

Abstract
Purpose
Despite the use of 5-FU-based adjuvant treatments, a large proportion of high risk stage II/III colorectal cancer (CRC) patients will relapse. Thus, novel therapeutic strategies are needed for early stage CRC. Residual micrometastatic disease from the primary tumour is a major cause of patient relapse.
Experimental Design
In order to model CRC tumour cell invasion/metastasis, we have generated invasive (KRASMT/KRASWT/+chr3/p53-null) CRC cell subpopulations. Receptor tyrosine kinase (RTK) screens were used to identify novel proteins which underpin the migratory/invasive phenotype. Migration/invasion was assessed using the XCELLigence system. Tumours from patients with early stage CRC (N=336) were examined for AXL expression.
Results
Invasive CRC cell subpopulations showed a transition from an epithelial-to-mesenchymal like phenotype with significant increases in migration, invasion, colony-forming ability and an attenuation of EGFR/HER2 autocrine signalling. Receptor tyrosine kinase arrays showed significant increases in AXL levels in all invasive sub-lines. Importantly, 5-FU treatment resulted in significantly increased migration and invasion and targeting AXL using pharmacologic inhibition or RNAi approaches, suppressed basal and 5-FU-induced migration and invasion. Significantly, high AXL mRNA and protein expression were found to be associated with poor overall survival in early stage CRC tissues.
Conclusions
We have identified AXL as poor prognostic marker and important mediator of cell migration/invasiveness in CRC. These findings provide support for the further investigation of AXL as a novel prognostic biomarker and therapeutic target in CRC, in particular in the adjuvant disease where EGFR/VEGF-targeted therapies have failed.
INTRODUCTION
Approximately 65% of all colorectal cancer (CRC) patients are initially diagnosed with early stage (stage II/III) CRC. 5-Fluorouracil (5-FU) or 5-FU/oxaliplatin chemotherapy following surgery is the current standard-of-care for patients with high risk stage II and stage III CRC respectively (1), however a large proportion of these patients will not derive any benefit from these treatments. The goals of adjuvant treatment are to eradicate residual local disease not removed by surgery and circulating micrometastatic disease. Epithelial-mesenchymal transition (EMT) has been suggested to play an important role in the initial invasion step during cancer metastasis (2). The success of standard adjuvant chemotherapy treatment may be limited as chemotherapeutic drugs preferentially kill proliferating cells, whereas residual micrometastatic disease may be non-proliferative or dormant (3).
Recent data from adjuvant phase III trials using the anti-VEGF monoclonal antibody (mAb) bevacizumab, or the EGFR mAb cetuximab have shown that, in contrast to the metastatic setting (4, 5), these agents do not prolong disease free survival (DFS) in stage II/III CRC (6, 7). These results suggest that clinical advances and drug development in the metastatic setting cannot necessarily be directly translated to early stage disease and suggest that primary tumours and metastatic lesions may represent distinct diseases driven by different tumour biology.
A number of studies have been focussing on the identification of the promoters and suppressors of invasion/metastasis and therapeutic modulation of these pathways. A recent study reported the acquisition of autocrine FGFR- or PDGFR-signalling in NSCLC cells that have undergone an EMT transition, supporting the development of FGFR- or PDGFR-targeted therapies to reduce NSCLC tumour metastasis and progression (8). Other studies have identified salinomycin and the HDAC inhibitor SAHA as potential novel therapies for eradicating EMT-like/cancer stem cell-like cells (9, 10), however none of these studies have yet been translated into the clinical setting.
AXL is a member of the TAM (Tyro3, AXL, MER) receptor tyrosine kinase (RTK) family and was originally isolated as a transforming gene in cells from patients with chronic myeloproliferative disorders (11, 12). This subfamily is characterized by an extracellular domain, consisting of two immunoglobulin-like domains and two fibronectin type III (FNIII) motifs, a single transmembrane domain and an intracellular tyrosine kinase domain (13). AXL and MER share the vitamin K-dependent ligand GAS6 (growth-arrest-specific 6). Binding of GAS6 to AXL results in receptor dimerization, autophosphorylation of the tyrosine residues 779, 821 and 866 and recruitment of adaptor molecules. In other cases, ligand-independent dimerization and activation can also occur (14). AXL is ubiquitously expressed and detected in a wide variety of cells such as macrophages, platelets and endothelial cells (15). Subsequent to its identification in chronic myelogenous leukemia, overexpression of AXL has been reported in a wide variety of cancers, such as breast, lung and brain tumours (16-18).
Herein, we have generated progressively more invasive/metastatic cell populations from the KRASMT HCT116, KRASWT HCT116 (HKH-2), HCT116+Chr3 and HCT116-p53-null CRC cell lines, which exhibited morphological, phenotypic and molecular characteristics consistent with EMT. Using a RTK array, RNAi and small molecule inhibitor studies, we have identified AXL as a major regulator of migration/invasion and EMT. In addition, AXL expression was found to be a prognostic biomarker of poor overall survival (OS) in early stage CRC tissues. Taken together, AXL-targeted therapies may represent a promising novel approach to prevent CRC recurrence and disease progression in early stage CRC.
MATERIALS AND METHODS
Materials
AZ13032202 was obtained from AstraZeneca (Macclesfield, UK), R428 (BGB324) (19) from BergenBio (Bergen, Norway) and GAS6 from R&D systems (Abingdon, UK). siRNAs targeting AXL (AXL_9, _10, _12 and _13), were purchased from Qiagen (Crawley, UK).
Cell culture
Authentication and culture of HCT116, HKH-2 and LoVo CRC cells have previously been described (20, 21). DLD-1 cells, provided by Senji Shirasawa in 8/2008, were maintained in DMEM and authenticated by SNP profiling in January 2011. The LoVo (2004) cells were obtained from the European Collection of Cell Cultures. HCT116-p53-null and HCT116+chr 3 cells were provided by B. Vogelstein (Johns Hopkins University School of Medicine, Baltimore) and CR. Boland (University of Michigan Medical School, Ann Arbor) respectively. COLO205 cells (2012) were obtained from the American Type Culture Collection (Authentication by short tandem repeat (STR) profiling/karyotyping/isoenzyme analysis) and maintained in RPMI.
Selection of Invasive Cells using Boyden Chambers
BD BioCoat Matrigel Invasion Chambers (BD Biosciences) were used to isolate invasive subpopulations from CRC cells. 10% DMEM was added in the lower chamber. CRC cell suspension in serum-free DMEM was seeded into the upper chamber.
Western blotting
Western blot analysis have previously been described (21, 22). Anti-E-cadherin (BD transduction laboratories), anti-SNAIL (Cell Signaling) anti-STAT3 (Cell Signaling) and anti-CD44 (Invitrogen) mouse monoclonal antibodies were used in conjunction with a HRP-conjugated sheep anti-mouse secondary antibody. Anti-AXL (Cell Signaling) and anti-p-STAT3 (Y705, Cell signaling) rabbit polyclonal antibodies were used in conjunction with an HRP-conjugated anti-rabbit secondary antibody.
Immunofluorescence
3×105 cells were seeded overnight onto sterile coverslips. Cells were washed for 3×5min in PBS and fixed in ice cold 50:50 MeOH/Acetone. Cells were permeabilised and blocked in AB buffer (0.1% triton X-100, 5% goat serum, 0.2% milk in PBS) for 1h followed by incubation in primary antibody (E-Cadherin BD, 1:1000) for 1h. Slides were then washed in AB buffer 3 times and incubated with goat anti-mouse Alexa Fluor 488 (Invitrogen Molecular Probes, 1: 1000) for 1 h at RT, followed by washing as before. Counterstaining was with DAPI DNA stain (Sigma Aldrich, 0.1μg/ml) before mounting in Vectashield.
Real-time reverse transcription-PCR analysis
Total RNA and reverse transcription was carried out as previously described (23).
Cell Viability and Clonogenic Survival assays
Cell viability assays were done as previously described (22, 23). Representative results of at least 3 independent experiments are shown.
ELISA
TGF-α, amphiregulin and GAS6 ELISA assays were carried out as previously described (21).
siRNA transfections
siRNA transfections were done as previously described (20).
In vitro Migration and Invasion assays
Cell migration and invasion rates were assessed using the CIM-plate 16 and the XCELLigence system (Roche Applied Sciences), according to the manufacturer’s instructions.
Generation of inducible AXL-silenced CRC cell lines
HCT116 cells were transfected with 1μg of pTRIPZ plasmid encoding Tet-inducible shRNA against AXL (Open Biosystems, Lafayette, United States) using GeneJuice transfection reagent (Novagen). Stably transfected cells were selected, maintained in medium supplemented with 0.5μg/mL puromycin (Life Technologies, Inc.) and induced with 2μg/ml doxycycline (Sigma-Aldrich, UK).
Analysis of publicly-available CRC datasets
To assess the statistical association between AXL gene expression and clinical outcome, a publicly-available CRC microarray dataset (NCBI GEO database, accession GSE17536 (24)) was accessed. The GSE17536 cohort consists of 177 patients with overall survival, disease-free survival and disease-specific survival times and event/censoring status. Patients had a median age of 65.5 ± 13.1 years (25). There were 54.2% and 45.8% male and female respectively. 13.6% (N=24), 32.2% (N=57), 32.2% (N=57) and 22% (N=39) had stage I, stage II, stage III and stage IV CRC tumours respectively, and 9.0% (N=16), 75.7% (N=134) and 15.3% (N=27) had well, moderately and poorly differentiated tumours respectively. Normalised/transformed expression intensities for the AXL gene (probeset 202686_s_at) were extracted. Median OS was 134.86 months (73 events), disease-specific survival: 134.86 months (55 events); and DFS: undefined (36 events, 32 patients with no information). The expression data was in normalised, log-transformed format. Tertile ranges for AXL expression values were identified. All patient samples were then allocated to one of three categories: AXL-LOW, AXL-MEDIUM and AXL-HIGH. Survival curves, comparing AXL-LOW (black) with AXL-HIGH (grey) expression groups for stage II/III were estimated with the Kaplan Meier method and compared by the log-rank test, using GraphPad Prism version 5 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com.
Clinicopathological data from the patients for tissue microarray
The study cohort consisted of 509 CRC cases who received resection of the primary tumour at the National University Hospital of Singapore between 1990 and 1999 (26). The available clinical and pathological details are displayed in Supplementary table S1. There were 7% (N=37), 66% (N=337) and 27% (N=135) patients with stage I, stage II/III and stage IV disease. This work was approved by the ethics committee of the National University of Singapore (NUS-IRB 131–05-017). The construction of the tissue microarray was previously described (26). Methods of immunohistochemistry (IHC) have previously been described for CD133 (26), p53 (26), KI67 (26), CD44 (27) and LGR5 (28). KRAS (codon 12/13) and BRAF (codon 600) mutational status was carried out as previously described (29, 30). In this study, we used anti-AXL antibody (Rabbit polyclonal, Sigma, 1:1000). Staining intensity was graded as 0 (no staining), 1 (weak staining), 2 (moderate staining) and 3 (strong staining). Analysis of staining in the normal colonic epithelium showed predominant absence or mild staining. In the CRC samples, grade 0-1 stain was classified as low expression, and grade 2-3 as high expression. Scoring was done independently by Tingting Wang and Supriya Srivastava.
Statistical analysis
All statistical analyses were performed using the SPSS package (version 15.0 for Windows, SPSS Inc., USA) with significance set at the 5% level. To test for correlations between the clinical features and the incidence of a respective mutation or immunohistochemical expression, we used the Kendall tau-b test for non-parametric parameters and Fisher’s exact test. Survival curves were plotted using the Kaplan-Meier method and compared using the log-rank test. The unpaired two-tailed t-test was used to determine statistically significant differences between treatment effects. Significance was defined as p<0.05. For univariate/multivariate analysis of Singapore data-set, see supplementary methods.
RESULTS
Selection and characterization of invasive CRC sub-populations
In order to identify targets which control invasion and metastasis in CRC, we generated a preclinical invasive CRC model using Matrigel Invasion Chambers (MIC). After 72-96h incubation, the cells that invaded through to the bottom chamber were collected and designated as ‘Invasive 1’ (I1). These cells were propagated and repeatedly passed through the MIC until highly invasive HCT116-I6 cells were selected (Supplementary Fig. S1). Using the XCELLigence system, we found significant increases in migration and invasion rates in the invasive HCT116 sub-lines, with a 4-fold and 20-fold increase in migration and invasion rate of HCT116-I6 cells compared to the parental HCT116 cells respectively (Fig. 1A-B). Importantly, the increased migratory/invasion capacity seen in HCT116-I6 cells was a stable event and was not due to increased proliferation rates (Supplementary Fig. S2A-B).
Figure 1. Characterization of invasive sub-populations of CRC cell lines.
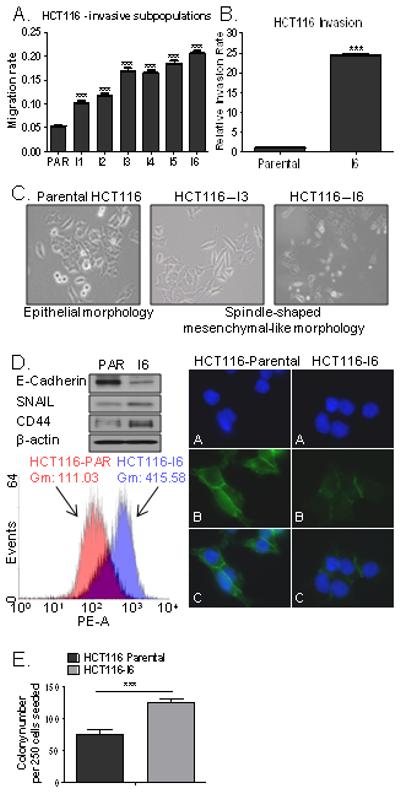
BD BioCoat MIC were used to isolate invasive subpopulations (I1-I6) from CRC cells. A. Migration assay of invasive HCT116 sublines compared to the parental HCT116 cells using the XCELLigence system. B. Real-time monitoring of HCT116 and HCT116-I6 invasion using the xCELLigence System. C. Images of morphology of the HCT116 parental and invasive HCT116-I3 and HCT116-I6 sublines. D. Left upper panel: Expression levels of E-cadherin, SNAIL and CD44 in parental (PAR) and invasive (I6) HCT116 cells. Left lower panel: Flow cytometry analysis of CD44 cell surface expression in HCT116 parental and invasive (I6) cells. Gm represents value of geometric mean. Right panel: Immunofluorescent images of HCT116 and HCT116-I6 cells. Green staining indicates E-Cadherin (B and C), DAPI staining (blue) indicates nuclei (A and C). E. Colony formation in HCT116 parental cells and invasive subline (HCT116-I6).
Notably, in contrast to the parental HCT116 cells, which displayed an epithelial morphology, we found that the majority of invasive HCT116 (I3-I6) cells had transitioned to a spindle-shaped mesenchymal-like morphology (Fig. 1C; S2C). In keeping with the morphological changes of EMT, we observed a loss of the epithelial marker E-cadherin and gain of the mesenchymal marker SNAIL in HCT116-I6 cells compared to the parental HCT116 cell line (Fig. 1D). Interestingly, we found that the expression of CD44, a marker associated with CRC stem cells (31), was ~4 fold higher in invasive HCT116-I6 cells than in control cells (Fig. 1D). In order to assess the tumour initiating abilities of the selected invasive sub-lines, we carried out colony-forming assays and found a ~2 fold increased colony number in HCT116-I6 cell line compared to the parental HCT116 cells (Fig. 1E).
The AXL receptor tyrosine kinase is overexpressed in CRC cells with increased migratory and invasive potential
In order to identify the signalling mechanisms which drive the EMT phenotype in our CRC models, we assessed the phosphorylation status of 42 RTKs and found increased phosphorylation levels of the RTK AXL in the invasive HCT116-I3 cells compared to its parental cell line (Supplementary Fig. S2D). Validation of our array results by co-immunoprecipitation and Western blotting showed that constitutive phosphorylation of AXL was significantly increased in the invasive HCT116-I6 cells compared to its parental cell line, and this was associated with increased AXL protein and mRNA transcript levels (Fig. 2A-B). We next assessed activation of EGFR family and IGF-1R and also measured release of the prototypical EGFR-ligands TGF-α and amphiregulin in the culture medium of parental and invasive HCT116 cells. Interestingly, TGF-α and amphiregulin levels and basal activity of EGFR, HER2 and IGF-1R were markedly decreased in the invasive HCT116 I1-I6 sub-lines (Fig. 2B; S2E), suggesting that a switch away from epithelial RTK-signalling is an early event in the acquisition of an invasive, mesenchymal phenotype in CRC and that this is accompanied by increased expression of AXL. Notably, expression of GAS6 was undetectable in parental and invasive HCT116 cells (Supplementary Fig. S2F).
Figure 2. AXL is up-regulated in invasive CRC sublines.
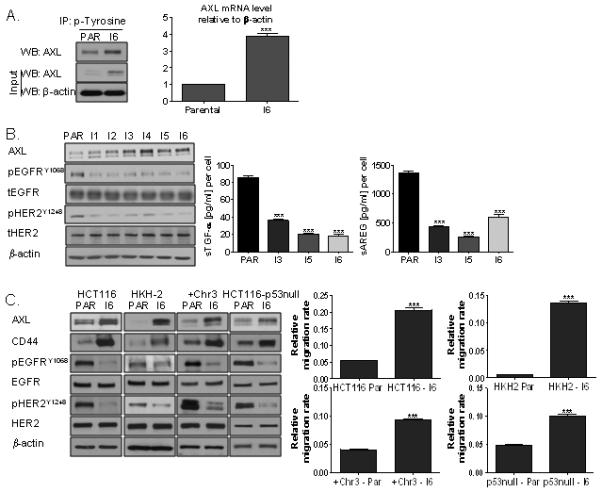
A. Left panel: Lysates from HCT116 parental and invasive (I6) cells were immunoprecipitated (IP) with anti-phosphotyrosine antibody and then immunoblotted (WB) for AXL. Right panel: SYBR Green Q-PCR analysis of AXL mRNA expression in parental and invasive (I6) HCT116 cells. Relative mRNA expression was calculated using the DDCt method with normalisation to β-actin. B. Left panel: AXL, pEGFRY1068, EGFR, pHER2Y1248 and HER2 expression levels in HCT116 parental and invasive sublines. Right panel: Shedding of TGF-α and amphiregulin (AREG) into the culture medium of parental and invasive HCT116 cells was measured. C. Left panel: AXL, CD44, pEGFRY1068, EGFR, pHER2Y1248 and HER2 expression levels in HCT116, HKH-2, HCT116+Chr3, and HCT116-p53-null parental (PAR) and invasive sublines (I6). Right panel: Migration assay in HCT116, HKH-2, HCT116+Chr3 and HCT116-p53-null parental and invasive sublines was performed using the XCELLigence system.
In order to rule out genotype-specific effects, invasive daughter lines were generated from KRASWT-HCT116 (HKH-2) (32), hMLH1-reconstituted MMR proficient HCT116 (HCT116+Chr3) (33) and isogenic p53-null (HCT116-p53-null) (34) cells (Supplementary Fig. S1). Similar to the data obtained in the KRASMT, p53WT, MMR deficient HCT116 cell line, we found significant increases in the migration rates of invasive HKH-2, HCT116+Chr3 and HCT116-p53-null sub-lines compared to their parental cells, and this was associated with marked increased AXL and CD44 expression and decreases in EGFR and HER2 activity (Fig. 2C).
AXL regulates CRC cell migration and invasion
To further investigate the involvement of AXL in CRC cell migration and invasion, we used the small molecule AXL inhibitors AZ13032202 and R428. AZ13032202 and R428 inhibited AXL tyrosine phosphorylation in both HCT116 parental and invasive I6 cells (Fig. 3A) and resulted in potent decreases in STAT3, Src family kinase (SFK) and Akt activity, basally and in response to the AXL ligand GAS6 (Fig. 3A; S3A). Treatment with AZ13032202 also resulted in potent decreases in STAT3, SFK and Akt activity in the COLO205, CACO-2 and LoVo cell lines (Supplementary Fig. S3B). Importantly, AZ13032202 not only resulted in potent decreases in migration rates of parental HCT116, LoVo, DLD-1, CACO-2 and COLO205 cell lines, but also of the invasive HCT116 cells (Fig. 3B; S3C).
Figure 3. Pharmacological inhibition and silencing of AXL reduces migration of parental and invasive CRC cells.
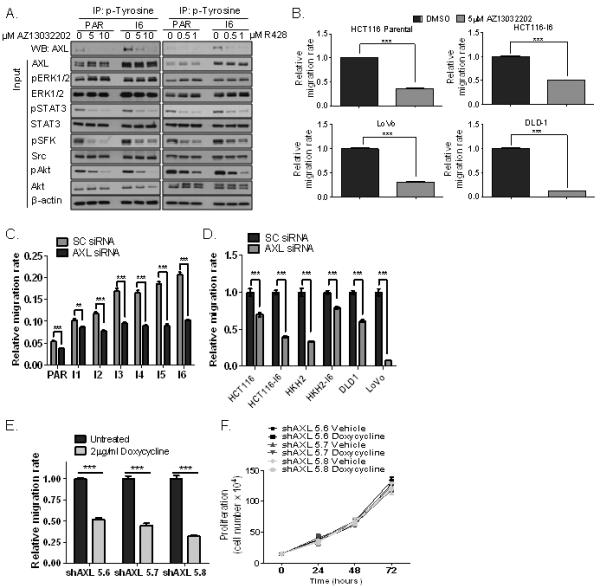
A. Lysates from HCT116 parental and invasive (I6) cells, treated with vehicle, AZ13032202 or R428 were immunoprecipitated (IP) with anti-phosphotyrosine antibody and then immunoblotted (WB) for AXL. Protein expression of AXL, pERK1/2, ERK1/2, pSTAT3, STAT3, pSFK, Src, pAkt and Akt were also analysed. B. Migration rates of parental and invasive HCT116 cells, LoVo and DLD-1 CRC cells in absence and presence of AZ13032202. C. HCT116 parental and invasive sublines (I1-I6) were transfected with 5nM AXL siRNA for 24h and the effect on migration was determined using the XCELLigence system. D. Migration of HCT116 parental and invasive (I6), HKH2 parental and invasive (I6), DLD-1 and LoVo cells, transfected with 5nM SC siRNA or 5nM AXL siRNA for 24h. E. Migration of HCT116 cells stably transfected with the AXL lentiviral pTRIPZ vector system in absence and presence of doxycycline for 72h. shAxl 5.6, 5.7, 5.8 denote different clones. F. Cell counts in HCT116 cells stably transfected with the AXL lentiviral pTRIPZ vector, 24h, 48h and 72h following incubation with vehicle or 2μg/ml doxycyclin.
To exclude any off-target effects from AZ13032202 that may affect cell migration, we analysed the effect of AXL-targeting siRNAs on migration. Silencing of AXL significantly reduced basal migration rates in parental and all invasive HCT116 cells (Fig. 3C; S4A), and these effects were not due to increased cell death or decreased cell proliferation following AXL silencing (Supplementary Fig. S4B-C). Similar data were obtained in parental and invasive HKH-2 cells, DLD-1 and LoVo CRC cell lines (Fig. 3D). We also established doxycycline-inducible AXL shRNA clones (Fig. 3E), and assessed the effect of AXL targeting on proliferation and migration. Treatment of 3 individual AXL shRNA clones with doxycycline (Dox) for 72h, resulted in potent decreases in migration rates similar to those observed with AZ13032202 and siRNAs (Fig. 3E; S4D) and this effect was not due to decreased cell proliferation in these clones (Fig. 3F). Collectively, all these data provide strong evidence that AXL is an important mediator of migration and invasion in CRC.
AXL regulates chemotherapy-induced migration and invasion
5-FU is the cornerstone of adjuvant treatment strategies for patients with early stage CRC. In addition, our group has previously shown that exposure to 5-FU can activate a number of pro-survival pathways in CRC tumours (20, 21). In view of this, we examined the effect of 5-FU treatment on CRC cell migration and invasion and found dose-dependent, significant increases in both migration and invasion in the parental HCT116 cells (Fig. 4A; S5A-B). These effects were not due to increased cell proliferation following 5-FU treatment (Supplementary Fig. S5B). Furthermore, a significant increase in invasion following 5-FU treatment was also observed in the invasive HCT116-I6 cells, but the migration rate was not further enhanced in this already highly migratory cell line (Fig. 4A; S5A and S5C). 5-FU treatment also resulted in significant increases in migration of LoVo, COLO205, CACO2 and HCT116+chr3 cells, indicating that the effect of 5-FU on cell migration is not cell line-specific and not dependent on the microsatellite instability status (Fig. 4B; S5D). Treatment with oxaliplatin resulted in further increases in migration when added to 5-FU (5-FU/oxaliplatin) in HCT116 cells (Supplementary Fig. S5E). Importantly, we found that the increased invasion and/or migratory potential following 5-FU treatment was abrogated following AXL gene silencing in parental and invasive HCT116 cells (Fig. 4A). Silencing of AXL also attenuated 5-FU-induced cell migration in LoVo and COLO205 cells (Fig. 4B). Furthermore, treatment of 2 individual AXL shRNA clones with doxycycline completely abrogated the increased migratory and invasion potential following 5-FU treatment (Fig. 4C-D). Western blot analyses demonstrated acute increased activity of STAT3, SFK and Akt following treatment with 5-FU, and this was completely inhibited upon co-treatment with AZ13032202 (Fig. 4E). Importantly, AZ13032202 treatment also abrogated the increased migration following 5-FU treatment in HCT116 CRC cells (Fig. 4E). These results highlight a central role for AXL in regulating both an intrinsic and 5-FU-induced invasive/migratory phenotype in CRC.
Figure 4. AXL regulates basal and 5-FU-induced migration and invasion in CRC cells.
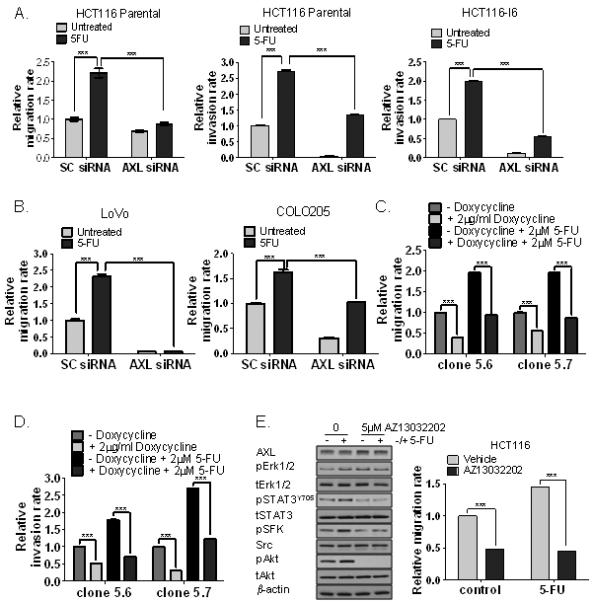
A. HCT116 parental and invasive (I6) sublines were transfected with 5nM SC siRNA or 5nM AXL siRNA for 24h and migration and/or invasion rate was determined in absence and presence of 2μM 5-FU using the xCELLigence System. B. Migration of LoVo and COLO205 cells transfected with 5nM SC siRNA or 5nM AXL siRNA in absence and presence of 2μM 5-FU. C. Migration of HCT116 cells stably transfected with the AXL lentiviral pTRIPZ vector system in absence and presence of doxycycline for 72h and treated with 2μM 5-FU. D. Invasion of HCT116 cells stably transfected with the AXL lentiviral pTRIPZ vector system in absence and presence of doxycycline for 72h and treated with 2μM 5-FU. E. Left panel: Expression of AXL, pERK1/2, ERK1/2, pSTAT3Y705, STAT3, pSFK, Src, pAkt and Akt in HCT116 treated with AZ13032202, 5-FU or co-treated AZ13032202 and 5-FU for 3h. Right panel: Migration rate of HCT116 cells treated with vehicle or AZ13032202 in the absence or presence of 2μM 5-FU.
AXL expression is a negative prognostic factor for survival in early stage CRC patients
To assess the clinical relevance of AXL expression in CRC, we initially accessed a publicly-available CRC microarray dataset (24). Importantly, analysis of AXL mRNA expression (LOW-AXL versus HIGH-AXL) in early stage (stage II/III) CRC within this dataset revealed a significant association between high AXL expression and decreased disease-specific survival (DSS, p=0.0265, N=76), disease-free survival (DFS, p=0.0336, n=74) and OS (p=0.0497, n=76) (Supplementary Fig. S6A). The multivariate analysis for AXL mRNA expression was not significant in this dataset (Supplementary Table S2).
We also investigated AXL expression in 509 (stage I-IV) CRC samples by IHC (26)(Supplementary Table S1). AXL was found to be highly expressed in CRC adenocarcinoma compared to normal colon tissue (Fig. 5A; S6B). There were no significant correlations with KRAS mutational status (p= 1.000) or BRAF mutational status (p=0.546) and AXL expression levels, supporting our preclinical findings (Supplementary Fig. S6C). There were also no significant associations with p53, Lgr5, CD133 or tumour cell proliferation by KI67 staining (Table 1A). However, in support of our preclinical invasive models, strong correlation was found between AXL expression and CD44 staining (p=0.0005) (Table 1A).
Figure 5. AXL is a strong negative prognostic factor in early stage CRC.
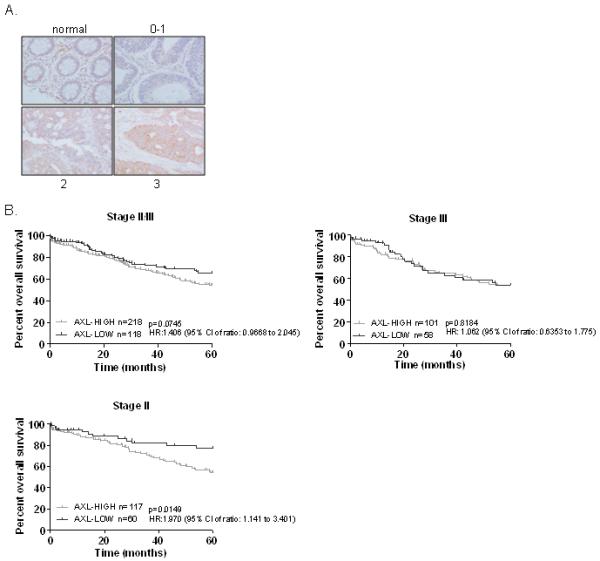
A. AXL expression in normal colon tissue and carcinoma, as determined by IHC. CRC tumours are stratified by low (0, 1) or high (2, 3) AXL expression. B. Univariate analysis of overall survival (Kaplan-Meier method) in stage II/III, stage III and stage II CRC patients using the Log-rank (Mantel-Cox) statistical test. OS was calculated from the date of surgery until date of death, censored were patients alive at date of last follow-up.
Table 1. AXL is an independent prognostic biomarker.
A. To test correlation between p53, CD44, LGR5, KI67, CD133 and AXL expression in CRC TMA, we used the Kendall taub test for non-parametric parameters and Fisher’s exact test.
B. Univariate and multivariate analyses in stage II CRC group using Cox proportional hazards regression.
| A. | ||||
|---|---|---|---|---|
|
| ||||
| Features | Low expression (n=180) |
High expression (n=329) |
Kendall’s tau |
p value |
| p53 | 0.018 | 0.711 | ||
|
| ||||
| Low expression | 92 | 162 | ||
| High expression | 88 | 167 | ||
|
| ||||
| CD44 | 0.145 | 0.0005* | ||
|
| ||||
| Low expression | 111 | 159 | ||
| High expression | 69 | 170 | ||
|
| ||||
| LGR5 | 0.079 | 0.087 | ||
|
| ||||
| Low expression | 64 | 92 | ||
| High expression | 116 | 237 | ||
|
| ||||
| KI67 | −0.035 | 0.445 | ||
|
| ||||
| Low expression | 108 | 209 | ||
| High expression | 72 | 120 | ||
|
| ||||
| CD133 | −0.080 | 0.072 | ||
|
| ||||
| Low expression | 99 | 208 | ||
| High expression |
81 | 121 | ||
| B. | ||||||||
|---|---|---|---|---|---|---|---|---|
| Stage II | Univariate | Multivariate | ||||||
| HR | CI | p-value | HR | CI | p-value | N (n) | ||
| Gender | Male | 1 | 1 | 90 (30) | ||||
| Female | 0.9787 | 0.5817-1.647 | 0.935 | 1.321 | 0.74934-2.33 | 0.3400 | 86 (27) | |
| Ethnic Group | Chinese | 1 | 1 | 152 (52) | ||||
| Non-Chinese | 0.7749 | 0.3094-1.941 | 0.586 | 0.797 | 0.30817-2.06 | 0.6400 | 24 (5) | |
| Tumour Size (mm) (Linear Term) |
Unit Increase | 1.0423 | 0.46 | 1.031962 | 0.6000 | 176 (57) | ||
| Vascular Invasion | No | 1 | 1 | 164 (54) | ||||
| Yes | 0.8992 | 0.281-2.878 | 0.858 | 1.112 | 0.30957-4.00 | 0.8700 | 12 (3) | |
| Lymphatic Invasion | No | 1 | 1 | 172 (56) | ||||
| Yes | 1.065 | 0.1472-7.709 | 0.95 | 0.507 | 0.22667-17.17 | 0.5400 | 4 (1) | |
| Perineural Invasion | No | 1 | 1 | 172 (54) | ||||
| Yes | 3.002 | 0.9324-9.662 | 0.0654 | 2.823 | 0.83632-9.53 | 0.0950 | 4 (3) | |
| AXL | Low | 1 | 1 | 59 (11) | ||||
| High | 2.217 | 1.148-4.281 | 0.0177 | 2.534 | 1.26255-5.09 | 0.0089 | 117 (46) | |
| Stage II | Multivariate (Final Model) | ||||
|---|---|---|---|---|---|
| HR | CI | P-value | N (n) | ||
| AXL | Low | 1 | 59 (11) | ||
| High | 2.217 | 1.148-4.281 | 0.0177 | 117 (46) | |
Concordance index: 0.571
Next, we assessed the prognostic value of AXL expression in the 336 early stage II/III CRC samples (subgroup of 509 CRC patients) with mature survival data. The Hazard Ratio (HR) for OS for all stage II and III patients with high AXL expression was 1.406 (95% CI: 0.9668 to 2.045), and showed a trend to statistical significance (p=0.0745) (Fig. 5B, left panel). However, when the stage II/III was broken down into individual stages, there was a significant correlation between high AXL expression and poor 5-years OS when the stage II individuals were considered alone (HR: 1.970; 95% CI: 1.141 to 3.401; p=0.0149) (Fig. 5B). In both univariate and multivariate analyses of stage II patients, the level of AXL expression was the only significant factor with higher expression correlating with poorer prognosis (Table 1B; Supplementary Table S3-S4). AXL expression levels were retained in the final multivariate model with a concordance index of 0.571 (Table 1B). Taken together, these data indicate that AXL expression is a predictor of poor clinical outcome in early stage (stage II) CRC.
DISCUSSION
Standard adjuvant therapies for patients with stage III and high risk stage II CRC consists of 5-FU/folinic acid/oxaliplatin, which also forms the control arms for most adjuvant trials. Recent phase III clinical trials failed to show any benefit from the addition of the biological agents Bevacizumab or Cetuximab to standard 5-FU/FA/oxaliplatin chemotherapy treatment in early stage CRC (6, 7). There is an urgent need to identify novel treatment strategies for early-stage CRC. In this study, we developed highly migratory/invasive CRC cell line models to identify potential novel targets/pathways driving colon cancer recurrence and metastasis. These invasive models displayed an EMT-like morphology and phenotype, high levels of the stem cell marker CD44 and increased colony-forming ability. Collectively, these data indicated that this in vitro invasive CRC model could provide new insights into understanding the biology of CRC cell invasion and metastasis.
Using a RTK array, AXL was identified to be highly activated in the invasive EMT-like CRC models. Furthermore, the increase in AXL activity in invasive/migratory CRC models was due to increase in AXL mRNA and protein expression. Consistent with a recent study in H358/doxZeb1, H358/doxSNAIL and H358/TGFβ inducible ‘metastable’ NSCLC cells (35), we found a shift from EGFR/HER2 activated autocrine signalling network to AXL signalling in KRASMT, KRASWT, +Chr3 and p53-null HCT116 cells, indicating that these effects are not dependent on the KRAS or p53 mutational or MSI-status. In contrast to recent studies in NSCLC, we found no evidence of decreased sensitivity to EGFR inhibitors in the invasive KRASWT, AXL overexpressing HKH-2 sub-line (Supplementary Fig. S7A) (36). High AXL expression has been reported in several tumours, including oesophageal (37), oral squamous cell (38), breast (16), lung (17) and malignant glioma cancers (18) and has been identified as a poor prognostic marker in these tumours. We demonstrate here for the first time a key role for AXL as a regulator of migration and invasion in CRC.
Using RNAi and shRNA against AXL and several AXL small molecule inhibitors, we further demonstrated potent inhibition of migration and invasion in a panel of parental and invasive CRC cell lines. In contrast to a previous study (9), we found no increased resistance to chemotherapy treatment, in particular 5-FU, in the EMT/stem cell like HCT116 invasive cells (Supplementary Fig. S7B). However, this is the first study showing that 5-FU treatment, the cornerstone of adjuvant treatment in CRC, significantly increases both the migratory and invasive potential of CRC cells and invasion of invasive CRC sub-lines. Importantly, we found that sh/siRNA against AXL and the small molecule inhibitor AZ13032202 abrogated the increased migratory and/or invasion potential following 5-FU treatment in parental and invasive CRC cells. Taken together, these studies would indicate that inhibitors of the AXL pathway alone or in conjunction with 5-FU chemotherapy could be a novel treatment strategy for early stage CRC. Nevertheless, other studies investigating the molecular changes associated with EMT transition have identified increased autocrine IL6-JAK2-STAT3 and PDGFR/FGFR signalling together with increased AXL/Tyro3 signalling, suggesting that pharmacological inhibition of this survival network, may result in reduced cancer progression and recurrence (35).
A number of studies have shown that AXL can signal through the MAPK, PI3K and NFκB pathways, resulting in increased cell proliferation, survival (39) and tumour growth (40) and resistance to chemotherapy (41). Consistent with previous studies in breast cancer, we found strong inhibition of basal and 5-FU-induced Akt activity following AXL inhibition (19). In addition, 5-FU treatment also resulted in increases in SFK and STAT3 activity and both basal and 5-FU-induced upregulation of pSTAT3 and SFK were potently abrogated following AXL inhibition. Several studies have shown key roles for both SFK and STAT3 signalling pathways in the growth of stem-cell like cancer cells (42, 43) and cancer cell migration (44, 45). In addition, a recent study showed that high STAT3 activity is positively associated with peritumoural lymphocytic reaction and shorter overall survival in colorectal cancer, suggesting its potential role as a therapeutic target in CRC (46). Taken together, these studies would suggest that activation of the SFK and/or STAT3 signalling pathways following 5-FU treatment leads to enhanced migratory/invasive potential, which can be reversed by AXL inhibition.
To investigate the clinical importance of AXL in CRC, we measured AXL expression in CRC tissues and found high levels of AXL in primary CRC tumours compared to normal tissue. Further investigation of AXL mRNA expression in public available datasets revealed that high AXL mRNA expression was significantly associated with poorer disease free survival (DFS), disease specific survival and overall survival in stage II/III CRC patients. Importantly, further univariate and multivariate analyses of our CRC TMA revealed also that stage II CRC patients with high AXL expression levels have a shorter 5 years OS compared to stage II patients with low AXL expression. We now plan a further retrospective validation of the prognostic role of AXL in a large cohort of stage II colon cancer samples as part of a clinical trial. The clinical management of patients with stage II CRC is a topic of debate with 5 year OS rates between 80%-85% (47). Currently, only stage II patients with poor prognostic clinical-pathological features such as lymphovascular invasion, peritoneal involvement, poor differentiation, obstruction and perforation, will receive adjuvant 5-FU treatment. There is an urgent need to identify the ~15-20% of patients who are most likely to relapse and will benefit from adjuvant treatments. A number of studies, including our own prognostic signature study (48, 49), have attempted to develop prognostic biomarkers in early stage CRC. The results herein obtained from both public available datasets and CRC TMA suggest that AXL may be a biomarker of poor prognosis in stage II CRC.
In conclusion, we have generated highly migratory/invasive CRC cell line models with EMT/stem cell like phenotype. Receptor tyrosine kinase screen identified AXL as a key regulator of CRC cell migration and invasion. Further functional validation showed that AXL regulates CRC migration and invasion in CRC cell line models, as single agent and in the context of 5-FU treatment. Importantly, AXL is highly expressed in CRC and is a poor prognostic biomarker in stage II CRC disease. The data suggest that inhibition of AXL alone or in combination with 5-FU treatment may represent a promising treatment strategy in CRC, in particular in adjuvant disease where EGFR-and VEGF-targeted therapies have failed.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Despite the use of 5-FU-based adjuvant therapies, a large proportion of high risk stage II/III (early stage/locally advanced) colorectal cancer (CRC) patients will relapse and die of metastatic disease. Therefore, novel drugs are needed that result in further increases in overall survival in early stage CRC patients. In this study, we have generated invasive/migratory CRC cell subpopulations with epithelial-to-mesenchymal like phenotype, which showed significant increases in AXL expression levels. We further show that 5-FU, the cornerstone of adjuvant treatment, results in significant increased migration and invasion, and targeting AXL, using RNAi and small-molecule inhibitor approaches, abrogated basal and 5-FU-induced migration/invasion. Importantly, high AXL expression levels were found to be associated with poor overall survival in early stage CRC tissues. Thus, these results provide evidence to support the further evaluation of AXL as a prognostic biomarker and novel therapeutic target in CRC.
Acknowledgments
Financial support from Cancer Research UK (C212/A7402); Cancer Research UK fellowship (C13749/A7261).
Footnotes
Conflicts of interest: P.G. Johnston is employed by Almac Diagnostics and has an ownership interest in both Almac Diagnostics and Fusion Antibodies. He is a consultant/advisor for, and has received honoraria from, Chugai pharmaceuticals, Sanofi-Aventis and Pfizer. All other authors have no conflicts of interest to declare.
REFERENCES
- 1.Kuebler JP, Wieand HS, O’Connell MJ, Smith RE, Colangelo LH, Yothers G, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol. 2007;25(16):2198–204. doi: 10.1200/JCO.2006.08.2974. [DOI] [PubMed] [Google Scholar]
- 2.Mego M, Mani SA, Lee BN, Li C, Evans KW, Cohen EN, et al. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int J Cancer. 2012;130(4):808–16. doi: 10.1002/ijc.26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pantel K, Schlimok G, Braun S, Kutter D, Lindemann F, Schaller G, et al. Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst. 1993;85(17):1419–24. doi: 10.1093/jnci/85.17.1419. [DOI] [PubMed] [Google Scholar]
- 4.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–17. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 6.Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol. 2011;29(1):11–6. doi: 10.1200/JCO.2010.30.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alberts SR, Sargent DJ, Nair S, Mahoney MR, Mooney M, Thibodeau SN, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. Jama. 2012;307(13):1383–93. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25(8):843–54. doi: 10.1007/s10585-008-9200-4. [DOI] [PubMed] [Google Scholar]
- 9.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One. 2011;6(8):e24099. doi: 10.1371/journal.pone.0024099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Molecular and cellular biology. 1991;11(10):5016–31. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen JW, Schulz AS, Steenvoorden AC, Schmidberger M, Strehl S, Ambros PF, et al. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6(11):2113–20. [PubMed] [Google Scholar]
- 13.Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert opinion on therapeutic targets. 2010;14(10):1073–90. doi: 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellosta P, Costa M, Lin DA, Basilico C. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Molecular and cellular biology. 1995;15(2):614–25. doi: 10.1128/mcb.15.2.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Advances in cancer research. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107(3):1124–9. doi: 10.1073/pnas.0909333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7(12):1058–64. doi: 10.1593/neo.05640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vajkoczy P, Knyazev P, Kunkel A, Capelle HH, Behrndt S, von Tengg-Kobligk H, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006;103(15):5799–804. doi: 10.1073/pnas.0510923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70(4):1544–54. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 20.Kyula JN, Van Schaeybroeck S, Doherty J, Fenning CS, Longley DB, Johnston PG. Chemotherapy-Induced Activation of ADAM-17: A Novel Mechanism of Drug Resistance in Colorectal Cancer. Clin Cancer Res. 2010;16(13):3378–89. doi: 10.1158/1078-0432.CCR-10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Schaeybroeck S, Kyula JN, Fenton A, Fenning CS, Sasazuki T, Shirasawa S, et al. Oncogenic Kras promotes chemotherapy-induced growth factor shedding via ADAM17. Cancer Res. 2011;71(3):1071–80. doi: 10.1158/0008-5472.CAN-10-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Schaeybroeck S, Kelly DM, Kyula J, Stokesberry S, Fennell DA, Johnston PG, et al. Src and ADAM-17-mediated shedding of transforming growth factor-alpha is a mechanism of acute resistance to TRAIL. Cancer Res. 2008;68(20):8312–21. doi: 10.1158/0008-5472.CAN-07-6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galligan L, Longley DB, McEwan M, Wilson TR, McLaughlin K, Johnston PG. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005;4(12):2026–36. doi: 10.1158/1535-7163.MCT-05-0262. [DOI] [PubMed] [Google Scholar]
- 24.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets--10 years on. Nucleic acids research. 2011;39:D1005–10. doi: 10.1093/nar/gkq1184. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138(3):958–68. doi: 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong CW, Kim LG, Kong HH, Low LY, Iacopetta B, Soong R, et al. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod Pathol. 2010;23(3):450–7. doi: 10.1038/modpathol.2009.181. [DOI] [PubMed] [Google Scholar]
- 27.Wang T, Ong CW, Shi J, Srivastava S, Yan B, Cheng CL, et al. Sequential expression of putative stem cell markers in gastric carcinogenesis. Br J Cancer. 2011;105(5):658–65. doi: 10.1038/bjc.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang T, Yeoh KG, Salto-Tellez M. Lgr5 expression is absent in human premalignant lesions of the stomach. Gut. 2012;61(12):1777–8. doi: 10.1136/gutjnl-2012-302372. [DOI] [PubMed] [Google Scholar]
- 29.Pang NK, Nga ME, Chin SY, Ismail TM, Lim GL, Soong R, et al. KRAS and BRAF mutation analysis can be reliably performed on aspirated cytological specimens of metastatic colorectal carcinoma. Cytopathology. 2011;22(6):358–64. doi: 10.1111/j.1365-2303.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 30.Krypuy M, Newnham GM, Thomas DM, Conron M, Dobrovic A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer. 2006;6:295. doi: 10.1186/1471-2407-6-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Todaro M, Francipane MG, Medema JP, Stassi G. Colon cancer stem cells: promise of targeted therapy. Gastroenterology. 2010;138(6):2151–62. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 32.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260(5104):85–8. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 33.Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61(13):5193–201. [PubMed] [Google Scholar]
- 34.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282(5393):1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 35.Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, et al. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis. 2011;28(2):137–55. doi: 10.1007/s10585-010-9367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–60. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hector A, Montgomery EA, Karikari C, Canto M, Dunbar KB, Wang JS, et al. The Axl receptor tyrosine kinase is an adverse prognostic factor and a therapeutic target in esophageal adenocarcinoma. Cancer Biol Ther. 2010;10(10):1009–18. doi: 10.4161/cbt.10.10.13248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee CH, Yen CY, Liu SY, Chen CK, Chiang CF, Shiah SG, et al. Axl Is a Prognostic Marker in Oral Squamous Cell Carcinoma. Annals of surgical oncology. 2012 doi: 10.1245/s10434-011-1985-8. [DOI] [PubMed] [Google Scholar]
- 39.Goruppi S, Ruaro E, Varnum B, Schneider C. Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene. 1999;18(29):4224–36. doi: 10.1038/sj.onc.1202788. [DOI] [PubMed] [Google Scholar]
- 40.Hafizi S, Dahlbäck B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine & growth factor reviews. 2006;17(4):295–304. doi: 10.1016/j.cytogfr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 41.Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008;268(2):314–24. doi: 10.1016/j.canlet.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 42.Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, et al. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011;71(23):7226–37. doi: 10.1158/0008-5472.CAN-10-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–35. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agudelo-Garcia PA, De Jesus JK, Williams SP, Nowicki MO, Chiocca EA, Liyanarachchi S, et al. Glioma cell migration on three-dimensional nanofiber scaffolds is regulated by substrate topography and abolished by inhibition of STAT3 signaling. Neoplasia. 2011;13(9):831–40. doi: 10.1593/neo.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen YS, Wu MJ, Huang CY, Lin SC, Chuang TH, Yu CC, et al. CD133/Src axis mediates tumor initiating property and epithelial-mesenchymal transition of head and neck cancer. PLoS One. 2011;6(11):e28053. doi: 10.1371/journal.pone.0028053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morikawa T, Baba Y, Yamauchi M, Kuchiba A, Nosho K, Shima K, et al. STAT3 Expression, Molecular Features, Inflammation Patterns, and Prognosis in a Database of 724 Colorectal Cancers. Clin Cancer Res. 2011;17(6):1452–62. doi: 10.1158/1078-0432.CCR-10-2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnston PG. Stage II colorectal cancer: to treat or not to treat. Oncologist. 2005;10(5):332–4. doi: 10.1634/theoncologist.10-5-332. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy RD, Bylesjo M, Kerr P, Davison T, Black JM, Kay EW, et al. Development and Independent Validation of a Prognostic Assay for Stage II Colon Cancer Using Formalin-Fixed Paraffin-Embedded Tissue. J Clin Oncol. 2011 doi: 10.1200/JCO.2011.35.4498. [DOI] [PubMed] [Google Scholar]
- 49.Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29(1):17–24. doi: 10.1200/JCO.2010.30.1077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.