

Mild stress treatments applied early in adult life, such as heat, irradiation, or reactive oxygen species (ROS) stress, can sometimes increase lifespan, a phenomenon often referred to as “hormesis.” In a recent study, we compared gene expression changes caused by heat, ionizing radiation, hyperoxia, and hydrogen peroxide to changes observed during normal aging.1 The results revealed that each stress condition and aging shared a core set of 18 upregulated genes, including the heat shock protein genes Hsp70, Hsp83 (Hsp90-family member), and l(2)efl (shsp-family member), and the mitochondrial unfolded protein response (mUPR) genes CG5966 (an apparent mitochondrial triacylglycerol lipase) and ref(2)P (ortholog of mammalian p62). ref(2)P encodes a protein implicated in marking mitochondria with mUPR for autophagy.2 This shared set of 18 upregulated genes indicates that one common feature of aging and hormesis-type stress is an unfolded protein response in the cytoplasmic and mitochondrial compartments. The fact that hormesis-type stress gene expression patterns are similar to the normal aging pattern is consistent with the general model for hormesis, in which mild stress causes upregulation of stress-response genes that protect the animal from subsequent stresses, such as those associated with aging. The results also confirmed and extended our previous observations that aging is associated with a failure in mitochondrial maintenance, as the data showed that aging is associated with downregulation of numerous mitochondrial genes, including electron-transport-chain (ETC) genes and mitochondrial metabolism genes, and a subset of these changes was also observed under the stress conditions.1
Overexpression of the mitochondrial gene MnSOD in Drosophila can extend lifespan, and this causes upregulation of many genes that are normally upregulated during aging.3 These targets include the mitochondrial chaperone gene Hsp22 (shsp-family) and 4 members of the core set described above: Pepck, CG32103, and the mUPR genes CG5966 and ref(2)P.1 These results suggest that MnSOD lifespan extension proceeds through a hormesis-type mechanism involving the mitochondria. In another recent study, we reported that the upregulation of Hsp22 during aging is particularly dramatic in a subset of the oenocytes (liver-like cells) indicating an age-related mUPR in these cells.4 MnSOD overexpression caused Hsp22 upregulation specifically in the oenocytes, consistent with the idea that MnSOD overexpression increases lifespan through a hormesis-type mechanism involving a mUPR in the oenocytes. Moreover, overexpression of Hsp22 itself is reported to increase lifespan, and this was also associated with Hsp22 induction in the oenocytes.4 Taken together our results indicate that Drosophila lifespan extension caused by MnSOD overexpression involves a mUPR response that is particularly dramatic in the liver-like oenocytes and a hormesis-type mechanism (Fig. 1).
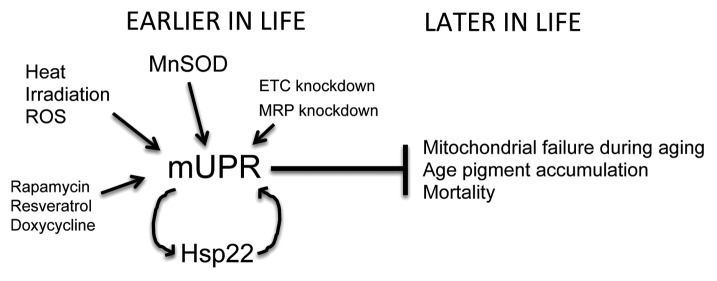
Figure 1. Model for mUPR in aging interventions. Several environmental and genetic interventions that can extend lifespan have in common the activation of the mUPR, preferentially in liver-like cells (the oenocytes of Drosophila, the intestinal tissue of C. elegans, and the hepatocytes of mouse). The mUPR is characterized by the induction of mitochondrial chaperone genes, including Hsp22 in Drosophila (as indicated). The induction of the mUPR in young animals can extend lifespan, and in Drosophila, this has been shown to correlate with reduced accumulation of age pigment.
The involvement of mUPR and hormesis-type mechanisms in lifespan extension is supported by recent results with additional model organisms. In both Drosophila and C. elegans, knockdown of expression of an ETC component can extend lifespan.5 In C. elegans, this intervention was found to activate the mUPR preferentially in gut tissue, including induction of mitochondrial chaperone genes hsp-6 (Hsp70-family member) and hsp-60, and these effects were dependent upon function of the mUPR pathway gene ubl-5.6 Interestingly, in C. elegans the gut is thought to be the liver-like tissue. Another study of C. elegans revealed that extension of lifespan caused by MnSOD overexpression is associated with upregulation of the mUPR chaperone gene hsp-6, again consistent with a hormesis-type mechanism involving the mUPR.7 Finally, a recent study of mice identified a strong correlation between longevity and a polymorphism near the mitochondrial ribosomal protein (MRP) gene Mrps5, and expression levels for Mrps5 were negatively correlated with mouse strain lifespan.8 Knockdown of the homologous mrps-5 gene in C. elegans increased C. elegans lifespan, and this intervention was again associated with induction of the mUPR genes hsp-6 and hsp-60 and required function of the mUPR pathway gene, ubl-5. These investigators also provided evidence that the drugs doxycycline, rapamycin, and resveratrol increase C. elegans lifespan by activating the mUPR, and can also activate the mUPR in cultured mammalian hepatocytes, suggesting possible conserved mechanisms acting on liver-like cells.
In summary, the recent studies suggest that several interventions that can increase lifespan across species, including MnSOD overexpression, hormesis-type environmental stress, and knockdown of ETC and MRP genes, have in common the activation of the mUPR and may act preferentially in liver-like cells (Fig. 1). Because our data indicate that normal Drosophila aging is associated with an age-dependent mUPR in liver-like cells,4 these studies support a hormesis-type model, in which a mUPR early in life buffers against mitochondrial failure during aging, and implicate the liver as a critical target tissue. One possible mechanism is that the mUPR early in life favors mitochondrial turnover through autophagy (“mitophagy”), and this provides the animal with a pool of newly synthesized and better-functioning mitochondria that favor longevity; additional possibilities include reduced production of toxic metabolites such as age pigment. In the future, it will be important to further explore the role of mUPR in hormesis and lifespan, and to determine the precise mechanisms by which an early mUPR favors subsequent mitochondrial maintenance and animal survival.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26354
References
- 1.Landis G, et al. Aging (Albany NY) 2012;4:768–89. doi: 10.18632/aging.100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pimenta de Castro I, et al. Cell Death Differ. 2012;19:1308–16. doi: 10.1038/cdd.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtis C, et al. Genome Biol. 2007;8:R262. doi: 10.1186/gb-2007-8-12-r262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tower J, et al. J Gerontol A Biol Sci Med Sci. 2013 [Google Scholar]
- 5.Hur JH, et al. Aging (Albany NY) 2009;1:881–3. doi: 10.18632/aging.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durieux J, et al. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabreiro F, et al. Free Radic Biol Med. 2011;51:1575–82. doi: 10.1016/j.freeradbiomed.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houtkooper RH, et al. Nature. 2013;497:451–7. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]