

Abstract
Autophagy is the primary cellular catabolic program activated in response to nutrient starvation. Initiation of autophagy, particularly by amino acid withdrawal, requires the ULK kinases. Despite its pivotal role in autophagy initiation, little is known about the mechanisms by which ULK promotes autophagy. Here we describe a molecular mechanism linking ULK to the pro-autophagic lipid kinase VPS34. Upon amino acid starvation or mTOR inhibition the activated ULK1 phosphorylates Beclin-1 on S14, thereby, enhancing the activity of the ATG14L-containing VPS34 complexes. The Beclin-1 S14 phosphorylation by ULK is required for full autophagic induction in mammals and this requirement is conserved in C. elegans. Our study reveals a molecular link from ULK1 to activation of the autophagy specific VPS34 complex and autophagy induction.
Macroautophagy, hereafter referred to as autophagy, is a lysosomal-dependent cellular degradation process capable of generating nutrients and energy to maintain essential cellular activities upon nutrient starvation1. Accumulating evidence suggests that the ULK1 protein kinase and the VPS34 lipid kinase are key regulators of autophagy initiation and progression2-5. Mammals have two homologues of the yeast autophagy initiating ATG1 kinase, ULK1 and ULK2 (jointly referred to hereafter as ULK kinases)6. ULK1 is regulated by the nutrient and energy sensitive kinases TORC1 and AMPK7-11. The tight regulation of ULK activity by intracellular energy and nutrient levels is in keeping with a central role for autophagy in the protection of cells from starvation. Notably, mouse embryonic fibroblasts (MEF) lacking FIP200, an essential component of the ULK complex, exhibit a defect in the induction of ATG14L-ATG16-WIPI puncta upon starvation, indicating that ULK regulates localization of VPS34 to the phagophore3, 12. To date, the mechanisms leading to ULK1-mediated autophagy induction remain largely undiscovered.
VPS34 is the only class III phosphoinositide 3-kinase (PI3K) in mammals, phosphorylating phosphatidylinositol to produce phosphatidylinositol 3-phosphate (PI3P)13. Cellularly, PI3P production has been implicated in promoting autophagy and retrograde trafficking from the endosomes to Golgi5. The VPS34 kinase forms a stable complex with p150 (VPS15 ortholog). VPS34-p150 can also associate with Beclin-1, which serves as a binding partner for several proteins that are capable of either promoting (ATG14L, UVRAG, Bif1, and AMBRA-1) or inhibiting (Bcl2, BclxL, Rubicon) the autophagic function of VPS3414-22. A series of studies have identified ATG14L, to be essential for autophagic initiation and VPS34 activity at the phagophore12, 14, 15, 21, 23. Recently AMPK was described to differentially regulate VPS34 complexes in response to glucose withdrawal24. However, it is not known if or how these distinct complexes are regulated by amino acid withdrawal, a powerful inducer of autophagy in mammalian cells.
Results
ULK is required for the activation of ATG14L-associated VPS34 complexes upon amino acid withdrawal
In order to study the regulation of VPS34 kinase activity we performed PI3P lipid kinase assays on complexes immunoprecipitated using antibodies against VPS34, Beclin-1 or ATG14L from cells grown in nutrient-rich or amino acid-starved conditions. The activity of VPS34 complexes immunoprecipitated by VPS34 or Beclin-1 was reduced upon starvation, while the activity of VPS34 complexes immunoprecipitated by ATG14L was significantly increased upon amino acid withdrawal (Fig.1a). This is consistent with previous reports of decreased VPS34 kinase activity under amino acid starvation using VPS34 immunoprecipitation25-27. ATG14L levels were low in Beclin-1 and VPS34 immunoprecipitates explaining the starvation-induced decrease in VPS34 activity in these complexes despite their ability to bind ATG14L (Fig.1a,b). These data indicate that a differential regulation of VPS34 complexes exists in response to amino acid starvation. It has been reported that the autophagy specific function of VPS34 can be regulated by the disruption of the VPS34 kinase complex, under extended starvation. Starvation with Hanks’ balanced salt solution for several hours results in a phosphorylation of the nonstructured loop of Bcl-2 and dissociation from Beclin-128. Under the starvation conditions used in this study we observed no significant change in Beclin-1-VPS34 interaction, consistent with previous observations25. The activation of ATG14L-containing VPS34 complexes provides a description of a unique mode of VPS34 regulation that does not necessitate the destabilization of the VPS34-Beclin-1 interaction.
Fig.1. ULK is essential for activation of the ATG14L-associated VPS34 upon amino acid starvation.
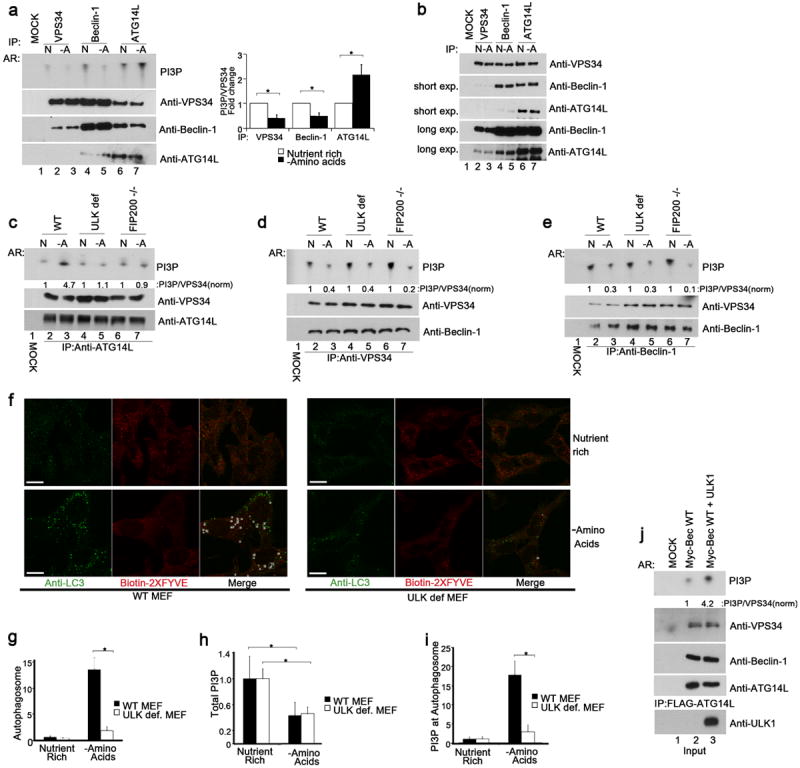
Unless otherwise stated experiments were repeated three times, data shown is representative. (a) Different VPS34 complexes were immunoprecipitated (IP) from MEF in the presence (N) or absence (-A) of amino acids using indicated antibodies and assayed for kinase activity (left, top panel). Inputs were immunoblotted using antibodies indicated (left, lower panels). Quantification of VPS34 activity is from 3 biological repeats (right panel, error bars denote S.D). (b) IP of three VPS34 complexes, normalized for VPS34, was performed using the indicated antibodies under nutrient rich or starvation conditions. VPS34 binding partners were analyzed by Western blot. (c) ATG14L-containing VPS34 complexes were immunopurified from wild-type, ULK1-/- 2KD (ULK def), and FIP200 -/- MEFs, and measured for lipid kinase activity similar to panel a. (d) VPS34 was immunopurified from wild-type, ULK def, and FIP200 -/- MEFs. Kinase assay was performed as in panel a. (e) IP of Beclin-1-containing VPS34 complexes from wild-type, ULK-def, and FIP200 -/- MEFs. Kinase assay was performed as in panel a. (f) LC3B puncta and PI3P levels were analyzed with anti-LC3B and Biotin-2XFYVE domain probe. Representative immunofluorescent images of LC3B and 2XFYVE domain binding were shown (scale bars,10μm). (g) Quantification of LC3B puncta from panel f. Details of quantification are provided in the methods section, data shown is mean -/+ S.D. Error bars in g-i are the standard deviation from a minimum of 6 unique fields of view from a representative experiment (see statistical source data). (h) Total PI3P was quantified from the experiment in panel f. Error bars was calculated as in g. (i) Quantification of PI3P that colocalizes with LC3B upon amino acid withdrawal from the experiment described in panel f. (j) VPS34 activity with or without ULK1 overexpression was assayed. A representative experiment of four repeats is shown. * in panels a,g,h,i denote a p-value <0.05 as determined by Student’s T-Test (see statistical source data and methods section). Quantification of PI3P/VPS34 provided under the PI3P panel in c-e,j nutrient rich conditions were normalized to 1.
In order to determine the requirement for ULK kinase in this activation we performed lipid kinase assays in wild-type MEF or two MEF lines deficient for ULK activity. Both ULK1 knockout MEFs with ULK2 stable knockdown (ULK-deficient MEF, described previously10) and FIP200 knockout MEF are defective for ULK activity. FIP200 is an essential cofactor for both ULK1&2, which perform redundant roles in autophagy3, 29. Interestingly, ATG14L-containing VPS34 complexes were not activated upon starvation in both ULK-deficient cell lines (Fig.1c). However, the starvation-induced repression of VPS34 complexes purified by VPS34 (Fig.1d) or Beclin-1 (Fig.1e) antibody was retained in FIP200 null and ULK deficient MEF. Therefore, ULK kinase is only required for the activation of the pro-autophagic ATG14L-containing VPS34 complex and not the repression of non-autophagic VPS34 complexes. The inability of ATG14L-containing VPS34 complexes to be activated was accompanied by a reduction in autophagy induction in ULK and FIP200-deficient cells (Fig.S1a) as well as a reduction of endogenous LC3B puncta (Fig.S1b,c).
In order to measure PI3P at the autophagosome we performed co-staining for endogenous LC3B and PI3P using an anti-LC3B antibody and a biotinylated 2XFYVE domain probe, respectively. Biotin-2XFYVE specifically binds PI3P through its two FYVE domains and has been used previously to monitor PI3P on endosomes when costained with an endosomal marker30. Under starvation the total number of PI3P puncta decreased in wild-type cells (Fig.1f,h). Conversely, the number of PI3P puncta co-localizing to autophagosomes, as marked by the LC3B staining, increased (Fig.1f,i). Similarly to wild-type MEF, ULK deficient cells showed a decrease in total PI3P puncta under starvation conditions (Fig.1f,h). In contrast ULK deficient cells showed a clear defect in the induction of PI3P-positive autophagosomes under starvation conditions (Fig.1f,i). The defect of ULK-deficient MEFs in LC3B puncta accumulation and PI3P-LC3B double positive puncta upon starvation were similarly observed in FIP200-/- MEF (Fig.S1b,c,e), which retained the ability to repress total PI3P (Fig.S1b,d). Importantly, pharmacological inhibition of VPS34 completely abolished both 2XFYVE probe labeling and amino acid starvation-induced LC3B puncta, showing the specificity of these staining (Fig.S1f). Together, these data support a critical role of ULK in regulating the autophagy specific ATG14L-containing VPS34 complex activity.
To determine if ULK1 is sufficient to activate ATG14L-containing VPS34 complexes, ATG14L was immunoprecipitated from HEK293 cells co-transfected with myc-Beclin-1, Flag-ATG14, HA-VPS34 and empty vector or HA-ULK1 under nutrient-rich conditions. Activity of the ATG14L-containing VPS34 complexes was dramatically increased upon ectopic expression of ULK1 indicating that ULK1 activates the ATG14L-containing VPS34 complexes (Fig.1j). These results demonstrate that distinct VPS34 kinase complexes are differentially regulated in response to nutrient starvation and ULK activity is critical for activation of ATG14L-containing VPS34 complexes in response to amino acid starvation.
Beclin-1 phosphorylation by ULK1 is required for activation of ATG14L bound-VPS34 in response to amino acid starvation
We sought to determine if ULK1 could directly phosphorylate any member of the VPS34 complex. ATG14L-containing VPS34 complexes were immunopurified from transfected cells and subjected to an in vitro ULK1 kinase assay using [γ32P]ATP. Autoradiography (AR) showed a single predominant band of approximately 60kDa (Fig.2a, left panel). Western blot confirmed co-migration of the AR band with Beclin-1 but not ATG14L (Fig.2a). To map the phosphorylation site on Beclin-1 we performed ULK1 in vitro kinase assays with [γ32P]ATP on various Beclin-1 deletions. ULK1 was capable of phosphorylating all truncations that shared the N-terminal 85 amino acids (Fig., S2a).
Fig.2. Beclin-1 S14 is phosphorylated by ULK1 and required for VPS34 activation in response to amino acid withdrawal.
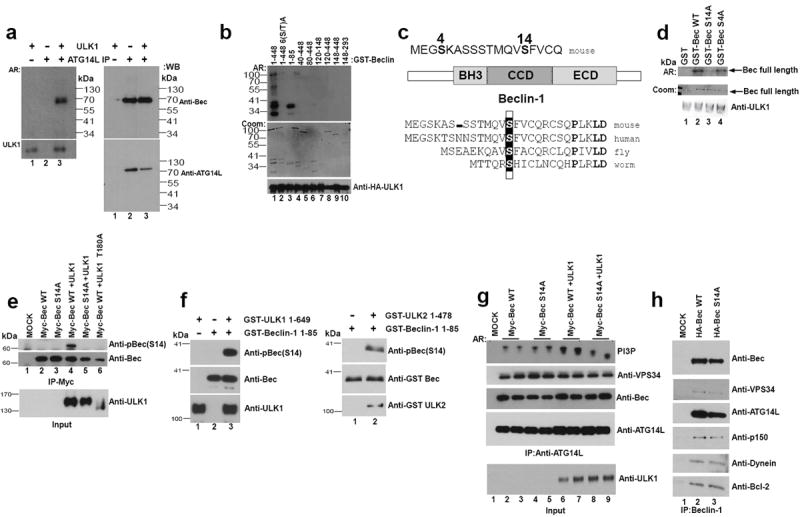
Unless otherwise stated all experiments were repeated three times and data shown is representative. (a) HEK293 cells were transfected with ATG14L, VPS34, and Beclin-1. ATG14L-containing VPS34 complexes were immunopurified and subjected to an in vitro ULK1 kinase assay in the presence of 32-P-ATP. Bound ATG14L complexes and soluble ULK1 were separated and phosphorylation was detected by autoradiography (AR, left panels). Western blot was performed (right panels). Results are representative of two unique experiments. (b) Full length murine GST-Beclin-1 and various truncations (as labeled) were subjected to an in vitro HA-ULK1 kinase assay. GST-Beclin-1 6(S-T) A has serine-threonine residues 4,7,10,14,29,42 mutated to alanine. ULK1 inputs were determined by Western, Beclin-1 inputs by Coomassie (Coom) and target phosphorylation by AR. (c) GST-Beclin-1 (1-85) was subjected to an in vitro ULK1 kinase reaction and analyzed by mass spectrometry. S14, S15in humans, (boxed) is the major phosphorylation site identified (for mass spectrometry data, see Fig.S2b,c). Mass spectrometry was performed on a single experiment. (d) Beclin-1 S14 is the major in vitro ULK1phosphorylation site. Beclin-1 WT, S4A, and S14A mutant were subjected to an in vitro ULK1 kinase assay. The reaction developed by autoradiography and stained for Beclin-1 input levels by Coomassie stain. Results are representative of two unique experiments. (e) 293 cells were transfected with the indicated plasmids under nutrient rich conditions. Beclin-1 was IP’d and immunoblotted with pBeclin-1 (S14), or anti-Beclin-1 as a loading control. ULK1 inputs are included below IP samples. (f) Purified GST-Beclin-1 (1-85) was subjected to in vitro phosphorylation by GST-ULK1 (left panel) and GST-ULK2 (right panel). Reactions were immunoblotted with the indicated antibodies. (g) 293 cells were transfected with ATG14L, VPS34, and Beclin-1 and grown under nutrient rich conditions. ATG14L-containing VPS34 complexes were IP’d and lipid kinase activity was assayed as described in Fig.1j. Inputs were immunoblotted with the indicated antibodies. Representative of four unique experiments. (h) Stable lines containing Beclin1 (WT or S14A) were used for Beclin-1 IP. Binding partners were determined by SDS-PAGE analysis and Western blot using the indicated antibodies.
We next sought to identify putative ULK1 phosphorylation sites in the N-terminus of Beclin-1 by mutagenesis and truncations. Deletion of the N-terminal 40 amino acids largely abolished ULK1-mediated phosphorylation (Fig.2b). Conserved serine and threonine residues in the N-terminus of mouse Beclin-1 were mutated to alanine (S-T(4,7,10,14,29,42)A). The Beclin-1 S-T(4,7,10,14,29,42) A mutant was not phosphorylated by ULK1 (Fig.2b, lane 2), indicating that one or more of the six residues are ULK1 phosphorylation sites. In conjunction we performed mass spectrometry analysis on an N-terminal fragment of Beclin-1 after performing an in vitro ULK1 kinase reaction. Two phosphorylation sites were detected (Fig.2c and S2b,c), one with low confidence, serine 4, and one with high confidence, serine 14, which is conserved to C. elegans (Fig.2c bottom).
The peptide encompassing conserved serine 63 was not detected by mass spectrometry so the GST-Beclin-1 1-85 S-T(4,7,10,14,29,42,63) A mutant was made. In this background alanine 4 and 14 were singly mutated back to serine. Recovery of serine 14 restored ULK-mediated phosphorylation, while recovery of serine 4 had no effect (Fig.S2d). In order to confirm the major phosphorylation site for ULK1, serine 4 and 14 were singly mutated to alanine in mouse Beclin-1. Mutation of serine 14 abolished ULK1-mediated phosphorylation while mutation of serine 4 had no effect, indicating that serine 14 (corresponding to S15 in human) is the primary ULK1 phosphorylation site in Beclin-1 (Fig.2c,d).
To determine if ULK1 phosphorylates Beclin-1 S14 in vivo we generated a phospho-specific antibody. To test the specificity of the antibody cells were transfected with Beclin-1 (wild-type or S14A) with or without ULK1 (wild-type or kinase inactive). Co-expression of the wild-type ULK1, but not a catalytically inactive mutant, induced Beclin-1 S14 phosphorylation (Fig.2e)31. As expected no phosphorylation was observed in Beclin-1 S14A (Fig.2e, lane 5). These data indicate that ULK1 can phosphorylate Beclin-1 in cells and validate the specificity of the phospho-antibody. To exclude the possibility that an ULK-associated kinase was responsible for Beclin-1 phosphorylation, we used ULK1 purified from insect cells for an in vitro kinase assay using recombinant Beclin-1 from E. coli. Immunoblot of the resulting kinase reaction showed robust Beclin-1 phosphorylation indicating that Beclin-1 is a direct target of ULK1 (Fig.2f and S2e). We asked if ULK2 is similarly capable of phosphorylating Beclin-1. ULK2 kinase purified from insect cells also phosphorylated Beclin-1 at S14, indicating a redundancy for the ULK kinases in promoting Beclin-1 phosphorylation (Fig.2f, right panel).
We next sought to determine if Beclin-1 (S14) phosphorylation was required for ULK1-mediated activation of the ATG14L-associated VPS34 lipid kinase. ATG14L was immunoprecipitated from transfected cells and an in vitro PI3P-lipid kinase assay was performed. As previously shown ULK1 cotransfection enhanced VPS34 kinase activity (Fig.2g, compare lanes 2&3 with 6&7); however, ATG14L VPS34 complexes containing mutant Beclin-1 did not respond to ULK1 co-transfection (Fig.2g, compare lanes 4&5 with 8&9). Importantly, we found that abrogation of the ULK1 phosphorylation site in Beclin-1 had no discernible effect on its ability to bind VPS34, ATG14L, p150, dynein, and Bcl2 (Fig.2h). These data indicate that direct phosphorylation of Beclin-1 on S14 by ULK1 is required for activation of the autophagy specific VPS34 kinase complex.
Serine 14 of Beclin-1 is phosphorylated by ULK kinase in response to amino acid withdrawal and mTOR inhibition
In order to determine if Beclin-1 is a physiological target of ULK1, ATG14L-associated Beclin-1 was immunopurified from wild-type MEF. Western blot analysis showed that endogenous Beclin-1 is phosphorylated upon amino acid starvation, while phosphatase treatment completely abolished Beclin-1 phospho-S14 signal (Fig.3a). ULK1 activity is potently repressed by TORC1 phosphorylation. To test if there is a correlation between Beclin-1(S14) phosphorylation and TORC1 signaling to ULK1, an amino acid withdrawal time course was performed. As expected, phosphorylation of ULK1 (S757, the TORC1 target site) decreased upon amino acid withdrawal, although more slowly than the dephosphorylation of S6K (another mTORC1 substrate) (Fig.3b). Interestingly, ULK1-mediated Beclin-1 phosphorylation inversely correlated with the inhibitory phosphorylation on ULK1 (Fig.3b). In order to determine if inhibition of TORC1 was sufficient to activate ULK1-mediated Beclin-1 phosphorylation, wild-type or FIP200-/- and ULK deficient MEF were treated with mTOR catalytic inhibitor, Torin-1, under nutrient-rich conditions or with amino acid withdrawal. Inhibition of mTOR resulted in a clear induction of Beclin-1 S14 phosphorylation only in the wild-type MEF, indicating that relief of mTOR-mediated inhibition of ULK1 stimulates downstream target phosphorylation (Fig.3c,d).
Fig.3. Beclin-1 is a physiological target of ULK kinase in response to amino acid withdrawal and mTOR inhibition.
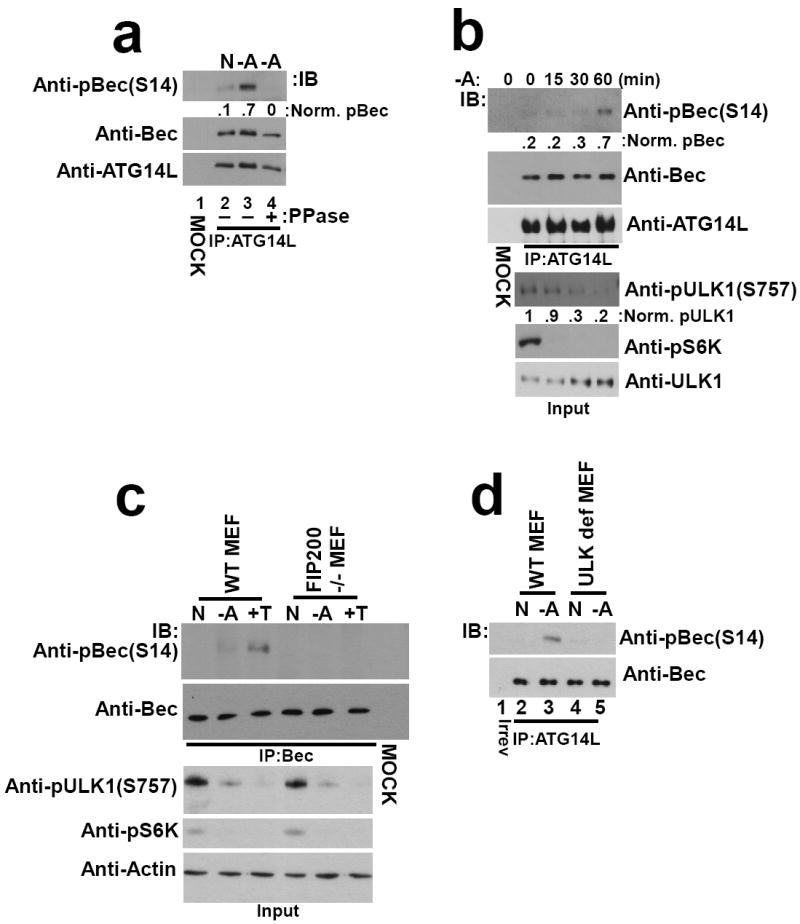
Unless otherwise stated all experiments were repeated three times and data shown is representative. (a) Wild-type MEF were cultured with or without amino acids. ATG14L-associated Beclin-1 was immunoprecipitated and treated with lambda phosphatase treatment (PPase) as indicated. Western blot was performed with the indicated antibodies. Beclin-1 S14 phosphorylation was quantified (shown under top panel) normalized to total Beclin-1. (b) Wild-type MEF were starved for the indicated time points. Beclin-1 was immunopurified by ATG14L IP and immunoblotted as indicated (top panels). Whole cell lysates were immunoblotted with pULK1 S757 (mTORC1-target site), pS6K and ULK1 antibodies (bottom panels). Two unique experiments were performed. (c) Wild-type or FIP200 -/- MEF were incubated under nutrient rich, amino acid deprived or Torin-1 (+T, an mTOR inhibitor) conditions. Beclin-1 was purified and immunoblotted as in Fig.3b. (d) Wild-type or ULK def MEF were incubated with or without amino acids. Beclin-1 was purified and immunoblotted as in Fig.3a. Two unique experiments were performed.
ATG14L promotes Beclin-1 phosphorylation by enhancing association with ULK1
The regulation of VPS34 lipid kinase activity by ULK1 is limited to the ATG14L-containing complex (Fig.1c); therefore, we asked whether ULK1 would exhibit a preference for phosphorylation of the ATG14L-bound Beclin-1. Beclin-1 alone or ATG14L-bound Beclin-1 was immunoprecipitated from transfected cells. Endogenous ATG14L co-precipitated from cells overexpressing Beclin-1 alone was negligible compared to the levels from complexes obtained from cells transfected with both Beclin-1 and ATG14L (Fig.4b). Both complexes were used as substrates for ULK1 in vitro kinase assays. We found that ATG14L-containing Beclin-1 was efficiently phosphorylated by ULK1 while Beclin-1 alone was a comparatively poor substrate (Fig.4a, b), indicating that ATG14L makes Beclin-1 a better substrate for ULK1 (Fig.4b). In order to determine if ATG14L plays a role in promotion of Beclin-1 phosphorylation in vivo we utilized an ATG14L-inducible cell line. Induction of ATG14L overexpression promoted Beclin-1 phosphorylation (Fig.4c), further supporting that ATG14L-bound Beclin-1 is the preferred target of ULK1 in vivo.
Fig.4. ATG14L stimulates Beclin-1 S14 phosphorylation by promoting association with ULK1.
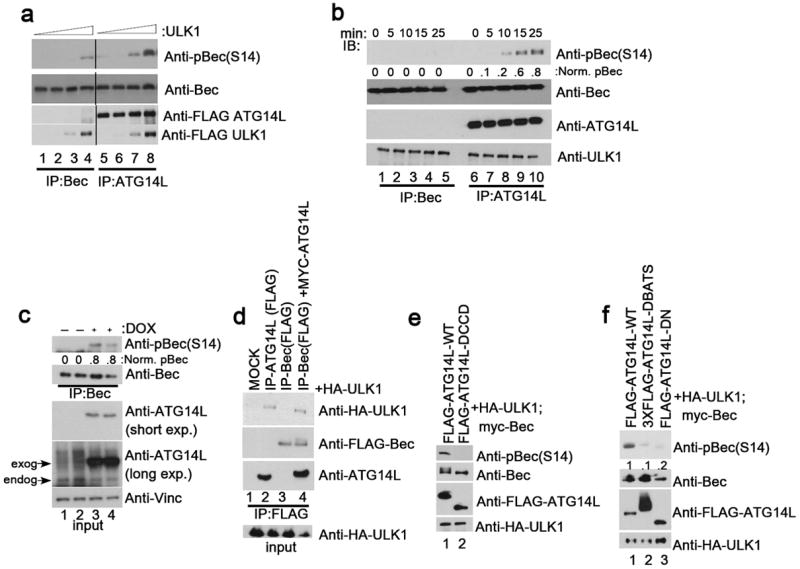
Unless otherwise stated all experiments were repeated three times and data shown is representative. (a) Beclin-1 alone (lanes 1-4) or Beclin-1 and ATG14L (lanes 5-8) were overexpressed in 293 cells. Beclin-1 was purified either by direct immunoprecipitation (lanes 1-4) or by ATG14L IP (lanes 5-8). IP samples were subjected to an in vitro ULK1 kinase assay with increasing amounts of ULK1. Reactions were immunoblotted with the indicated antibodies. Black line denotes discontinuous lanes from the same gel. Two unique experiments were performed. (b) Beclin-1 alone or bound to ATG14L was purified as described in panel a. Equal amounts of ULK1 were added to each complex and reactions were quenched at the indicated time points. Western blot was performed with the indicated antibodies. (c) An ATG14L-FLAG-6His inducible U2OS cell line was induced for 16 hours in the presence of amino acids. Endogenous Beclin-1 was immunoprecipitated and immunoblotted as in Fig.3a. ATG14L input levels are detected by immunoblotting. Two unique experiments were performed. (d) 293 cells transfected with either ATG14L or Beclin-1, or both, in conjunction with ULK1 were immunoprecipitated as indicated and blotted with the indicated antibodies. (e) 293 cells were transfected with Beclin-1 and ULK1 in the presence of ATG14L-WT or ATG14LΔCCD, which is defective in Beclin-1 binding, under nutrient rich conditions. Lysates were resolved by SDS-PAGE and blotted with the indicated antibodies. (f) 293 cells were transfected with ULK1 and Beclin-1 in conjunction with either ATG14L-WT, or one of two mutants (ΔBATS, ΔN) that are defective in phagophore localization. Samples were handled as in panel e.
To understand the mechanism underlying the ATG14L-mediated promotion of Beclin-1 phosphorylation we performed immunoprecipitation assays to determine the interaction of ATG14L and Beclin-1 to ULK1 when expressed alone or together. Interestingly, we found that immunoprecipitation of Beclin-1 pulled down ULK1 only when cotransfected with ATG14L (Fig.4d, compare lanes 3 and 4). Conversely, immunoprecipitation of ATG14L could pull down ULK1 in the absence of Beclin-1 suggesting that ATG14L may recruit Beclin-1 to ULK1 for phosphorylation (Fig.4d, lane 2). To confirm that ATG14L stimulates Beclin-1 phosphorylation by promoting an ULK1-ATG14L-Beclin-1 complex, we cotransfected ATG14L WT or ATG14LΔCCD (a Beclin-1-binding deficient mutant) with Beclin-1 and ULK114, 15. We found that the ability of ATG14 to stimulate Beclin-1 phosphorylation was completely lost in ATG14LΔCCD (Fig.4e). The mechanistic model where ATG14L acts as an adaptor to recruit ULK1 to Beclin-1 is further supported by the fact that promotion of Beclin-1 phosphorylation is preserved in the in vitro kinase reaction (Fig.4b).
Recruitment of ATG14L-containing VPS34 complexes to the phagophore requires ULK1. Therefore, we asked whether localization of ATG14L to the phagophore took place upstream or downstream of Beclin-1 S14 phosphorylation. We found that removal of the BATS (Barkor/Atg14L autophagosome targeting sequence) domain or extreme N-terminus of ATG14L, both of which are necessary for the localization of ATG14L to the phagophore 23, 32, but not binding to Beclin-1, severely compromised Beclin-1 phosphorylation when compared to WT control (Fig.4f). Previously, phosphorylation of AMBRA-1 by ULK1 was shown to be required for localization of Beclin-1 to the phagophore18, making the activation of the ATG14L-containing VPS34 kinase a downstream event. While the reported role for ULK1 in the regulation of ATG9 cycling from the trans-golgi network to endosomes can be blocked by inhibition of VPS34, putting ATG9 regulation downstream of ULK-mediated activation of ATG14L-VPS34 complexes2.
This complex and multifaceted regulation of VPS34 kinase by ULK likely underscores the pivotal role for autophagy induction in the cellular response to starvation.
UVRAG promotes phosphorylation of Beclin-1 by ULK1
Beclin-1 also binds UV radiation resistance-associated gene protein (UVRAG) to promote the maturation of the autophagosome and this complex is known to be free of ATG14L. Therefore, we asked whether UVRAG might play a similar role in promoting Beclin-1 phosphorylation as ATG14L. Indeed UVRAG bound Beclin-1 preferentially associated with ULK1 and overexpression of UVRAG promoted Beclin-1 phosphorylation (Fig.5a,b). UVRAG binds only a small minority of the total Beclin-1 and represents a minor fraction of total Beclin-1-associated VPS34 activity24. Therefore, in addition to regulating autophagy initiation through ATG14L-complexes it is possible that Beclin-1 phosphorylation may also play a role in the autophagosome maturation through regulation of the UVRAG containing VPS34 complex.
Fig.5. UVRAG promotes Beclin-1 S14 phosphorylation and association with ULK1.
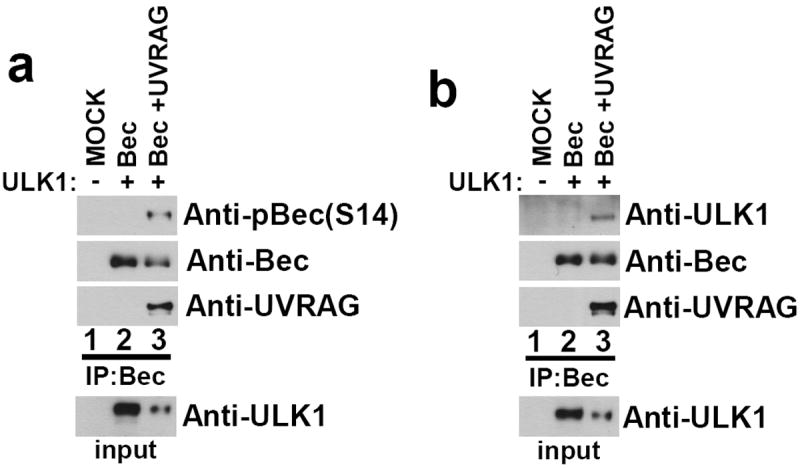
(a) 293 cells were transfected with Beclin-1, with or without UVRAG, in conjunction with ULK1 as indicated in the presence of amino acids. Lysates were immunoblotted with the indicated antibodies. A representative experiment of three repeats is shown. (b) UVRAG bridges the interaction between Beclin-1 and ULK1. 293 cells were transfected with Beclin-1, with or without UVRAG, in conjunction with ULK1 as indicated. Lysates were immunoprecipitated with anti- HA(Beclin-1) antibody and blotted with the indicated antibodies. A representative experiment of three repeats is shown.
Phosphorylation of Beclin-1 is required for induction of autophagy in response to amino acid starvation
We previously showed that ATG14L-associated VPS34 complexes containing Beclin-1 S14A could not be activated by ULK1 (Fig.2g). We tested if this lack of activation results in an overall deficiency in autophagy. Cotransfection of ULK1 with wild-type Beclin-1 and ATG14L strongly increased autophagic flux as indicated by the increase of LC3B II accumulation (Fig.6a). Comparatively, cotransfection of phospho-defective Beclin-1-S14A attenuated ULK-mediated activation of autophagic flux (Fig.6b), indicating that activation of the ATG14L-containing VPS34 complex may be required for autophagy induction.
Fig.6. Beclin-1 S14 phosphorylation plays a critical role in autophagy induction by amino acid starvation.
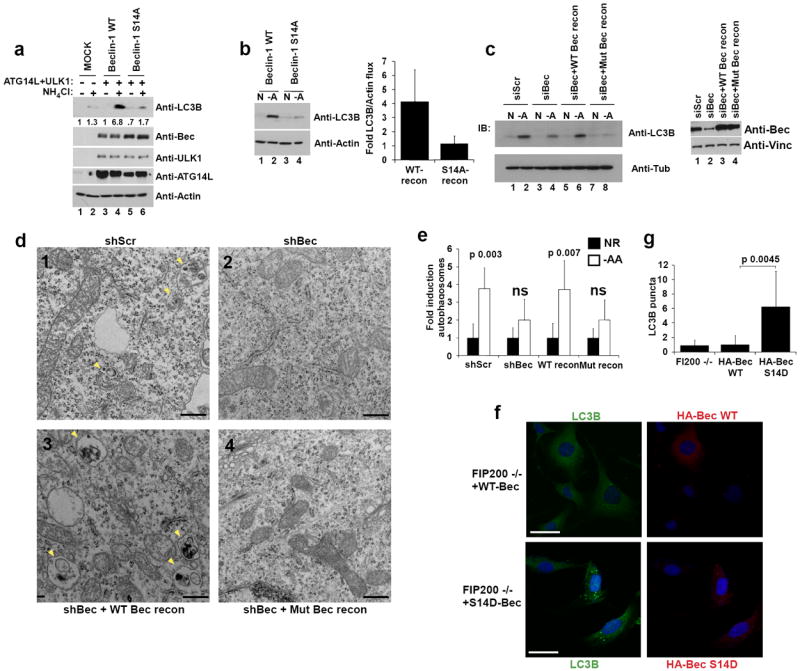
Unless otherwise stated all experiments were repeated three times and data shown is representative. (a) 293 cells were transfected with the indicated plasmids. Cells grown in the presence of amino acids and treated with NH4Cl to block autophagic turnover where indicated. Lysates were immunoblotted with the indicated antibodies. (b) 293 cells transfected with Beclin-1 ATG14L were grown in the presence or absence of amino acids. Lysates were immunoblotted with the indicated antibodies (left panel) and quantified by densitometry (right panel). Error bars represent the SD of three unique experiments. (c) shBeclin-1 reconstituted lines (wild-type or mutant) and controls (shScramble or shBeclin-1) were grown with or without amino acids were assessed for autophagy (left panel) and Beclin-1 levels (right panel). (d) Autophagosome (denoted by arrow heads) generation upon amino acid withdrawal was analyzed by electron microscopy. Cell lines and conditions from panel c were used and representative images from the indicated amino acid starved lines are shown; scale bar in bottom right represents 0.4μm. (e) Quantification of panel d. Fold induction was determined by arbitrarily making the nutrient rich condition 1 (solid bars) for each line. Error bars represent the standard deviation on the mean value over an average of 20 fields of view within a representative experiment. (f) HA-Beclin-1 WT or S14D was transiently expressed in FIP200 -/- MEF grown under nutrient rich conditions. Indirect immunofluorescence was performed using antibodies against endogenous LC3B and HA-Beclin-1. Scale bars represent 20μm. (g) Quantification of LC3B puncta from confocal displayed in panel f. In the HA-Beclin-1 or HA-Beclin-1-S14D transfected samples, only the HA staining positive cells were counted for LC3B puncta. Error bars were processed as in e. Mean value displayed, p-value determined by Student’s T-Test using 10 unique fields of view from panel f.
In order to further elucidate the role of ULK1-Beclin-1-S14 phosphorylation in the promotion of autophagy we generated knockdown-reconstitution stable cell lines (see material and methods). Cell lines expressing a scrambled non-targeting shRNA or Beclin-1 shRNA without reconstitution served as controls. Reconstituted Beclin-1 levels were comparable to that of endogenous Beclin-1 (Fig.6c). The wild-type and mutant Beclin-1 showed similar localization (Fig.S3c). Starvation of cells containing Beclin-1 knockdown or with mutant Beclin-1 reconstitution showed a reduction in LC3B II accumulation compared to shRNA-scramble or wild-type Beclin-1 reconstituted control (Fig.6c). In order to validate these findings we performed a similar experiment and analyzed the cells by electron microscopy. Consistent with previous observations we found that Beclin-S14A and shBeclin-1 lines did not produce a significant increase in autophagosomes upon amino acid starvation (Fig.6e, 6d). Conversely, shScramble and WT-Beclin-1 reconstituted lines showed a significant accumulation of autophagosomes when starved for amino acids (Fig.6d, 6e). The lack of autophagosome induction in mutant Beclin-1 reconstituted HeLa cells was also determined quantitatively by scoring of LC3B puncta accumulation (Fig.S3a,b). These data indicate that phosphorylation of Beclin-1 by ULK kinase is required for an appropriate autophagic response upon amino acid withdrawal.
We next sought to determine if autophagy could be driven by introduction of a phospho-mimetic residue at S14 of Beclin-1. HA-Beclin-1 (WT and S14D) were transiently expressed in the FIP200 -/- background. Induction of autophagy was determined by quantification of LC3B puncta in Beclin-1 expressing cells versus their non-expressing neighbors. Overexpression of wild-type Beclin-1 had no significant effect on inducing LC3B puncta (Fig.6f, top panels). However, the majority of cells expressing Beclin-1 –S14D had significantly more LC3B puncta than the FIP200 -/- background (Fig.6f, bottom panels). The induction of autophagy by the Beclin-1 S14D indicates that at least some of the ATG14L-containing complexes were capable of getting to the phagophore, possibly as a result of transient overexpression, to induce autophagy.
The ULK phosphorylation site in Beclin-1 is conserved in C. elegans and required for proper autophagy
In order to extend our observations at the organismal level and across species we used a C. elegans model system. During worm embryogenesis, the germline P granules (PGL granules) are selectively degraded by the autophagic machinery in somatic cells33. In autophagy mutant embryos, PGL granules remain in somatic cells during early embryonic divisions due to failed removal by autophagy34. To ascertain if the conserved serine 6 of BEC-1 (Beclin-1 worm homologue, Fig.2c) was also required for proper autophagic induction, we generated transgenic lines with pbec-1∷bec-1(WT or S6A)∷gfp in the bec-1(ok700) background. In wild type worms, PGL-1, a key component of PGL granules, was exclusively expressed in the two germline precursor cells (Fig.7a, arrows mark germline cells). In contrast, bec-1 and unc-51 (ULK/ATG1 homologue) worms displayed somatic cell PGL-1 staining, indicating a defect in autophagic clearance (Fig.7a). Importantly, bec-1 (WT) transgene expression fully rescued the defective degradation of the PGL-1 granules in the bec-1 background (Fig.7a). In contrast, bec-1 (S6A) transgenic embryos contained PGL-1 body staining in the somatic cells (Fig.7a,b,c). Comparable expression levels of the bec-1 transgene was validated by Western blot (Fig.7b). The PGL-1 staining phenotype in bec-1 S6A lines varied between embryos with punctuate staining in 2/3rd of embryos and diffuse staining in 1/3rd of embryos; Fig.7c). We also noted that the increased PGL-1 staining is less pronounced in bec-1 (S6A) worms than in the bec-1 worm, indicating a partial rescue of PGL body clearance by BEC-1(S6A). These data collectively demonstrate a role for both unc-51 and the conserved serine 6 of bec-1 in the autophagic clearance of protein aggregates in C. elegans embryogenesis.
Fig.7. The conserved ULK phosphorylation site in C. elegans Bec-1 is required for autophagy.
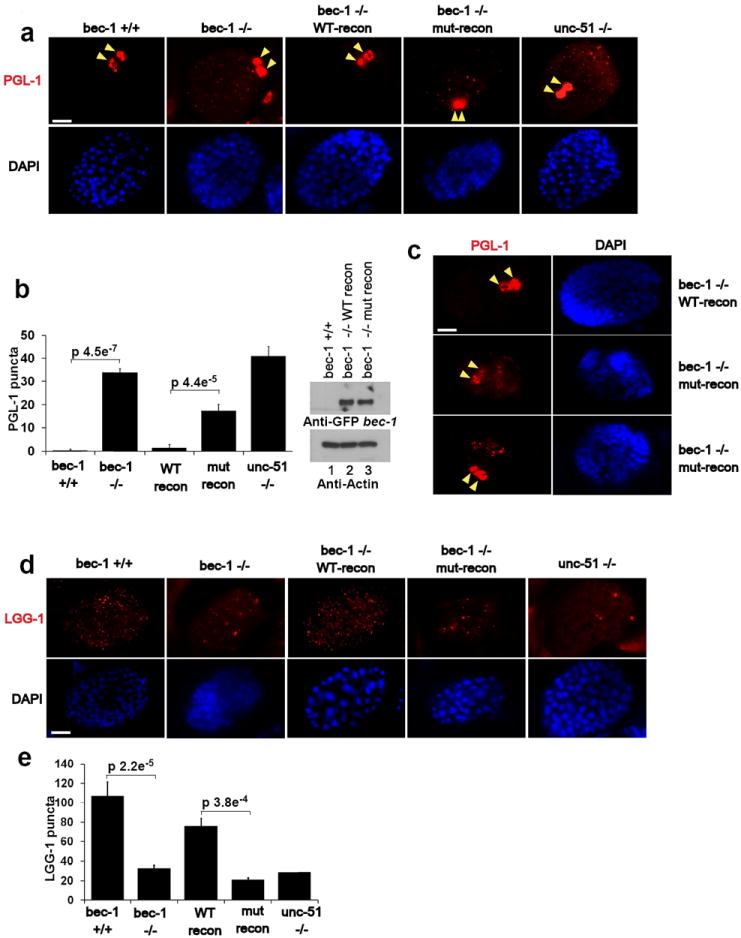
Unless otherwise stated all experiments were repeated three times and data shown is representative. (a) Bec-1 C. elegans were reconstituted with either wild-type or mutant GFP-BEC-1. Stable worm lines with Bec-1 rescue were obtained and embryos were stained with anti-PGL-1 antibody. Arrow indicates normal PGL-1 staining in germline cells. Scale bars represent 10μm. (b) Quantification of PGL-1 puncta outside germline cells (left panel). Error bars represents standard deviation between 3 unique embryos in a representative experiment. Reconstituted Bec-1 (WT and mut) levels in Bec -/- stable worms were compared by Western blot. Mean value presented. (c) Spectrum of defects in PGL granule degradation in bec-1 mutant rescue embryos. Mutant embryos displayed either high levels of diffuse PGL-1 staining (middle-left panel, 1/3 of the embryos), or large punctuate PGL-1 structures in somatic cells (bottom-left panel. 2/3 of the embryos). Both diffuse or punctuate PGL-1 staining in somatic cells have been described in autophagy deficient embryos. (d) Embryos from the lines described in Fig.6a were labeled with anti-LGG-1, along with wild-type and unc-51 worms. Representative embryos at ~100 cell stage are shown. (e) Quantification of LGG-1 per embryo from labeling in panel d. Error bars generated as in panel b. Mean value presented.
We next investigated autophagosome biogenesis in bec-1 (WT) and bec-1(S6A) rescued transgenic lines. The use of anti-LGG-1 (LC3B/ATG8 homologue) antibody in worms to distinguish levels of autophagy has been established in previous studies35. In bec-1 worms, the number of LGG-1 punctate structures was dramatically decreased, whereas the size and intensity were increased compared to the wild-type (Fig.7d,e). This phenotype was rescued by a transgenic line containing bec-1(WT) but not bec-1(S6A) (Fig.7d,e). Failure to rescue the autophagic defect in bec-1(S6A) transgenic worms is consistent with phosphorylation of S6 being crucial for the initiation of autophagosome formation. Together, our data indicate that the bec-1 (S6A) displays an autophagic defect largely overlapping that of unc-51 worms, indicating that this residue is required for robust unc-51-dependent induction of autophagy.
Discussion
Generation of PI3P at the phagophore by VPS34 is a critical early event in autophagosome formation36. In yeast, PI3P is enriched in the inner lumen of the nascent autophagosome serving as an anchor recruiting PI3P binding proteins such as ATG18 (mammalian homologues are WIPI 1,2), which may play a role in the formation of the early autophagosomal membrane37,38, 39. Interestingly, VPS34 activity has been reported to be repressed by amino acid starvation, an observation at odds with our understanding of the genesis of the autophagosomal membrane25-27. Here we provide the mechanistic understanding for the paradoxical decrease in total cellular PI3P with concomitant induction of PI3P at the autophagosomal membrane upon amino acid withdrawal. VPS34 complexes devoid of pro-autophagic adaptors ATG14L and UVRAG are repressed by the removal of amino acids in an ULK-independent manner. Conversely, the activity of ATG14L-containing VPS34 complexes is strongly induced by starvation in an ULK-dependent manner that, along with changes in localization, mediates the upregulation of PI3P at the autophagosomal membrane. Interestingly a recent report showed that AMPK is responsible for both the activation of pro-autophagic VPS34 and repression of non-autophagic complexes upon glucose starvation24. The ability of amino acid withdrawal to potently inhibit total VPS34 in the absence of ULK means that the regulation of these complexes is performed by another nutrient-sensitive pathway. Future study into the repression of total VPS34 will undoubtedly shed light on the functions of VPS34 in the starvation response.
Our study supports a model that TORC1 inhibition by amino acid starvation leads to de-repression of ULK kinase activity. The active ULK directly phosphorylates Beclin-1 S14 and activates the pro-autophagy VPS34 complexes to promote autophagy induction and maturation (Fig.8). This model reveals a continuous signaling pathway amino acids-TORC1-ULK1-VPS34-Beclin-1. The identified ULK1 phospho-site on Beclin-1 has no obvious conservation in yeast ATG6. It will be interesting to see if the link between ATG1 and ATG6 is functionally maintained in yeast, one could look at the functional conservation of TOR-ATG1 as an example. Given the complex nature of autophagy biology, the regulation of VPS34 is likely to be complex and future studies are required to have a comprehensive understanding of VPS34 regulation in response to the wide range of autophagy inducing signals.
Fig.8. A working model of VPS34 complex regulation by ULK upon amino acid withdrawal.
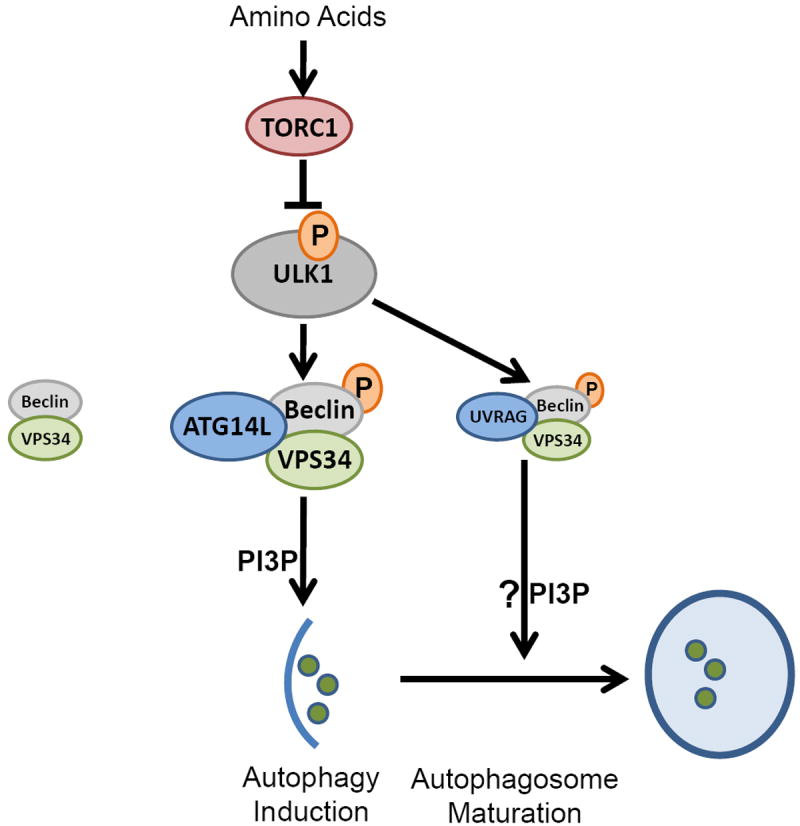
Amino acid starvation inactivates TORC1, de-repressing ULK1. ULK1 is recruited to VPS34-Beclin-1 complexes via binding to ATG14L, phosphorylating Beclin-1, activating the VPS34 kinase and PI3P production at the nascent autophagosome. Additionally, UVRAG-bound Beclin-1 is phosphorylated by ULK1, which may promote autophagosome maturation.
Online methods
Antibodies
Anti-phospho-Beclin-1 (S14) was obtained from Abbiotec, San Diego, CA (Cat#254515). Anti- LC3B (Cat#2775 for WB), Vps34 (Cat#4263 for WB), Beclin-1 (Cat#3738 for WB), ULK1(Cat#8054), pULK1 S757 (Cat#6888), and phospho-S6K (Cat#9205) were purchased from Cell Signaling Technology. Anti-Atg14L (Cat#PD026 for IP), UVRAG (Cat#M160-3) and LC3B (Cat#PM036 for immunostaining) antibodies were obtained from MBL. Immunoprecipitation was performed using Anti-Vps34 (Echelon, Cat#ZR015) and Beclin-1 (Bethyl Cat#A302-567A) antibodies. Anti-Vinculin (Cat#V9264), alpha-tubulin (Cat#T6199), Flag (Cat#F3165), and Atg14L (Cat#A6358) antibodies were obtained from SIGMA and used for Western blot. Anti-HA (Clone 16B12, Cat#MMS-101P) and Myc (Clone 9E10, Cat#MMS-150P) antibodies were obtained from Covance. Anti-actin (Cat# Ab3280) antibody was obtained from Abcam.
Cell Culture
Immortalized MEFs were cultured in high glucose DMEM supplemented with 10% FBS (Hyclone), 50ug/ml pen/strep, 1mM sodium pyruvate, 55um 2-mercaptoethanol, and 0.1mM non-essential amino acids (each obtained from Invitrogen). Amino acid free medium was made according to Gibco standard recipe omitting all amino acids and supplemented as above without addition of non-essential amino acids and substitution with dialyzed FBS (Invitrogen). 293 cells were grown in similar media omitting the 2-mercaptoethanol. Media was replenished the night before starvation experiments and again three hours before withdrawal of amino acids or addition of mTOR inhibitors. FIP200-/- MEF were a kind gift from Dr. Jun-Lin Guan, University of Michigan. ULK1 -/- ULK2 knockdown were described previously10. ATG14L-FLAG-6His inducible U2OS cell line was a kind gift from Dr. Qing Zhong, University of California Berkeley.
Generation of knock-down reconstitution lines
HeLa cells were stably infected with shRNA against Beclin-1 (target 5’ CTCAGGAGAGGAGCCATTTAT 3’) or scrambled control in the pLKO.1 lentiviral background. Lentiviral particles were made by co-transfection with psPAX2 and pMD2.G plasmids into HEK293T cells. Viral supernatants were harvested from 48-60 hours. Cleared supernatant was filtered through a 0.45 μm filter and applied every 12hrs on target cells for 3 rounds. 4 μg/ml Polybrene (Sigma) was supplemented to viral supernatants. 24 hours after final infection stable shRNA containing populations were obtained by selection with 2μg/ml puromycin (Invivogen). shBeclin-1 HELA cells were then infected with retroviral particles expressing shRNA resistant HA-Beclin-1 (wild-type or mutant) in the pQCXIH background. Retroviral particles were generated by transfection of HA-Beclin-1 constructs into Phoenix packaging cell lines. Following the same infection protocol described for lentiviral particles. Secondary selection was performed with 1μg/ml Puromycin and 250 μg/ml Hygromycin (Invitrogen). Polyclonal populations were screened until wild-type and mutant lines were generated that had near endogenous Beclin-1 reconstitution levels.
Western blot and Immunoprecipitation
Whole cell extracts were generated by direct lysis with 1X denaturing Laemmli sample buffer. Samples were boiled for 10min at 100C and resolved by SDS-PAGE. Immune complexes were obtained from cells lysed in mild lysis buffer [10mM Tris pH 7.5, 2mM EDTA, 100 mM NaCl, 1% NP-40, 50 mM NaF, supplemented at time of lysis with 1 mM Na3VO4 and protease inhibitor cocktail -EDTA (Roche)]. Antibodies were prebound to protein A beads (Repligen) overnight in MLB with 1%BSA. Beads were washed 1X with MLB and mixed with cleared cell lysates for 2 hours followed by 5 washes with MLB. Beads were boiled in 1X denaturing Laemmli sample buffer for 5 min before resolving by SDS-PAGE.
Quantification of Western blot
Statistical analysis was performed on biological repeats using Image J and application of Student’s 2 tailed T-Test. Error bars represent the standard deviation in normalized fold changes in observed induction or repression.
Mass Spectrometry
A gel band corresponding to Beclin-1 GST fusion protein was cut from a SDS-PAGE gel after an in vitro kinase reaction. The gel band was subjected to in-gel trypsin digestion and the resulting peptide mixture was subjected to liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Protein and modification identification was performed with the database search algorithm X!Tandem and peptide identifications were validated with Peptide Prophet.
Indirect Immunofluorescence
Fibronectin coated coverslips were placed in 24 well plates and cells were seeded the night before starvation or treatment. Cells were fixed by 4% paraformaldehyde in PBS for 15min, followed by permeabilization with 50ug/ml digitonin in 2%BSA, and 2% goat serum in PBS for 10 min. Cells were blocked in 2%BSA, and 2% goat serum in PBS for 30 min then incubated with primary antibodies in the same buffer for one hour at room temperature. Biotin-2XFYVE domain probe was incubated separately from other primary antibodies to reduce background. Slides were washed 3X in PBS followed by incubation of secondary antibody in 2%BSA, and 2% goat serum in PBS for 1hr at room temperature. Slides were washed 3X in PBS and mounted. Images were captured with an Olympus FV1000 confocal microscope.
Quantification of Indirect Immunofluorescence
Confocal microscopy images were used to determine colocalization using an automated protocol built in the Velocity 3D imaging software to reduce bias. The same protocol was applied to each field of view and across samples. Quantification was performed on representative experiments with an average of 7 unique fields of view. For C. Elegans experiments each embryo was scored as n=1, in a representative experiment. Puncta and staining patterns of embryos were quantified using a blind manual scoring method.
EM
HeLa lines were fixed in modified Karnovsky’s fixative (1.5% glutaraldehyde, 3% paraformaldehyde and 5% sucrose in 0.1 M cacodylate buffer, pH 7.4) for 15min at room temperature followed by overnight at 4C. The following day they were treated with 1% osmium tetroxide, 0.1 M cacodylate buffer for 1hr. Samples were stained in 1% uranyl acetate and dehydrated with ethanol. Epoxy resin embedded samples were sectioned (60 to 70 nm), and placed on Formvar, carbon-coated copper grids. Grids were stained with uranyl-acetate and lead nitrate. Images were obtained using a JEOL 1200EX II transmission electron microscope and photographed on a Gatan digital camera.
Statistical analysis
Error bars for microscopy (with the exception of C.elegans data) was presented as the standard deviation between unique fields of view within a representative experiment. In C.elegans microscopy the error bars represent the standard deviation between unique embryos. Error bars for Western blot analysis represent the standard deviation between densitometry data collected from 3 or more unique biological experiments. Statistical significance was determined using unpaired Student’s two-tailed T-test for two data sets.
In vitro ULK1 kinase assay
ULK1 proteins were immunoprecipitated and extensively washed with MLB (once) and RIPA buffer (50 mM Tris at pH 7.5, 150 mM NaCl, 50 mM NaF, 1 mM EDTA, 1 mM EGTA, 0.05% SDS, 1% Triton X-100 and 0.5% deoxycholate) once, followed by washing with MLB buffer once followed by equilibration with ULK1 assay buffer [KBB supplemented with 0.05 mM DTT 10 μM cold ATP and 2 μCi 32PATP per reaction. Reaction was quenched by direct addition of 4X Laemmli buffer followed by boiling for 5min and resolution by SDS-PAGE. The analysis of some kinase reactions necessitated the separation of the kinase and substrate. In these cases one of the components (either kinase or substrate) was left on beads and the eluant and washed beads were loaded on separate gels. Substrate and kinase were separated in kinase assays displayed in Fig.2a,b,d. Fractions of the total kinase reaction are shown in Fig. S2a,d. All in vitro kinase reactions that were analyzed by Western with the phospho-Beclin-1 antibody were fractions from the total reaction.
in vitro VPS34 lipid kinase assay
Immuno-purified complexes were equilibrated in 1X kinase base buffer [20 mM HEPES pH7.4, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2]. One half volume was taken for input, the remainder was centrifuged and excess 1X KBB was removed. 40ul of 1X kinase assay buffer was added to the beads [ 1XKBB supplemented with 0.1 mg/ml phosphatidylinositol, 50 μM cold ATP, 5 μCi 32P-ATP, 5 mM MnCl2, and 50 μM DTT] followed by incubation at 37C for 30 min with vigorous shaking. Reaction was quenched by direct addition of 10ul 1M HCl, followed by lipid extraction with 2 volume of MeOH:CHCl3 (1:1). Samples were vortexed for 1min and centrifuged at max rpm for 2min. Aqueous phase was discarded and organic loaded on a thin layer chromatography plate (Whatman). Resolution of phospho-lipid was achieved using a buffer composition of CHCl:MeOH(99%):NH4OH(30%):Water (129:100:4.29:24). Resolved plated were analyzed by autoradiography.
Mutagenesis
Site-directed mutagenesis was performed following Stratagene Quikchange kit instructions. With the following modifications. Primer length designed to manufacturer’s specifications were generally reduced by 4mer on each end. Primers were purified using standard desalt quality. Template amount was increased to 50ng. Cycles were increased to 25. Specificity of mutagenesis was confirmed by direct sequencing.
Protein purification
Bacterial GST-fusion proteins
Beclin-1 (including truncations and mutations) were cloned into the pGEX-KG vector and transformed into BL21 E.coli. Depending on protein stability induction was either performed on cultures (OD600 0.5-0.8) 16C overnight with 0.5mM IPTG or at 37C for 2hr. Bacteria were pelleted and resuspended in PBST (PBS, 0.5% Triton X-100, PMSF, and 2mM b-mercaptoethanol) and lysed by ultrasonication at 4C. Cleared lysates was subjected to Glutathione sepharose. Beads were washed with PBST and bound proteins were eluted using reduced glutathione according to the manufacturer’s instructions. Eluants were dialyzed against 20mM Tris-HCl pH 8.0, 10% glycerol.
ULK1 kinase production in mammalian/insect cells
To generate immunoprecipitated ULK1 kinase, 3XHA-ULK1 was transfected into 293 cells and immunoprecipitated with anti-HA antibody. Immunoprecipitated HA-ULK1 was washed as described in the kinase section of the methods. To generate liquid ULK1 kinase, FLAG-ULK1 was transfected into 293 cells and immunoprecipitated using anti-DYKDDDDK antibody (Agilent Technologies #200474). Immune-complexes were washed as described above and eluted in 4 bead volumes of PBS, 400ug/ml 3XFLAG peptide for 30min. Eluant was dialyzed (50 KDa mw cutoff) for 3hours in 1X kinase assay buffer, 10%glycerol. Active GST-ULK1 (1-649) and GST-ULK2 (1-478) from insect cells was purchased from CQuential Solutions (Moraga, CA).
C. elegan Strains
The following strains were used in this work: unc-51(e1189), VC424 bec-1(ok700)/nTl (qls51). ok700 is an allele that has 1377bp deletion and is considered equivalent to a null allele of bec-1. Homozygous bec-1(ok700) is lethal so bec-1(ok700/+) worms were used to generate transgenic lines. Bristol N2.
C. elegan Reporter Construction
Both bec-1 genomic DNA and bec-1 cDNA driven by bec-1 endogenous promoter were cloned into pPD95.79 vector. The mutant S6A was made based on the wild type constructs. The constructs were co-injected with pRF4(rol-6(su1006)) into VC424 worms and two stable transgenic lines were analyzed for each reporter.
C. elegan Immunofluorescence
Embryos were obtained from well-fed adult hermaphrodites of transgenic lines by the Freeze-Cracking methods. The slides were fixed, blocked, and incubated with diluted antibodies at 4 degree over night. The worms were then washed three times with PBST (0.5%) and incubated with Rhodamine-conjugated secondary antibody. Slides were viewed using an Epitome microscope.
Bec-1 has a maternal effect that rescues the autophagy defect in the bec-1 null embryos arising from bec-1(-/+) worms. Therefore, bec-1 null embryos shown are from the bec-1 null /pBEC-1∷BEC-1∷GFP non integrated transgenic lines, by selecting embryos that do not express the GFP transgene. Both Anti-PGL-1 and Anti-LGG-1 antibodies were gifts from Dr. Hong Zhang’s lab in NIBS.
Supplementary Material
Acknowledgments
We would like to thank Dr. Hong Zhang for LGG-1 and PGL-1 antibodies; Dr. Jenna Jewell, Dr. Carsten Hansen, and Ms. Karen Tumaneng for critical reading of this manuscript; Dr. Marilyn Farquhar on electron microscopy. Phosphosite identification by mass spectrometry was performed by the Proteomics Facility at the Fred Hutchinson Cancer Research Center. Confocal analysis was performed at UCSD Neuroscience Microscopy Shared Facility (Grant P30 NS047101). This work was supported by National Institutes of Health (NIH) grants GM51586, GM62694, CA108941, and Department of Defense (W81XWH-0901-0279). RCR is supported by a Canadian Institutes of Health Research (CIHR) postdoctoral fellowship.
Footnotes
Author Contributions
R.C.R. planned and performed experiments, Y.T. planned and performed experiments, H.Y., H.W.P., Y.Y.C and H.K. performed experiments, J.M.K., T.P.N., A.D., and K.L.G planned experiments, R.C.R and K.L.G wrote the manuscript.
Competing Financial Interest
The author(s) declare no competing financial interests.
References
- 1.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Young AR, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. Journal of cell science. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 3.Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 6.Yan J, et al. Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene. 1999;18:5850–5859. doi: 10.1038/sj.onc.1202988. [DOI] [PubMed] [Google Scholar]
- 7.Ganley IG, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volinia S, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995;14:3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Q, et al. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Bartolomeo S, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–168. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pattingre S, et al. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zalckvar E, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO reports. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 23.Matsunaga K, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- 26.Gulati P, et al. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobukuni T, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee EJ, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy. 2011;7:689–695. doi: 10.4161/auto.7.7.15450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gillooly DJ, et al. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440:283–291. doi: 10.1042/BJ20101894. [DOI] [PubMed] [Google Scholar]
- 32.Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L) Proc Natl Acad Sci U S A. 2011;108:7769–7774. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell. 2009;136:308–321. doi: 10.1016/j.cell.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Tian E, Zhang H. Selective autophagic degradation of maternally-loaded germline P granule components in somatic cells during C. elegans embryogenesis. Autophagy. 2009;5:717–719. doi: 10.4161/auto.5.5.8552. [DOI] [PubMed] [Google Scholar]
- 35.Tian Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 36.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 37.Obara K, Ohsumi Y. PtdIns 3-Kinase Orchestrates Autophagosome Formation in Yeast. Journal of lipids. 2011;2011:498768. doi: 10.1155/2011/498768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes to cells : devoted to molecular & cellular mechanisms. 2007;12:209–218. doi: 10.1111/j.1365-2443.2007.01050.x. [DOI] [PubMed] [Google Scholar]
- 39.Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes to cells : devoted to molecular & cellular mechanisms. 2008;13:537–547. doi: 10.1111/j.1365-2443.2008.01188.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.