The excised livers were immediately frozen at -70° C and analyzed for glutathione (GSH) within 24 hours. Briefly, samples of liver (200 mg) were homogenized in 8 ml of 20 mM EDTA. The GSH contents were assessed by determining non-protein sulphydryl contents with the Ellman reagent.15
Lipid peroxidation
Level of lipid peroxidation was measured in different experimental groups. Briefly, reaction mixture consist of 0.2 ml 8% SDS, 1.5 ml 20% trichloro acetic acid, and 0.6 ml distilled water. 0.2 ml of tissue homogenate was added to the reaction mixture. Reaction was initiated by adding 1.5 ml of 1% thiobarbituric acid (TBA) and terminated by 10% trichloroacetic acid (TCA). Samples were centrifuged (3000 g for 5 minutes) and the absorbance of developed color was read at 532 nm using an Ultrospec 2000® UV spectrophotometer.16
Statistical analysis
Results are shown as Mean±SE. Comparisons between multiple groups were made by a one-way analysis of variance (ANOVA) followed by Turkey’s post hoc test. Differences were considered significant when P<0.05.
Results
Mice were treated with 100 mg/kg of methimazole, which was reported as a hepatotoxic dose of this drug in previous investigations.3 Serum alanine aminotransferase (ALT) and liver histopathological changes were used as indicators for occurrence of hepatotoxicity induced by methimazole. Serum ALT levels were measured in different time points after methimazole administration (Figure 1). It was found that the maximum serum ALT levels occurred at 5 hours after drug administration (Figure 1), and gradually declined within the next 24 hours (Figure 1). Hence, all experiments (Serum transaminase levels in other groups, hepatic glutathione contents, lipid peroxidation, and histopathological evaluation of liver) were carried out five hours after methimazole administration to mice.
Figure 1 .
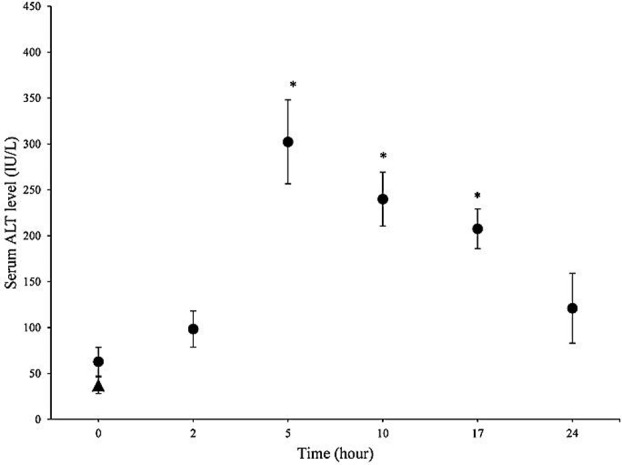
Serum ALT levels after methimazole (100 mg/kg) administration to mice. The peak serum ALT level was observed at 5 hours after methimazole administration.
▲: Control (vehicle-treated) animals. ●: Methimazoletreated animals.
Data are expressed as Mean±SE for at least five animals.
*Significantly higher than control levels (P<0.05).
Methimazole (100 mg/kg) caused a significant elevation in serum ALT levels (P<0.05) as compared with the control animals (Figure 2). This might indicate the liver damage caused by this drug. To investigate the impact of glutathione reservoirs as a basic defense mechanism against xenobiotics-induced hepatic damage, hepatic glutathione content (GSH) was depleted with BSO,12 then the toxicity profile of methimazole was investigated in glutathione-depleted animals. The liver/animal weight ratio was assessed in different experimental groups (Table 1). When glutathione-depleted animals were treated with methimazole, a dramatic elevation in serum ALT levels was observed (Figure 2) (P<0.05). NAC (300 mg/kg) administration effectively reduced (P<0.05) methimazole-induced ALT elevation in both intact and/or glutathione-depleted animals (Figure 2).
Figure 2 .
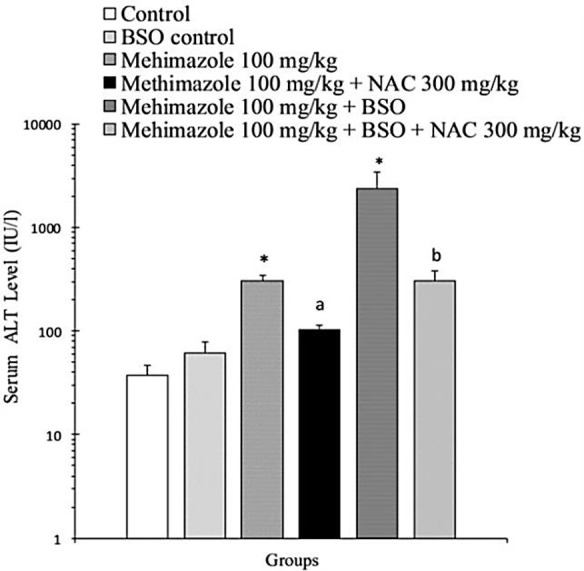
The effect of methimazole (100 mg/kg) on serum ALT level in mice. The role of glutathione reservoirs and protective effects of NAC administration.
BSO: L-buthionine sulfoximine. NAC: N-acetylcysteine.
Data are given as Mean±SE for five animals as measured after 5 hours of drug administration.
*Significant difference as compared to the control animals (P<0.05).
a Significant difference as compared to the methimazole-treated animals (P<0.05).
Table 1. Liver/Animal weight in the study on methimazole-induced hepatotoxicity.
| Groups | Parameters assessed | ||
| Animal weight (gram) | Liver weight (gram) | Liver/animal weight Ratio (%) | |
| Control (Vehicle-treated animals) | 38±0.77 | 2.46±0.13 | 6.45±0.31 |
| + Methimazole 100 mg/kg | 36±1.34 | 2.25±0.12 | 6.38±0.50 |
| + L-buthionine sulfoximine (BSO) 1 g/kg § | 35±1.80 | 1.64±0.27 | 4.77±0.29 |
| + Methimazole 100 mg/kg | 35±2.30 | 1.27±0.86 | 3.90±0.33 |
| + ß-naphtoflavone (Enzyme-induced animals) §§ | 35±0.38 | 3.24±0.30* | 9.27±1.04* |
| + Methimazole 100 mg/kg ▲ | 35±0.24 | 3.36±0.23* | 9.72±0.71* |
| + Phenobarbital (Enzyme-induced animals) # | 37±2.50 | 2.85±0.32 | 7.70±0.39 |
| + Methimazole 100 mg/kg | 38±2.32 | 2.5±0.42 | 6.60±0.92 |
| Data are given as Mean±SE for at least five animals. | |||
| *Significantly different from control group (P<0.05). | |||
| § Mice were treated with BSO (1g/ kg, i.p).One hour later; the BSOtreated animals were given methimazole (100mg/kg i.p). Food was removed at 15 hours before dosing with BSO, and supplied again at 2 hours after methimazole administration. | |||
| ▲Three out of fiveanimals were death when they were treated with methimazole (100 mg/kg, i.p). | |||
| §§ Mice were pre-treated with ß-naphtoflavone (40 mg/kg, i.p) for three consecutive days. On the 4th day, animals were given methimazole (100mg/kg i.p) and the mentioned parameters were measured after five hours. | |||
| # Mice were pre-treated with phenobarbital (80 mg/kg, i.p) for three consecutive days. On the 4th day, animals were given methimazole (100mg/kg i.p) and parameters were measured after five hours. | |||
To evaluate the role of reactive metabolites, the effect of enzyme-induction on methimazole-induced hepatotoxicity was investigated using serum ALT levels as an indicator. It was found that methimazole (100 mg/kg) caused a dramatic raise (P<0.05) in serum ALT activities in β-naphtoflavone treated animals as compared to the control groups (Figure 3). Three out of five animals were death after methimazole administration in β-naphtoflavone treated group (Table 1). Moreover, phenobarbital-induced animals showed higher serum ALT activity when treated with methimazole (Figure 3) (P<0.05). These findings might reveal the role of metabolic pathways and reactive intermediary metabolites in methimazole-induced hepatotoxicity. NAC (300 mg/kg) administration alleviated serum ALT elevation in enzyme-indiced animals, which were treated with methimazole (P<0.05) (Figure 3).
Figure 3.
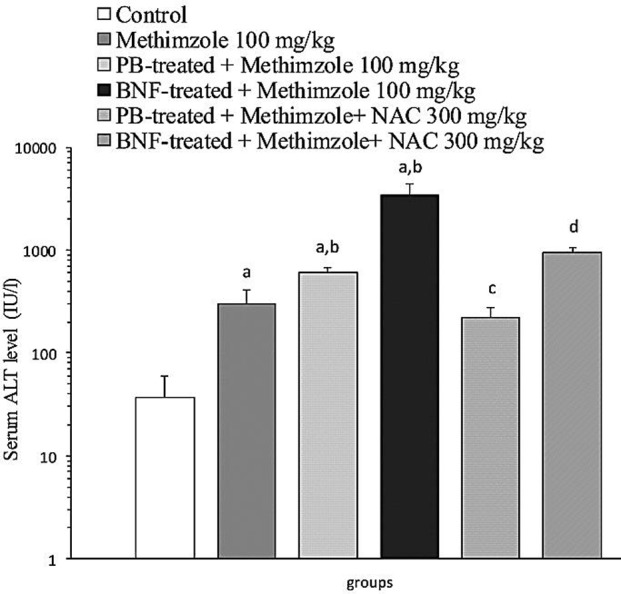
The effect of methimazole (100 mg/kg) on serum ALT level in mice. The role of enzyme-induction. BNF: β-naphtoflavone, PB: Phenobarbital.
Data are expressed as Mean±SE for five animals.
a Significantly higher than control animals (P<0.05).
b Significantly higher than methimazoletreated animals (P<0.05).
c Significantly lower than phenobarbital-induced animals which were treated with methimazole (P<0.05).
d Significantly lower than β-naphtoflavone-induced animals which were treated with methimazole (P<0.05).
As methimazole caused more ALT elevation in glutathione-depleted animals (Figure 2), hepatic glutathione levels were assessed to investigate the effect of methimazole on hepatic glutathione reservoirs. It was found that, methimazole (100 mg/kg) administration caused a decrease in hepatic glutathione (GSH) contents (P<0.05) as compared with control groups (Figure 4). When enzymeinduced mice were treated with methimazole the decline in glutathione reservoirs was more significant (P<0.05) (Figure 4). These findings might indicate that methimazole metabolites conjugated with glutathione in mice liver.
Figure 4.
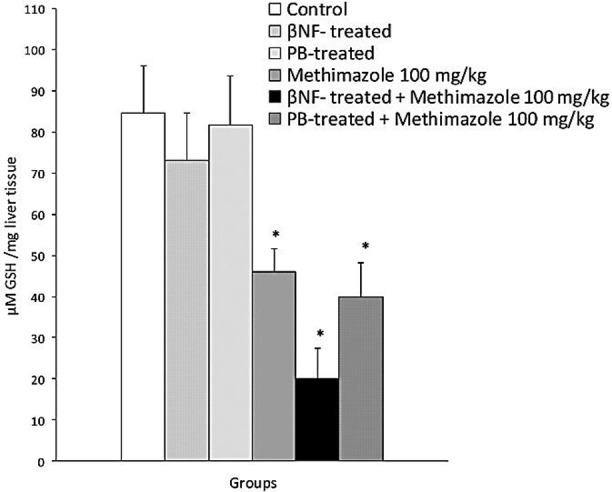
Methimazole-induced reduction in hepatic glutathione (GSH) content in mice.
Data are showed as Mean±SE for at least five animals. BNF: β-naphtoflavone, PB: Phenobarbital.
* Significantly lower than control animals (P<0.05).
The probability of lipid peroxidation in liver tissue was investigated. We found that methimazole caused increase in thiobarbituric acid reactive substances (TBARS) in mice liver (Figure 5) (P<0.05), which indicates the occurrence of lipid peroxidation. Methimazoleinduced lipid peroxidation was more severe in glutathione-depleted (BSO-treated) animals (P<0.05) (Figure 5). NAC administration effectively reduced methimazole-induced lipid peroxidation in intact and/or glutathione-depleted mice (Figure 5) (P<0.05). Enzyme induction using β-naphtoflavone and/or phenobarbital deteriorated (P<0.05) methimazole-induced lipid peroxidation (Figure 6), which might indicate the role of reactive metabolites. NAC (300 mg/kg) reduced (P<0.05) the level of methimazole-induced lipid peroxidation in enzyme-induced animals (Figure 6) (P<0.05).
Figure 5 .
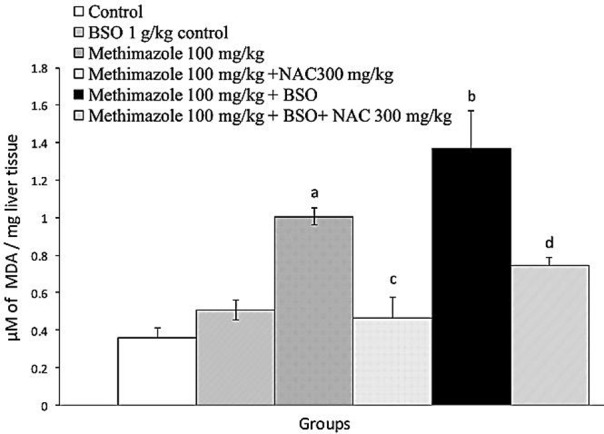
Methimazoleinduced lipid peroxidation: the role of glutathione reservoirs and the effect of NAC administration. BSO: L-buthionine sulfoximine; NAC: N-acetyl cysteine.
Data are given as Mean±SE for five animals.
a Significantly higher than control animals (P<0.05).
b Significantly higher than methimazoletreated animals (P<0.05).
c Significantly lower than methimazoletreated animals (P<0.05).
dSignificantly lower than methimazoletreated animals which were depleted of glutathione (P<0.05).
Figure 6 .
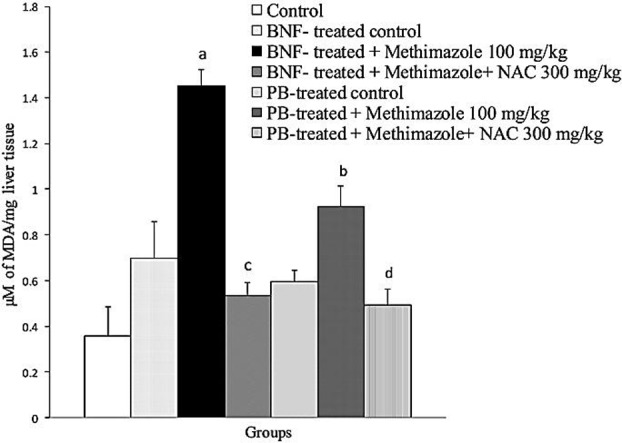
The effect of Enzyme induction on methimazole-induced lipid peroxidation in mice liver.
Data are expressed as Mean±SE for five animals.
a Significantly higher than control (BNF-treated) animals (P<0.05).
b Significantly higher than methimazoletreated animals (P<0.05).
Histopathological evaluation of mice liver revealed that inflammatory cell a mild inflammatorycell infiltration (Figure7, Part C). When glutathione-depleted animals were treated with methimazole (100 mg/kg), a severe inflammatory cells infiltration, and widespread bridging necrosis of liver was observed after 5 hours of drug administration (Figure 7, Part D).
Figure 7 .

Histopathological evaluation of mice liver treated with methimazole (100 mg/kg). The effect of glutathione reservoirs depletion on methimazole-induced hepatotoxicity. Hematoxylin and eosin (H&E) staining. A: Control, B: BSO (1g/kg) control, C: Methimazole (100 mg/kg), D: Methimazole (100 mg/kg) + BSO (1g/kg), E: Methimazole (100 mg/kg) + NAC (300 mg/kg), F: Methimazole (100mg/kg) + BSO (1g/kg) + NAC (300 mg/kg). Methimazole caused a mild Inflammatory cell infiltration (C). A severe inflammatory cells infiltration, and widespread bridging necrosis of liver (D) was occurred when glutathione-depleted animals were treated with methimazole. NAC (300 mg/kg) administration, alleviated methimazole-induced changes in normal (E) and/or glutathione-depleted (F) animals.
The effect of enzyme-induction on mice liver pathology was studied. Histologically, the enzyme induction was evident as an expansion of the surface area of hepatocytes (Figure 8, Parts C&D). It was seen that, when enzyme-induced animals were treated with methimazole, the extensive necrosis of liver parenchymal cells and inflammatory cells infiltration was occurred (Figure 8, Parts C&D). The adverse effect of methimazole in β-naphtoflavone-induced animals seems to be more severe than phenobarbital-induced ones (Figure 8, Part C). The protective effects of NAC against methimazole-induced hepatotoxicity and its role in alleviating pathologic lesions were evaluated (Figure 7 and 88. It was found that NAC alleviated methimazole-induced histopathological changes in mice liver, even in glutathione depleted (Figure 7, Parts E & F) and/or enzyme-induced (Figure 8, Parts E & F) animals.
Figure 8.

Effect of enzyme-induction on mice liver histopathological changes, caused by methimazole (100 mg/kg). A: normal control, B: Methimazole (100 mg/kg), C:BNF-induced + Methimazole (100 mg/kg), D: Phenobarbital-induced + Methimazole (100 mg/kg), E: BNFinduced + Methimazole (100 mg/kg) + NAC (300 mg/kg), F: Phenobarbital-induced + Methimazole (100 mg/kg) + NAC (300 mg/kg). When enzyme-induced animals were treated with methimazolea severe and widespreadnecrosis of liver parenchymal cells and inflammatory cells infiltration was occurred (C & D), mitigation of methimazole-induced changes in enzyme-induced animals was observed by NAC (300 mg/kg) administration, which reduced necrosis and inflammatory cells infiltration (E&F).
Discussion
Methimazole (100 mg/kg) caused hepatotoxicity in mice as revealed by elevation in serum ALT activities, a decrease in liver glutathione (GSH) reservoirs, lipid peroxidation, and histopathological changes of the liver. The toxic effects of methimazole toward mice liver were more severe in glutathione-depleted and/or enzyme-induced animals. NAC (300 mg/kg) administration effectively ameliorated all aspects of methimazole-induced hepatotoxicity in different conditions such as intact, glutathione-depleted, and/or enzyme-induced animals.
It has been shown in previous studies that cellular glutathione play a fundamental role in preventing the adverse effects of methimazole even in olfactory mucosa4 and/or liver.3,6 Our study on methimazole in glutathione-depleted animals is in line with previous investigations. Furthermore, in this study we showed that a significant amount of lipid peroxidation and a considerable reduction in hepatic glutathione (GSH) content was occurred as a consequence of methimazole administration.
Lipid peroxidation and/or serum ALT activities were more severe in glutathione-depleted animals which are another indicator that endorses the pivotal role of glutathione in preventing methimazole-induced toxicity. The role of glutathione in methimazole-induced hepatotoxicity might predict the risk of hepatotoxicity induced by this drug in human cases. Hence, in clinical situations where liver glutathione reservoirs are interrupted for example in malnutrition17 or alcoholism,18 the risk of hepatotoxicity induced by methimazole might be highest.
NAC, a protective agent which replenish glutathione reservoirs19 and/or directly scavenges reactive species,20 could protect mice liver against methimazole-induced hepatotoxicity in different situations. Furthermore, we previously showed the protective role of NAC against methimazole-induced cytotoxicity in a model of isolated rat hepatocytes.21 These findings might suggest this protective agent as a potential therapeutic choice in methimazole-induced hepatotoxicity cases in humans.
The role of metabolic pathways and reactive intermediates in methimazole-induced hepatotoxicity was evaluated by investigating the effects of this drug in enzyme-induced animals. Methimazole showed a severe hepatotoxic profile in β-naphtoflavone-induced animals. Phenobarbital-induced animals showed an enhanced profile of toxicity too. These findings revealed that metabolism and reactive metabolites formation play an important role in methimazole-induced hepatotoxicity.
Different CYP450 types5,8 as well as flavin containing monooxygenase (FMO)2,8 are involved in methimazole metabolism and converting it to reactive intermediates. As previously proposed, N-methylthiourea is suspected to be the reactive and toxic metabolite of methimazole,3,8,22 but in an in vitro study on methimazole-induced cytotoxicity toward isolated rat hepatocytes,6 we showed that in addition than N-methylthiourea, glyoxal as another metabolite of methimazole,8,22 might had a role in the hepatotoxic effects of this drug. A part of the protective effects of NAC on methimazole-induced hepatotoxicity might be attributed to the glyoxal trapping properties of N-acetyl cysteine.23 However, other properties of NAC such as its glutathione replenishment activity might be included.
β-naphtoflavone induces different CYP enzymes, mainly CYP1A family.24 Phenobarbital causes different type of CYP enzyme induction such as CYP 2C9 and 1AC.25 Since a wide range of metabolic enzymes are induced by these agents, it is difficult to distinguish the specific enzyme which is involved in methimazole metabolism. Hence, more investigations are needed to elucidate the role of the exact CYP and/or other enzymes, which are responsible for converting methimazole to its reactive metabolites. Furthermore, using other glyoxal trapping agents such as metformin and/or aminoguanidine23 against methimazole-induced hepatotoxicity could be the subject of future investigations.
Conclusion
Methimazole-induced hepatotoxicity seems to be mediated through its reactive intermediary metabolites. Hepatic glutathione reservoirs play a critical role in preventing methimazole-induced hepatotoxicity. NAC as a thiol containing hepatoprotective agent alleviated methimazole-induced hepatotoxicity in mice due to its effects on reactive metabolites.
Acknowledgments
This study was carried out at Drug Applied Research Center of Tabriz University of Medical Sciences. The authors thank Drug Applied Research Center for providing financial supports and facilities to carry out this investigation. Students’ research committee in Tabriz University of Medical sciences supported this investigation. The authors thank students’ research committee for their support.
Conflict of Interest
The authors report no conflicts of interest.
References
- 1.Woeber KA. Methimazole-induced hepatotoxicity. Endocr Pract . 2002;8(3):222–4. doi: 10.4158/EP.8.3.222. [DOI] [PubMed] [Google Scholar]
- 2.Genter MB, Deamer NJ, Blake BL, Wesley DS, Levi PE. Olfactory toxicity of methimazole: dose-response and structure-activity studies and characterization of flavin-containing monooxygenase activity in the Long-Evans rat olfactory mucosa. Toxicol Pathol . 1995;23(4):477–86. doi: 10.1177/019262339502300404. [DOI] [PubMed] [Google Scholar]
- 3.Mizutani T, Murakami M, Shirai M, Tanaka M, Nakanishi K. Metabolism-dependent hepatotoxicity of methimazole in mice depleted of glutathione. J Appl Toxicol . 1999;19(3):193–8. doi: 10.1002/(sici)1099-1263(199905/06)19:3<193::aid-jat553>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Bergstrom U, Giovanetti A, Piras E, Brittebo EB. Methimazole-induced damage in the olfactory mucosa: effects on ultrastructure and glutathione levels. Toxicol Pathol . 2003;31(4):379–87. doi: 10.1080/01926230390201101. [DOI] [PubMed] [Google Scholar]
- 5.Xie F, Zhou X, Genter MB, Behr M, Gu J, Ding X. The tissue-specific toxicity of methimazole in the mouse olfactory mucosa is partly mediated through target-tissue metabolic activation by CYP2A5. Drug Metab Dispos . 2011;39(6):947–51. doi: 10.1124/dmd.110.037895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heidari R, Babaei H, Eghbal M. Mechanisms of methimazole cytotoxicity in isolated rat hepatocytes. Drug Chem Toxicol. 2013;36(4):403–11. doi: 10.3109/01480545.2012.749272. [DOI] [PubMed] [Google Scholar]
- 7.Bergman U, Brittebo EB. Methimazole toxicity in rodents: covalent binding in the olfactory mucosa and detection of glial fibrillary acidic protein in the olfactory bulb. Toxicol Appl Pharmacol . 1999;155(2):190–200. doi: 10.1006/taap.1998.8590. [DOI] [PubMed] [Google Scholar]
- 8.Mizutani T, Yoshida K, Murakami M, Shirai M, Kawazoe S. Evidence for the involvement of N-methylthiourea, a ring cleavage metabolite, in the hepatotoxicity of methimazole in glutathione-depleted mice: structure-toxicity and metabolic studies. Chem Res Toxicol . 2000;13(3):170–6. doi: 10.1021/tx990155o. [DOI] [PubMed] [Google Scholar]
- 9.Huq F. Molecular Modelling Analysis of the Metabolism of Methimazole. J Pharmacol Toxicol . 2008;3(1):11–9. [Google Scholar]
- 10.Sahu S, Rani Sahoo P, Patel S, Mishra BK. Oxidation of thiourea and substituted thioureas: a review. J Sulfur Chem. 2011;32(2):171–97. [Google Scholar]
- 11.Mansuy D, Dansette PM. Sulfenic acids as reactive intermediates in xenobiotic metabolism. Arch Biochem Biophys . 2011;507(1):174–85. doi: 10.1016/j.abb.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Drew R, Miners JO. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem Pharmacol . 1984;33(19):2989–94. doi: 10.1016/0006-2952(84)90598-7. [DOI] [PubMed] [Google Scholar]
- 13.Valoti M, Fusi F, Frosini M, Pessina F, Tipton KF, Sgaragli GP. Cytochrome P450-dependent N-dealkylation of L-deprenyl in C57BL mouse liver microsomes: effects of in vivo pretreatment with ethanol, phenobarbital, beta-naphthoflavone and L-deprenyl. Eur J Pharmacol . 2000;391(3):199–206. doi: 10.1016/s0014-2999(00)00078-9. [DOI] [PubMed] [Google Scholar]
- 14.Moezi L, Heidari R, Amirghofran Z, Nekooeian AA, Monabati A, Dehpour AR. Enhanced anti-ulcer effect of pioglitazone on gastric ulcers in cirrhotic rats: the role of nitric oxide and IL-1beta. Pharmacol Rep . 2013;65(1):134–43. doi: 10.1016/s1734-1140(13)70971-x. [DOI] [PubMed] [Google Scholar]
- 15.Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman's reagent. Anal Biochem . 1968;25(1):192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 16.Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem . 1978;86(1):271–8. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- 17.de Oliveira IMV, Fujimori E, Pereira VG, Lima AR. Relationship between liver gamma-glutamyltranspeptidase activity and glutathione content in chronic-malnourished pups of adolescent rats. Nutr Res . 2000;20(1):103–11. [Google Scholar]
- 18.Vogt BL, Richie JP, Jr. Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem Pharmacol . 2007;73(10):1613–21. doi: 10.1016/j.bcp.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masubuchi Y, Nakayama J, Sadakata Y. Protective effects of exogenous glutathione and related thiol compounds against drug-induced liver injury. Biol Pharm Bull . 2011;34(3):366–70. doi: 10.1248/bpb.34.366. [DOI] [PubMed] [Google Scholar]
- 20.Gillissen A, Scharling B, Jaworska M, Bartling A, Rasche K, Schultze-Werninghaus G. Oxidant scavenger function of ambroxol in vitro: a comparison with N-acetylcysteine. Res Exp Med (Berl) . 1997;196(6):389–98. doi: 10.1007/BF02576864. [DOI] [PubMed] [Google Scholar]
- 21.Heidari R, Babaei H, Eghbal MA. Cytoprotective effects of organosulfur compounds against methimazole-induced toxicity in isolated rat hepatocytes. Adv Pharm Bull . 2013;3(1):135–42. doi: 10.5681/apb.2013.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erve JC. Chemical toxicology: reactive intermediates and their role in pharmacology and toxicology. Expert Opin Drug Metab Toxicol . 2006;2(6):923–46. doi: 10.1517/17425255.2.6.923. [DOI] [PubMed] [Google Scholar]
- 23.Mehta R, Wong L, O'brien PJ. Cytoprotective mechanisms of carbonyl scavenging drugs in isolated rat hepatocytes. Chem Biol Interact . 2009;178(1-3):317–23. doi: 10.1016/j.cbi.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Iba MM, Fung J, Thomas PE, Park Y. Constitutive and induced expression by pyridine and beta-naphthoflavone of rat CYP1A is sexually dimorphic. Arch Toxicol . 1999;73(4-5):208–16. doi: 10.1007/s002040050608. [DOI] [PubMed] [Google Scholar]
- 25.Runge D, Kohler C, Kostrubsky VE, Jager D, Lehmann T, Runge DM. et al. Induction of cytochrome P450 (CYP)1A1, CYP1A2, and CYP3A4 but not of CYP2C9, CYP2C19, multidrug resistance (MDR-1) and multidrug resistance associated protein (MRP-1) by prototypical inducers in human hepatocytes. Biochem Biophys Res Commun . 2000;273(1):333–41. doi: 10.1006/bbrc.2000.2902. [DOI] [PubMed] [Google Scholar]