

Abstract
Cell migration is dependent on a series of integrated cellular events including the membrane recycling of the extracellular matrix receptor integrins. In this paper, we investigate the role of autophagy in regulating cell migration. In a wound-healing assay, we observed that autophagy was reduced in cells at the leading edge than in cells located rearward. These differences in autophagy were correlated with the robustness of MTOR activity. The spatial difference in the accumulation of autophagic structures was not detected in rapamycin-treated cells, which had less migration capacity than untreated cells. In contrast, the knockdown of the autophagic protein ATG7 stimulated cell migration of HeLa cells. Accordingly, atg3−/− and atg5−/− MEFs have greater cell migration properties than their wild-type counterparts. Stimulation of autophagy increased the co-localization of β1 integrin-containing vesicles with LC3-stained autophagic vacuoles. Moreover, inhibition of autophagy slowed down the lysosomal degradation of internalized β1 integrins and promoted its membrane recycling. From these findings, we conclude that autophagy regulates cell migration, a central mechanism in cell development, angiogenesis, and tumor progression, by mitigating the cell surface expression of β1 integrins.
Keywords: macroautophagy, cell migration, endocytosis, integrins, MTOR, lysosomes
Introduction
Cell migration is fundamental in embryo development, wound healing, and inflammation, and is also involved in various pathophysiological situations, including tumor progression and vascular diseases.1 Cell migration is a highly dynamic process that coordinates a series of events involving the front–to-back cell polarization that requires the disassembly of adhesive contacts or focal adhesions at the trailing edge, and the assembly of new adhesive contacts at the leading edge.2,3 Basically, the turnover of adhesion contacts depends on the attachment and detachment of the cell surface receptor integrins, which are α/β heterodimers, from the extracellular matrix.1,3,4 The redistribution of integrins during cell migration is controlled by internalization and recycling. Clathrin-mediated endocytosis plays a crucial role in the disassembly of focal adhesion in migrating cells.1,5,6 Integrin trafficking during cell migration also involves macropinocytose via circular dorsal ruffles, macropinosomes, and the recycling of endosomes and early endosomes to form new focal adhesion at the leading edge.1,7,8
Macroautophagy (hereafter referred to as autophagy) is a lysosomal degradation pathway for cytoplasmic components, including organelles.9,10 The process starts by the formation of a double-membrane bound vacuole or autophagosome by the hierachical intervention of Atg proteins.11 The process is initiated by the ULK1 (the mammalian ortholog of the yeast Atg1) complex and the PIK3C3/VPS34 complex, which contains ATG14L and Beclin 1 (the mammalian ortholog of the yeast Atg6).12 These complexes are located upstream of the two ubiquitin-like conjugation systems, ATG12-ATG5 and LC3 (one of the mammalian homologs of yeast Atg8)-phosphatidylethanolamine that elongate and close the autophagosomal membrane. The autophagosome merges with endocytic compartments to form an amphisome, and acquires acidic and degradative properties before fusing with lysosomes, where the final degradation of autophagic cargoes takes place. It has recently been demonstrated that the fusion of early endosomes with autophagosomes is required for autophagy to occur.13 In addition, the clathrin-dependent endocytosis machinery delivers ATG16L-containing vesicles to the assembly site of the phagophore .13-15
Autophagy plays a major role in various physiological and pathophysiological processes involving cell migration; these include development, inflammation, cancer and metastasis.16-20
However, little is known about the impact of autophagy on cell migration. In the present study, we show that cell migration is associated with a low level of autophagy. This low level of autophagy is in turn correlated with high activity of the mechanistic target of rapamacyn (MTOR) in migrating cells. Activation of this kinase in the MTOR complex 1 (MTORC1) is known to be a major repressor of autophagy.10,16,18 Treatment with rapamycin, a MTORC1 inhibitor, stimulates autophagy and reduces cell migration. In contrast, the migration of cells in which the autophagy genes (atg3−/− MEFs and atg5−/− MEFs) have been deleted occurs faster than that of their wild-type counterparts. We also show that the autophagic pathway merges with the endocytic route of the integrins, and reduces their membrane recycling. Thus, autophagy is a mechanism that regulates both the cell surface expression of integrins and their capacity for cell migration.
Results
MTOR is activated in HeLa-GFP-LC3 cells at the leading edge of migration
MTOR is a key regulator of autophagy.10 Several reports in the literature have shown that rapamycin, a MTORC1 inhibitor, and its analogs (rapalogs) inhibit cell motility in a panel of mouse and human cell lines.21,22 Since rapamycin is known to induce autophagy, we wondered whether autophagy could be involved in the modulation of cell migration. To explore the relationship between MTORC1 activity and autophagy during the cell migration process, we first observed the level of autophagy in HeLa-GFP-LC3 cells during a wound-healing assay (Fig. 1). LC3 is a protein that becomes associated with the autophagosomal membrane when autophagy is stimulated.23 Thus, the accumulation of GFP-LC3 puncta provides an effective way of detecting autophagic structures by fluorescence microscopy.
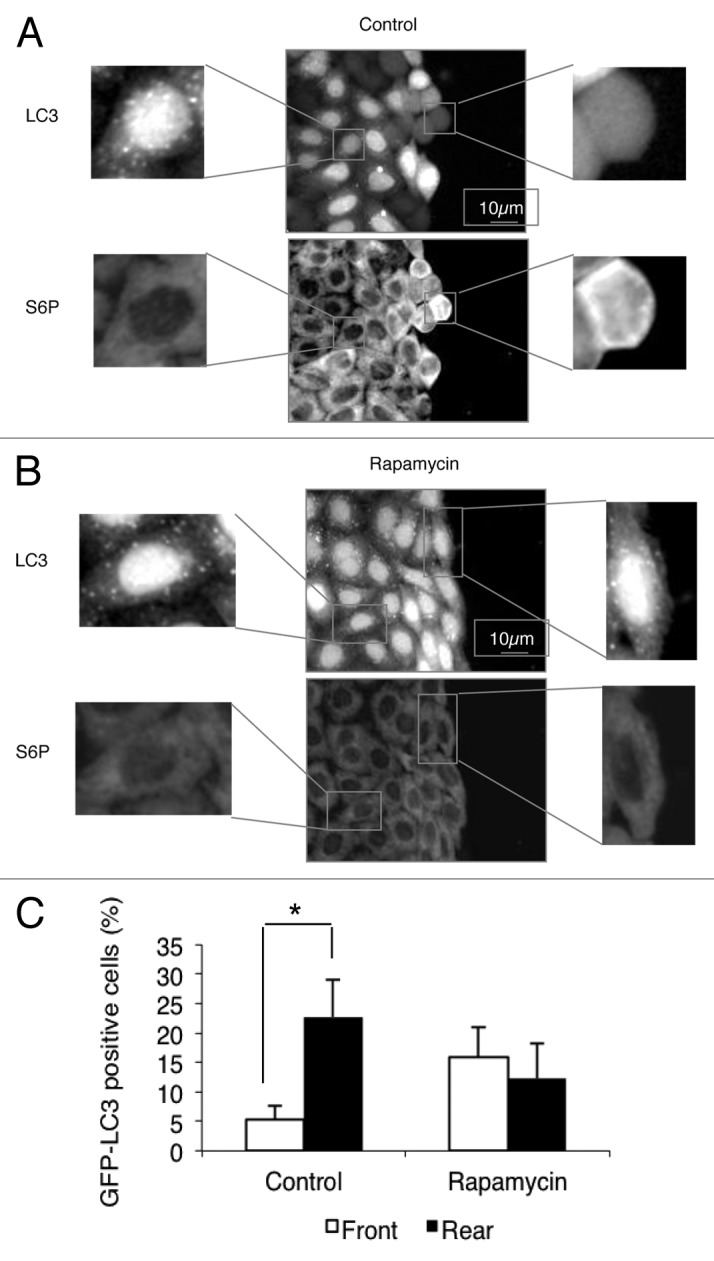
Figure 1. Activation of MTOR in HeLa cells at the leading edge is correlated with inhibition of autophagy. Wound healing assays were performed on an HeLa-GFP-LC3 monolayer. Cells were fixed with 4% paraformaldehyde and stained with S6-P antibody (1:100).(A) Untreated cells.(B) Cells treated with 200nM rapamycin for 4 h.(C) The graph indicates the percentage of GFP-LC3 puncta-positive cells (>30 GFP-LC3 puncta per cell) at the leading edge (front) and at the rear (rear) in untreated cells (Control) and in rapamycin-treated cells (Rapamycin). Results are representative of 3 independent experiments ± SD *P < 0.05
The migration of cohesive groups of cells subsequent to wound healing makes it possible to distinguish two cells populations: the leader cells in the front row, which provide cell guidance, and the cells following behind them.24 Cells located at the front of the wound had fewer LC3-positive vacuoles than cells at the rear (Fig. 1A and C). One of the downstream effectors of MTORC1 is the 70-kDa S6K1 frequently hyperactivated in cancer.25 We tested the MTOR activity through the level of phosphorylation of ribosomal protein S6, a substrate of S6K1, during the wound-healing assay, using immunofluorescence to detect the phosphorylated S6. Cells at the leading edge showed a higher level of phosphorylation of S6 than cells trailing further back (Fig. 1A). When the wound-healing assay was performed in the presence of rapamycin, the differences in the degree of phosphorylation of S6, and the accumulation of GFP-LC3 puncta between cells at the leading edge and rearward cells were abolished (Fig. 1B). These findings support the conclusion that differences observed in the wound-healing assay between cells at the leading edge and cells further back are not simply an effect of the scratch itself, but are also attributable to polarization of the cell populations during migration. It should be noted that the activation of MTOR (as revealed using an anti S6-P) was not entirely homogeneous in the confluent cell monolayer before wounding. A close correlation was observed between the intensity of S6-P-staining and the accumulation of LC3 puncta. Cells staining weakly for S6-P and which had a large number of GFP-LC3 puncta were randomly distributed in the monolayer (Fig. S1). In contrast, the S6-P staining was uniformly low, and the accumulation of GFP-LC3 puncta was homogeneously distributed in a confluent cell monolayer that had been exposed to rapamycin treatment for 4 h (Fig. S1).
Recent studies have shown that active MTORC1 is located on the surface of lysosomes.26,27 We therefore investigated the location of MTOR in the migrating cells. For this purpose we used HeLa cells, which express the lysosomal membrane protein GFP-CD63 in wound-healing assays.28 After 4 h of migration, the localization of MTOR was determined by indirect immunofluorescence. We observed more co-localization of MTOR with lysosomes in cells at the front of the wound than in rearward cells (Fig. 2A). A similar staining pattern of active MTOR was observed when the positioning of the autophosphorylated form of MTOR was studied (Fig. 2B). Thus, the low rate of autophagy in cells at the front of the wound correlated with the localization of active MTOR on the surface of the lysosomes.

Figure 2. Co-localization of MTOR and lysosomes in migrating cells. Wound healing assays were performed on an HeLa-GFP-CD63 monolayer. Cells were fixed with 4% paraformaldehyde.(A) Cells were stained with anti- MTOR antibody (1:200)(B) Cells were stained with anti- P-MTOR antibody (1:200).
Inhibition of autophagy stimulates cell migration
In a first series of experiments to find out whether the inhibition of migration is a general response to autophagy distinct from the inhibition of MTOR, we investigated the effect of trehalose. This disaccharide has been shown to trigger autophagy independently of the inhibition of MTOR.29 Like rapamycin, trehalose slowed down the migration of GFP-LC3-HeLa cells (Fig. S2). Following on from the results reported above, we wanted to determine the role of autophagy in the regulation of cell migration. For this purpose, we used two different assays to measure cell migration to appreciate its dependency on autophagy: a wound-healing assay and a Transwell migration assay, in control cells and in cells with a defective autophagy pathway (Fig. 3), respectively. Inhibition of autophagy in Hela-GFP-LC3 cells was obtained by knocking down ATG7, a protein involved in the synthesis of the two ubiquitin-liked conjugates required for the formation of autophagosomes.30 Confluent monolayers of HeLa-GFP-LC3 cells treated with control siRNA (Control) or with ATG7 siRNA (siRNA ATG7) were scratched, and migration into the wound area lasted for 24 h (Fig. 3A and B). The wound area was almost completely healed by the siRNA ATG7 HeLa cells, whereas a significant gap remained in the control. In another set of experiments, we used a Transwell migration assay to investigate the role of autophagy during cell migration (Fig. 3C and D). In line with the results obtained in the wound-healing assay, knockdown of ATG7 increased the rate of cell migration into the lower chamber. As ATG7 can influence cellular events independently of its activity in the autophagic pathway,31 we repeated migration experiments in atg3−/− MEFs and atg5−/− MEFs (see Fig. S3). ATG3 and ATG5 are involved in the production of the LC3-PE (LC3-II) and ATG12-ATG5 ubiquitin-like conjugates, respectively, which contribute to the elongation and sealing of the autophagosome membrane.11 In both atg3−/− MEFs and atg5−/− MEFs, we observed a higher rate of migration than in their wild-type counterparts. Overall, these results support the hypothesis that autophagy acts as a brake on cell migration.

Figure 3. Inhibition of the migration of HeLa cells is dependent on the expression of Atg7. HeLa-GFP-LC3 were transfected with 200 nM scrambled siRNA (Control) or with 200 nM Atg7 siRNA for 24 h (siRNA Atg7). Experiments were performed 48 h post transfection.(A) Cell monolayers were assayed for wound-healing migration in complete medium. Phase-contrast microscopy was performed to record the relative rate of wound closure immediately after scratching (0 h) and after migrating for 24 h (24 h).(B) The surface of the wound was evaluated using Image J software, and the percentage of the surface to have recovered was determined. Results are representative of four independent experiments ± SD ***P < 0.005.(C) Cells were plated at the top of a Transwell, and allowed to migrate for 6 h. They were then rinsed, fixed with paraformaldehyde, stained with crystal violet and counted. Images show the cells on the undersurface of a filter.(D) The percentage of migrated cells was evaluated in terms of the migration after 24 h, which was taken to be 100% migration. Results are representative of four independent experiments ± SD *P < 0.05
The endocytic route followed by β1 integrins is different after the induction of autophagy
Cell migration requires the formation of a membrane protusion, which in turn requires polarization from the rear to the front of the cell membrane and the formation of focal adhesions. The turnover of these adhesion sites determines the cell’s capacity for migration.24These sites of adhesion contain integrins that are transmembrane receptors of extracellular matrix molecules. It has been proposed that directional migration entails the recycling of these integrins via various loops associated with small Rab GTPases. Integrins are endocytosed and localized with early endosomes as well as multivesicular bodies, before being redirected to the plasma membrane to form new protusions required for the guidance and the motility of cells.1 We therefore hypothesized that the induction of autophagy, which converges with endocytic routes,32 could interfere with the trafficking of endocytosed integrins, and redirect them to the lysosomal compartment. To investigate this, we labeled HeLa-GFP-LC3 cells with a monoclonal antibody P5D2 directed against the β1 chain of integrin, and investigated the endocytosis of the antigen-antibody complex after various periods of incubation (60 to 120 min). HeLa-GFP-LC3 cells were cultured either in complete medium, to restrict autophagy to a low level, or in starvation medium to induce autophagy. Co-localization of β1 integrins and GFP-LC3-positive vesicles or with lysosomes was evaluated by calculating Pearson’s correlation coefficient (r). Co-localization between β1 integrin positive vesicles and GFP-LC3-positive vesicles was weak (r < 0.5) after 120 min in complete medium (Fig. 4A), and β1 integrin-positive vesicles moderately colocalized with lysosomes at all times (r around 0.6) (Fig. 4A). In contrast, greater co-localization of β1 integrin-positive vesicles with autophagosomes was detected in starvation medium from 60 min to 90 min (Fig. 4B). However, after 120 min the number of β1 integrin positive and GFP-LC3 vesicles had decreased significantly. In contrast after 90 min almost all β1 integrin-positive vesicles had colocalized with lysosomes (r = 0.9) (Fig. 4B). From these results we concluded that stimulating autophagy induces integrin trafficking toward the lysosomes and leads to integrin degradation. In order to investigate the autophagy-dependent sequestration of β1 integrin during migration, we performed wound healing assays on HeLa-GFP-LC3 cells and tracked β1 integrin and LC3-stained autophagic vacuoles by live fluorescence microscopy. In complete medium, fusion events between GFP-LC3 vesicles and vesicles containing β1 integrin were barely detectable in cells at the leading edge (Fig. 5A; see also Video S1). This was a consequence of the very small number of autophagosomes detected in these cells (see Fig. 1). In contrast, in rapamycin-treated cells, fusion events were easily detectable in cells at the leading edge (Fig. 5B; see also Video S2). This result is closely correlated with the rapamycin-dependent slowdown of cell migration. Similar results were observed in starved cells (data not shown).

Figure 4. Co-localization of β1 integrins with autophagosomes and with lysosomes. HeLa-GFP-LC3 cells were stained with anti-β1 antibody (1:50) and Lysotracker (1:30000). After 60, 90, and 120 min of β1 integrin endocytosis, the cells were fixed with 4% paraformaldehyde. Autophagosomes (AP: green), β1 integrin (Beta 1: gray) and lysosomes (Lys: red), were observed by confocal-fluorescence microscopy. The graph indicates the Pearson’s correlation coefficient (r), which is indicative of the degree of colocalization between autophagosomes and β1 integrin vesicles, and that between lysosomes and β1 integrin vesicles. (A) Experiments were conducted in complete medium. (B) Experiments were conducted in starvation medium.

Figure 5. Convergence of the β1 integrin endocytic pathway and the autophagic pathway in migrating cells during starvation. HeLa-GFP-LC3 cells were stained with anti-β1 integrin antibody (P5D2 1:50) and Alexa fluor 594 anti-IgG antibody (1:2000). After the immunocomplex had been subjected to endocytosis for 30 min, the cells were washed, and a scratch was performed on the monolayer. Autophagosomes (green) and β1 integrins (red) were observed by time-lapse videomicroscopy. Time in seconds corresponding to each frame is indicated.(A) Control cells (see also Video S1).(B) Cells treated with 200 nM rapamycin for 4 h (see also Video S2).
Induction of autophagy limits the levels of β1 integrins
In order to demonstrate directly that autophagy interferes with the β1 integrin endocytic/recycling pathway, we investigated the expression of integrins during starvation and its reversal when the medium was replenished with nutrients to block autophagy. We quantified autophagosomes and β1 integrin-containing vesicles after 90 min in starvation medium, and 30 min and 60 min after adding nutrients (RPMI 10% FBS). As expected, the number of autophagosomes decreased when nutrients were added to the starvation medium (Fig. 6). Interestingly, the number of vesicles containing of β1 integrin increased significantly under these conditions (Fig. 6). Similarly, the number of β1 integrin containing vesicles was significantly increased in starvation medium in the presence of lysosomal cathepsin inhibitors (E64d + pepstatin A) (Fig. S4). These results indicate that starvation-induced autophagy leads to the sequestration and degradation of β1 integrin in the lysosomes, but that this process is reversible as soon as cells are returned to conditions that inhibit autophagy.

Figure 6. Quantification of autophagosomes and β1 integrins after starvation and after replenishing.(A) HeLa-GFP-LC3 cells were first incubated in starvation medium for 4 h. The cells were then stained with anti-β1 integrin antibody (P5D2 1:50). After 90 min of endocytosis in starved medium (EBSS), the cells were incubated with complete medium for 30 or 60 min (RPMI). The cells were fixed with 4% paraformaldehyde, and then the autophagosomes and β1 integrins were observed by fluorescence microscopy.(B) The graph indicates the number of autophagosomes per cell and the number of β1-stained vesicles per cell. Results are representative of three independent experiments ± SD *P < 0.05, **P < 0.01, ***P < 0.005
To investigate the role of autophagy in membrane recycling of β1 integrins we used total internal reflection fluorescence (TIRF) microscopy to visualize events occurring in proximity of the cell membrane. The re-distribution of β1 integrins was analyzed in Hela cells treated with the P5D2 antibody in complete and starved medium. TIRF revealed a dynamic redistribution of β1 integrins suggesting continuous remodeling of adhesion contacts containing β1 integrins in complete and starved media (Fig. 7A; see also Video S3; and Fig. 7B; see also Video S4). However, the quantification of β1 integrin puncta indicated a marked reduction in the expression of these receptors at the membrane level in starved cells (Fig. 7C). This results suggested that β1 integrins were not only re-distributed, but also degraded in starved cells.

Figure 7. Starvation-induced autophagy inhibited the re-expression of β1 integrin on the adhesion sites at the plasma membrane after anti-β1 integrin specific antibody-mediated endocytosis. TIRF was used to analyze β1 integrin cell surface expression at the adhesion sites of non-confluent Hela cells. The cells were incubated with anti-β1 integrin antibody (P5D2 1:50) and Alexa fluor 555 anti-IgG antibody (1:2000) for 30 min at 4 °C, then washed and incubated in complete medium or in EBSS under the videomicroscope at 37 °C for the times indicated. (A and B) Representative TIRF montages showing the turn-over of β1 integrins at the cell surface of cells incubated with complete medium (A, see also Video S3) or EBSS (B, see also Video S4).(C) Quantification of puncta containing β1 integrin as the percentage surface expression of β1 integrin after various times of endocytosis. Results are representative of four independent experiments ± SEM.
In order to study the role of autophagy in damping the membrane recycling of β1 integrins, we used MCF7 cells transfected with a control shRNA (MCF7 shSC20) and MCF7 transfected with a shATG7 (MCF7 shATG7). We performed a FACS analysis in MCF7 shSC20 and MCF7 shATG7 cells treated with the P5D2 antibody in complete medium and in starvation medium to determine the cell surface level of β1 integrins (Fig. 8). In MCF7shSC20, cell surface β1 integrin expression had decreased in both complete medium and starvation medium after 120 min P5D2 binding. This decrease reflects the endocytosis of β1 integrins. After 180 min, cell surface β1 integrin expression returned to its initial level in complete medium, indicating membrane recycling. In contrast, the cell surface pool of β1 integrin was not replenished in starvation medium (Fig. 8, high panel). When the same experiment was conducted in starved MCF7 shATG7 cells, β1 integrin expression decreased after 120 min, and its membrane expression had almost recovered to its initial level after 180 min in both complete and starvation media (Fig. 8, high panel). These results indicated that autophagy prevents the cell surface re-expression of β1 integrins after endocytosis induction.

Figure 8. Starvation-induced autophagy inhibited the re-expression of β1 integrin on the plasma membrane after anti-β1 integrin specific antibody-mediated endocytosis. Flow cytometry was used to analyze β1 integrin cell surface expression of MCF-7 adherent cells stably transfected with shRNA-mediated scrambled (MCF7 shSC20) or shRNA-mediated silencing of ATG7 (MCF7 shATG7). The cells were or not starved in EBSS for 2 h before being incubated with anti-β1 integrin antibody (P5D2 1:75) for 30 min at 4 °C. The cells were then washed and incubated in complete medium or in EBSS at 37 °C for the times indicated . For the flow cytometry analysis by indirect immunofluorescence, the cells were fixed and incubated with anti-mouse IgG coupled with biotin and then with streptavidin coupled with allophycocyanin .(A) Representative histograms show surface expression of β1 integrin. Red line/curve, isotypic control (IC); blue line/curve, 0 min; yellow line/curve, 120 min; green line/curve, 180 min. The numbers indicate the median fluorescence intensity.(B) Graphs represent the ratio of the median fluorescence intensities of the anti-β1 integrin specific antibody and isotypic control antibody for each time indicated.
Discussion
Endocytosis and the membrane recycling of integrins contribute to the regulation of cell migration.1-3,8 Membrane recycling involves a short pathway, which depends on Rab4, and a long pathway, which depends on Rab11. Although lysosomal delivery is not the major route for internalized integrins, recent studies demonstrated that the lysosomal degradation of a membrane pool ubiquitinated α5β1 integrin with its ligand fibronectin is an important event during cell migration.33 In the work reported here, we show that autophagy is downregulated during cell migration. This downregulation prevents the sequestration of integrins in autophagic vacuoles and their subsequent lysosomal degradation. This degradation occurs because the autophagic pathway merges with the endocytic pathway.13,15,27 Thus, inhibition of autophagy would limit the sequestration and degradation of β1 integrins during migration. From our results and those previously reported on the role of lysosomal degradation of ubiquitinated β1 integrin in the regulation of cell migration,33 it can be hypothesized that bulk autophagy and selective lysosomal degradation have to be finely tuned to allow cells to migrate.
The downregulation of autophagy is associated with the robust activation of MTORC1 in migrating cells. The role of MTORC1 in controlling autophagy in migrating cells is supported by the fact that rapamycin, an established stimulator of autophagy,10 inhibits cell migration.21,22 Recently, Sabatini and colleagues have shown that MTORC1 association with the lysosomal membrane is required for its activation by amino acids.27 We observed that MTOR is more tightly associated with lysosomes in cells at the leading edge of wound healing than in cells at the trailing edge. This could contribute to the higher MTOR activity in migrating cells, and thus to the downregulation of autophagy observed in cells at the leading edge in wound-healing assay. We also observed that lysosomes associated with MTOR are more peripherally distributed in migrating cells. It is known that migrating cells have a modified morphology, with a reshaped cytoskeleton.1,2,24 This could suggest that autophagy can be tightly regulated by the cell shape and the location of organelles. Interestingly, it has been reported that the peripheral location of lysosomes is important in determining the activation of lysosomal-membrane bound MTORC1.26,32 The role of the MTORC2 complex, which has been reported to be involved in the stimulation of cell migration34,35 and to control autophagy,36 cannot be disregarded. However, we showed that triggering autophagy independently of MTOR inhibition by trehalose is sufficient to slow down cell migration. These findings point to autophagy as an important cellular event in regulating cell migration, whatever its upstream stimulatory pathways.
The downregulation of autophagy accelerated cell migration in two different migration assays, the wound-healing assay and the Transwell migration assay, which are in-vitro models of collective cell migration and single cell migration, respectively.24,36 These two modes of migration co-exist in vivo during embryogenesis, development, and cancer invasion. Interestingly, at least in vitro, autophagy modulates both types of migration. This would suggest that the downregulation of autophagy in migrating cells has a common function in active migrating cells. The downregulation of autophagy impairs the degradation of the cell surface integrins that need to be recycled to the cell surface to support migration. In collective cell migration, it would be interesting to find out whether the autophagic capacities of rearward cells have any function in controling migration related to change in cell morphology or metabolic adaptation to migration.37 Interestingly, the GTPases of the Ras superfamily (Rab, Rho, Rac, Ral),12,36,38,39 which are involved in both collective and individual cell migration, are also known to regulate autophagy.12,40,41
The role of autophagy during cell migration is an emerging field. Cell migration is observed in various different physiological and pathophysiological situations.1,24,39,42 Caution should be exercised before generalizing the inhibition of autophagy to every biological process where cell migration is involved. For example, autophagy has been reported to play a role in the migration of invasive breast cancer cells under hypoxic conditions39,43 where autophagy is probably required to maintain cell survival by providing ATP via lysosomal degradation. A further degree of complexity is probably associated with the involvement of autophagy regulators in cell migration. Recently, p62 and DRAM, which are both autophagy regulators,39,44,45 have been shown to be required for the migration of glioblastoma stem cells.39,46
Our study suggests that a selective form of autophagy plays a role during cell migration, and we propose a working model in which nonselective autophagy mitigates cell migration as a result of merging with the endocytic route of the β1 integrins. It would be interesting to find out whether the unselective and selective forms of autophagy have complementary roles during cell migration.
Materials and Methods
Reagents
BSA (Cat. No. A1595), crystal violet dye content >90% (Cat. No. C3886), rapamycin from Streptomyces hygroscopicus (Cat . No. R0395), E64d (Cat. No. E8640), pepstatin A (Cat. No. P5318), and paraformaldehyde (Cat. No. P58127) were all purchased from Sigma.
Antibodies and RNAi
Anti-LC3B (Sigma, Cat. No. L7543), anti-Atg 7 (kindly provided by Dr William A. Dunn, University of Florida at Gainsville), anti-β-actin (Millipore, Cat. No. MAB1501), anti-P-S6 ribosomal protein (Cell Signaling, Cat. No.2211), anti-mouse IgG-HRP (Bio-Rad, Cat. No. 170-6516), anti-rabbit IgG-HRP (Amersham, Cat. No. NA9340V), and anti-p62 (BD Transduction laboratories, Cat. No. 610833) were used for immunoblotting. Anti-P-MTOR (Cell Signaling, Cat. No. 2971), and anti-MTOR (Cell Signaling, Cat. No.2972) were used for immunocytochemistry. Cy5 or Rhodamine (TRITC)-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories. Alexa Fluor 555- and 594-conjugated anti-IgG antibodies (ref: A21424 and A11020) and Lysotracker D99 were from Molecular Probes, Invitrogen Co.
The anti-β1 integrin mouse monoclonal antibody P5D2, developed by Dr Wayner, was obtained from the Developmental Studies Hybridoma Bank maintained by the University of Iowa.
The AllStars control scrambled siRNA, and the predesigned siRNA oligos Atg7siRNA were purchased from Eurogentec. The sequence used was as follows Atg7 siRNA sense, 5′-CAGUUUGGCA CAAUCAAUA-3′.
Cell culture
Atg3+/+, atg3−/−, atg5+/+, and atg5−/− murine embryonic fibroblast (MEF) cells obtained from Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan) and Noboru Mizushima (Tokyo Medical and Dental University, Japan) respectively, were cultured in DMEM containing 10% fetal bovine serum (FBS) and antibiotics (complete medium).
MCF7 shSC20 and MCF7 shAtg7 cells obtained from Saieb Ghavami (Depts. of Physiology and Internal Medicine, University of Manitoba, Canada) were cultured in DMEM containing 10% fetal bovine serum (FBS) and 4 µg/mL of puromycin, a selective antibiotic (complete medium) in an atmosphere of 10% CO2.47
The HeLa-GFP-LC3 and HeLa-GFP-CD63 cell lines were obtained from Dr. Aviva M. Tolkovsky (University of Cambridge, UK), and cultured in RPMI 1640 with 10% fetal bovine serum (FBS), and 500 µg/mL G418 at 37 °C with 5% CO2 (complete medium). The starvation medium consisted of EBSS (Earle Balanced Salt Solution). The cells were seeded into 6-well plates (5 × 105 cells per well); the following day, siRNAs were transfected using Oligofectamine 2000, according to the Manufacturer's instructions (Invitrogen). With the exception of the dose–response experiments, siRNAs were transfected at 200 nM for 24 h. Forty-eight hours post transfection, the cells were submitted to a wound healing assay, Transwell migration assay or immunoblot, as detailed below. The AllStars Negative Control siRNA were used as control.
Wound healing assay
Confluent cell monolayers were wounded by scratching with p200 pipet tip in Labtek slides. Debris were removed by washing the cells with complete medium. The cells were then incubated at 37 °C for various periods of time. Images were acquired at 0 h, and at the end of the incubation time using a brightfield microscope. References points were used to match the image fields. The images acquired were further analyzed by comparing the surface recovery between the reference point at time 0 and the end of incubation in the same field using ImageJ software.
Transwell migration assay
Cell motility was measured using Transwell chambers with a suitable pore size for several cell lines: 8-μm pore (BD Biosciences). HeLa-GFP-LC3 cells, used after 48 h of transfection, or MEFs were harvested with trypsin/EDTA, resuspended in EBSS, and added to the top of the inserts at 1.25 × 105 cells per Transwell. RPMI or DMEM with 10% serum were added to the lower chamber of the 12-well format Transwells to provide a chemoattractant. After 5 or 6 h at 37 °C, depending on the cell lines, the Transwells were fixed with 4% paraformaldehyde, and stained with crystal violet solution (2.5%). Any cells that had not migrated were removed from the top of the membranes using cotton swabs. The membranes were detached from the Transwells, and then affixed to glass slides using mounting medium (Dako Fisher Scientific). To determine the number of cells that had migrated, ten random images of each Transwell were taken at 20× on a light microscope. The number of migrated cells per image was counted using the Particle analysis function in the ImageJ software. The percentage was expressed as a percentage of the number of cells that had crossed the filters after incubating for 24 h, which was taken to be 100%.
Integrin endocytosis assay
The cells underwent preliminary incubation in complete medium or starved medium for 4 h. Cell surface β1 integrin molecules and lysosomes were then labeled by incubating HeLa-GFP-LC3 cells with the anti-β1 integrin monoclonal antibody P5D2 and the lysotracker at 0 °C, when endocytosis is inhibited. Endocytosis was resumed by incubating the cells at 37°C for various periods of time (60, 90, and 120 min). After this period of endocytosis, the cells were fixed with 4% paraformaldehyde and then treated with NH4Cl (50 mM) for 10 min to quench the aldehyde groups, and blocked in 10% FBS in PBS for 30 min, before being permeabilized with 0.075% saponin in PBS. The cells were then labeled with a Cy5-conjugated anti-mouse antibody (to stain endocytosed β1 integrin). Confocal fluorescence microscopy was used to check for colocalization of β1 integrins with GFP-LC3 vesicles and with lysosomes. Confocal images were collected using a 63 × 1.4 Plan-Apochromat oil immersion objective on an LSM 510 Zeiss-Meta laser confocal microscope. The detector pinhole was set to give 0.6-µm optical slides. The images were processed using Zeiss LSM 510 and Adobe Photoshop software. Quantification was achieved by calculating Pearson’s coefficient using the Image J JACoP plugin as previously described.48
Time-lapse confocal-fluorescence microscopy
To visualize the connection between the endocytic and autophagic routes, coverslips of stably transfected Hela-GFP-LC3 were wounded with a p200 pipet tip, and then incubated with an anti-β1integrin monoclonal antibody (P5D2 1:100 and Alexa-Fluor 594 anti-IgG antibody) for 4 h at 37 °C, treated or not with rapamycin (200 nM for 4 h). After the incubation, the coverslips were placed in a temperature- and CO2-controlled incubator chamber mounted on the microscope stage, and examined using time-lapse fluorescent microscopy. Laser lines at 470 nm (for pEGFP constructs) and 590 nm (Alexa 594-conjugated secondary antibodies) were used; the power of the lasers was kept to a minimum in order to minimize photobleaching and photocytotoxicity. A total of approximatively 200 images were obtained; each had a timelapse of 1.2 s, and the images were taken with the green and red filters sets on the CDD HSm (High Speed) camera laser on the AxioObserver Z1_Colibri ZEISS fluorescence microscope with 63 × 1.4 Plan-Apochromat oil immersion lens. The videos obtained were analyzed in Zeiss LSM Image Browser 3.5 for double labeling, and deconvoluted with AutoDeblur X Gold-Edition (AutoQuant) algorith2D blind deconvolution software.
Visualization of the distribution of β1 integrin in focal adhesion by TIRF Microscopy
Hela-GFP-LC3 were seeded in µ-Dish glass bottom (ibidi) at 0.12 × 105 cells/0.22cm2, and cultured for 24 h in complete medium. Non-confluent cells were washed, and then incubated with anti-β1 integrin monoclonal antibody (clone P5D2 at 1:50) and Alexa fluor 555 anti-IgG antibody (1:2000) for 30 min at 4 °C; they were then washed and re-incubated with complete medium or EBSS. Cells were examined with an inverted microscope, AxioObserver Z1_Colibri (Zeiss) equipped with a Peltier-cooled (−40 °C) CoolSnap HQ2 CCD camera (Photometrics), an XL incubator, and the Laser TIRF 3 slider. The penetration depth was 76 nm (72.3 ° angle), making it possible to observe events at or near the plasma membrane. All images were acquired with a Plan-Apochromat 100 × /1.6 NA oil immersion objective lens using a 561-nm diode laser and a BP 616/57-nm emission filter. For TIRF microscopy-videos, frames were acquired every 3 min for two hours under physiological conditions, i.e., 5% CO2 and 37 °C. The focus was maintained by the Definite focus device.
Measuring the surface re-expression of β1 integrin after endocytosis by flow cytometry analysis
The cells were plated in a 10-cm tissue culture dish at density of 5 × 105 cells, and cultured overnight. The cells were or were not starved by being placed in EBSS for 2 h at 37 °C. The cells were incubated for 2 h at 37 °C either in complete medium or in EBSS starvation medium, and then incubated with P5D2 anti-β1 integrin monoclonal antibody (1:75) for 30 min at 4 °C. The cells were then washed and re-incubated in complete medium (control) or in EBSS for the times indicated at 37 °C. For flow cytometry analysis by indirect immunofluorescence, cells were fixed with 1% paraformaldehyde in PBS for 30 min before being scraped off. The cells were then incubated with anti-mouse IgG coupled with biotin (1:100) for 20 min at 4 °C, and then with streptavidin coupled with allophycocyanin to amplify the fluorescence intensity. The cells were analyzed on an LSRFortessa flow cytometer (BD Biosciences), and the data processed using BD FACSDiva and FlowJo (Tree Star) software.
Statistical analysis
Data are reported as the mean of 3 independent experiments ± standard deviation (SD). One-tailed t-tests were used to determine whether the differences in a given assay were statistically significant. A P value of < 0.05 was considered significant. For the Figure 7, data reported are the mean of 3 independent experiments ± standard (SEM)
Supplementary Material
Acknowledgments
We are grateful to Aviva M Tolkovsky (University of Cambridge, UK) for kindly sharing HeLa-GFP-LC3 cells and HeLa-GFP-CD63, to Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science, Japan) for sharing atg3−/− MEFs and to Noboru Mizushima (Tokyo Medical and Dental University, Japan) for sharing atg5−/− MEFs, to Saieb Ghavami (Depts of Physiology and Internal Medicine, University of Manitoba, Canada) for kindly sharing MCF7 shSC20 and MCF7 shAtg7 cells. The anti-Atg7 polyclonal antibody was generously provided by William A Dunn (University of Florida, USA). This work was supported by institutional funding from INSERM, University Paris-Sud 11, and grants from the ANR (Agence Nationale de la Recherche) and INCa (Institut National du Cancer). AH is a recipient fellowship of the Ligue contre le Cancer.
Glossary
Abbreviations:
- ATG
autophagy related gene
- DRAM
damage-regulated autophagy modulator
- EBSS
Earle balanced salt solution
- GFP-LC3
green fluorescent protein-microtubule-associated protein-1 light chain 3
- LAMP
lysosomal associated membrane protein
- MTOR
mechanistic target of rapamycin
- PBS
phosphate buffer saline
- PE
phosphatidylethanolamine
- PI3KC
phosphatidylinositol 3-kinase complex
- S6K1
ribosomal protein S6 kinase 1
- TIRF
total internal reflection fluorescence
- ULK-1
Unc-51-like kinase 1
- VPS34
vacuolar protein sorting 34
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26298
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26298
References
- 1.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 2.Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic. 2006;7:14–21. doi: 10.1111/j.1600-0854.2005.00362.x. [DOI] [PubMed] [Google Scholar]
- 3.Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–53. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Chao WT, Kunz J. Focal adhesion disassembly requires clathrin-dependent endocytosis of integrins. FEBS Lett. 2009;583:1337–43. doi: 10.1016/j.febslet.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ezratty EJ, Partridge MA, Gundersen GG. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol. 2005;7:581–90. doi: 10.1038/ncb1262. [DOI] [PubMed] [Google Scholar]
- 7.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8:835–50. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 8.Pellinen T, Arjonen A, Vuoriluoto K, Kallio K, Fransen JA, Ivaska J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of beta1-integrins. J Cell Biol. 2006;173:767–80. doi: 10.1083/jcb.200509019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 10.Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci. 2009;46:210–40. doi: 10.1080/10408360903044068. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 12.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–82. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–21. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tooze SA, Razi M. The essential role of early endosomes in autophagy is revealed by loss of COPI function. Autophagy. 2009;5:874–5. doi: 10.4161/auto.9066. [DOI] [PubMed] [Google Scholar]
- 16.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21:683–90. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol. 2010;298:C776–85. doi: 10.1152/ajpcell.00507.2009. [DOI] [PubMed] [Google Scholar]
- 18.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 19.Criollo A, Chereau F, Malik SA, Niso-Santano M, Mariño G, Galluzzi L, Maiuri MC, Baud V, Kroemer G. Autophagy is required for the activation of NFκB. Cell Cycle. 2012;11:194–9. doi: 10.4161/cc.11.1.18669. [DOI] [PubMed] [Google Scholar]
- 20.Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 2012;11:2022–9. doi: 10.4161/cc.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–51. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–40. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–57. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 25.Holz MK. The role of S6K1 in ER-positive breast cancer. Cell Cycle. 2012;11:3159–65. doi: 10.4161/cc.21194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–60. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–7. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 29.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 30.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 31.Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–8. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–8. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 33.Lobert VH, Brech A, Pedersen NM, Wesche J, Oppelt A, Malerød L, Stenmark H. Ubiquitination of alpha 5 beta 1 integrin controls fibroblast migration through lysosomal degradation of fibronectin-integrin complexes. Dev Cell. 2010;19:148–59. doi: 10.1016/j.devcel.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O’Connor KL, Gao T, et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71:3246–56. doi: 10.1158/0008-5472.CAN-10-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L, Parent CA. Review series: TOR kinase complexes and cell migration. J Cell Biol. 2011;194:815–24. doi: 10.1083/jcb.201102090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Carito V, Bonuccelli G, Martinez-Outschoorn UE, Whitaker-Menezes D, Caroleo MC, Cione E, Howell A, Pestell RG, Lisanti MP, Sotgia F. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11:3403–14. doi: 10.4161/cc.21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci. 2013;126:1059–69. doi: 10.1242/jcs.123075. [DOI] [PubMed] [Google Scholar]
- 39.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–41. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 40.Bejarano E, Girao H, Yuste A, Patel B, Marques C, Spray DC, Pereira P, Cuervo AM. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012;23:2156–69. doi: 10.1091/mbc.E11-10-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kadandale P, Kiger AA. Role of selective autophagy in cellular remodeling: “self-eating” into shape. Autophagy. 2010;6:1194–5. doi: 10.4161/auto.6.8.13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Indelicato M, Pucci B, Schito L, Reali V, Aventaggiato M, Mazzarino MC, Stivala F, Fini M, Russo MA, Tafani M. Role of hypoxia and autophagy in MDA-MB-231 invasiveness. J Cell Physiol. 2010;223:359–68. doi: 10.1002/jcp.22041. [DOI] [PubMed] [Google Scholar]
- 44.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 46.Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, Dinsdale D, Condorelli F, Brandner S, Campanella M, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699–712. doi: 10.1038/onc.2012.111. [DOI] [PubMed] [Google Scholar]
- 47.Ghavami S, Mutawe MM, Schaafsma D, Yeganeh B, Unruh H, Klonisch T, Halayko AJ. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2012;302:L420–8. doi: 10.1152/ajplung.00312.2011. [DOI] [PubMed] [Google Scholar]
- 48.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.