

Natural products constructed entirely or partially of N-methyl peptide building blocks display many interesting activities. Some, such as cyclosporine, are in use clinically. N-methylation has the effect of both stabilizing the peptide backbone towards proteolytic degradation and, by removing highly hydrated N-H bonds, generally improves the cell permeability and bioavailability of these compounds (Rezai et al., 2006a; Rezai et al., 2006b; Yu et al., 2005). N-methylation can also strongly stabilize one or a few particular conformations of the backbone (Goodman and Fried, 1967; Zhang et al., 2006), particularly in the context of cyclic structures (Chatterjee et al., 2009; White et al., 2011)
N-methylated peptides are a sub-family of peptide tertiary amides (PTAs) (Fig. 1A) in which the main chain nitrogen is alkylated. Another PTA sub-class that has garnered considerable attention is the peptoid scaffold (Simon et al., 1992) (Fig. 1, R1 = H). Peptoids are oligomers of N-alkylated glycines. Like N-methyl peptides, they are also stable to proteases and exhibit enhanced cell permeability relative to standard peptides (Kwon and Kodadek, 2007; Miller et al., 1994). In contrast to N-methyl peptides, there have been numerous reports of the construction of large synthetic combinatorial peptoid libraries that have been screened successfully for protein ligands (Kodadek, 2011; Kodadek et al., 2004; Sanchez-Perez et al., 2003; Zuckermann and Kodadek, 2009). This is largely due to the ease of constructing peptoid libraries via the “sub-monomer” route (Fig. 1B, R1 = H) (Figliozzi et al., 1996; Uno et al., 1999; Zuckermann, 1992). In this scheme, an activated ester of 2-bromoacetic acid (or the chloro analogue (Burkoth et al., 2003)) is added to an amine-displaying resin. The halide is then displaced by a nucleophilic primary amine, providing a peptoid monomer. Since hundreds of suitable primary amines are available commercially or synthesized readily, large one bead one compound (OBOC) libraries of peptoids can be made easily using the split and pool strategy (Lam et al., 1991). These libraries can be screened on bead against labeled, soluble proteins (Alluri et al., 2003; Lim et al., 2007; Wrenn et al., 2007; Xiao et al., 2007) or against cells (Aina et al., 2005; Lee et al., 2010a; Udugamasooriya et al., 2008) to identify novel protein ligands. However, these “hits” do not generally display high affinity, with rare exceptions (Zuckermann et al., 1994). This may be due, in part, to the “floppiness” of peptoids. N-substitution and the lack of a chiral center in peptoids results in the loss of the preference for the trans amide bond conformation characteristic of simple peptides. Moreover, there is little preference for particular conformations around the carbonyl-Cα or Cα-N bonds. Assuming that the peptoid binds its target protein in a defined conformation, this means a significant entropic penalty must be paid to form a complex, limiting affinity. Peptoid floppiness may also render hit optimization difficult.
Figure 1.
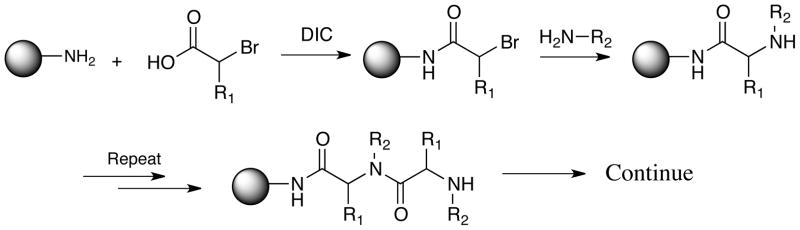
Synthesis of peptide-like oligomers. A. Basic structures of peptides, peptoids and PTAs (peptide tertiary amides). R = general substituent. B. Solid-phase sub-monomer synthesis of peptoids (when R1 = H).
Therefore, we, and others, have been interested in creating peptoid (Chongsiriwatana et al., 2008; Gorske and Blackwell, 2006; Huang et al., 2006; Kwon and Kodadek, 2008; Lee et al., 2011; Lee et al., 2010b; Shah et al., 2008; Shin et al., 2007; Stringer et al., 2011), or peptoid-like (Aquino et al., 2011; Sarma and Kodadek, 2011) molecules with greater conformational constraints.
Previous studies of N-methylated peptides have indicated that they are far more constrained conformationally than either peptoids or simple peptides. For example, CD spectra of poly-N-methyl-L-alanine indicated a helical structure (Goodman and Fried, 1967), though X-ray crystallography of (N-Me-Ala)6 shows a more extend, β-strand-like conformation (Zhang et al., 2006). A striking indication of the much greater conformational constraints in these molecules have been reports of conformational isomers of the same compound that can be separated by HPLC (Alfredson et al., 1994; Teixido et al., 2005). This can be rationalized on the basis of non-bonded steric interactions. For example, as shown in Fig. 2A, the trans amide bond geometry will be strongly favored in N-methylalanine in order to separate the two branched stereocenters, as is also true in simple peptides. Moreover, allylic 1,3 (A1,3) strain will act to restrict rotation about the carbonyl-Cα and the Cα-nitrogen bonds so as to keep the hydrogen atom at the chiral center in or near the plane of the substituted amide (Fig. 2B). These effects are seen clearly in the crystal structure of (N-Me-Ala)6 (Zhang et al., 2006).
Figure 2.

Conformational analysis of peptide tertiary amides, using a simple model compound in which all of the substituents are methyl groups. The bond whose conformation is considered is highlighted in red. A. amide bond conformations. B. Carbonyl-Cα bond conformations. C. Cα-N bond conformations. The conformations indicated as favorable are seen in the crystal structure of hexa-N-methylalanine (Zhang et al., 2006).
Of course, the same arguments would apply to molecules containing other N-substituents besides methyl. Thus it would be of great interest to construct combinatorial libraries of diverse N-alkylated peptides and investigate them as sources of bioactive molecules, but this has never been done. N-methylated, N-protected amino acid building blocks are available (Biron and Kessler, 2005), providing the building blocks required for the synthesis of OBOC libraries of N-methylated peptides by standard peptide bond couplings. But to apply a standard peptide synthesis approach to libraries with different N-substituents would entail the maintenance of a huge number of different N-substituted, Fmoc-protected amino acid building blocks. To address this important goal, we therefore considered a different solution: to create libraries of PTAs via sub-monomer synthesis in which chiral 2-bromo carboxylic acids are employed in place of 2-bromoacetic acid (Fig. 1B; R1 ≠ H).
We report here a relatively general procedure for the creation of high quality OBOC libraries of diverse N-substituted alanines and also demonstrate that the same chemistry can be applied to include residues other than methyl at the alpha carbon. Moreover, we demonstrate that these libraries are indeed a source of high affinity ligands for a protein target. Characterization of one of these PTA-protein complexes provides strong evidence for the importance of conformational constraints in supporting high affinity binding.
Result and Discussion
Synthesis of chiral sub-monomers
To construct PTAs via a peptoid-like, sub-monomer synthetic route (Zuckermann et al., 1992), chiral 2-bromo carboxylic acids must be employed in place of 2-bromoacetic acid (Figure 1B, R≠H). These building blocks can be prepared from α-amino acids by treatment with sodium nitrate in the presence of KBr and HBr (see supplemental Figure 1S.A) (Izumiya and Nagamatsu, 1952; Tanasova et al., 2009). This reaction proceeds with retention of stereochemistry, presumably because, after conversion of the amine to a diazonium group, a reactive three-membered lactone is formed that is then opened by bromide ion (Izumiya and Nagamatsu, 1952).
To make full use of both enantiomers of a given 2-bromo carboxylic acid in the construction and screening of PTA combinatorial libraries, a method to decode stereochemistry in each monomer is required. Since tandem mass spectrometry is the most convenient method to elucidate the structure of “hits” after screening one bead one compound (OBOC) combinatorial libraries (Astle et al., 2010; Paulick et al., 2006; Thakkar et al., 2009), isotope encoding of the absolute stereochemistry in PTAs is required. This meant accessing deuterated amino acids, which can be accomplished through transaminase-catalyzed hydrogen-deuterium exchange using D2O as the donor. Following the published procedure (Oshima and Tamiya, 1961), more than 90% of the L-alanine was labeled (with deuterium on both the α and β carbons. When the procedure was repeated three times, >98% isotopic purity was obtained (see supplemental Figures S1 and S2). This simple procedure can be performed on a large scale (>30g) since L-alanine, D2O and transaminase are all inexpensive.
Isotopically labeled L-alanine was then converted to the bromide as described above. After distillation under reduced pressure, the desired acid displayed high stereochemical purity (>95% e.e.) as determined by HPLC of the corresponding (+)-camphorsultam amide (see supplementary Figure S1 D).
Sub-monomer synthesis of optically pure N-substituted alanines
The peptide bond-forming step in the sub-monomer synthesis of peptoids usually employs a carbodiimide such as diisopropylcarbodiimide (DIC) to activate bromoacetic acid. While the use of this reagent can be problematic in peptide synthesis due to racemization at the α-carbon, this was not anticipated to be an issue in PTA synthesis via the sub-monomer route because amino acid racemization involves formation of an intermediate oxazalone that cannot form in this class of molecules (Goodman and Levine, 1964; Yuan et al., 2002). To confirm this, R and S bromopropionic acid were condensed with a chiral amine ((S)-(−)-1-Phenylethylamine) under these conditions. No evidence for racemization was observed in the NMR of the final products (see supplemental Figure S2).
To determine if oligomers prepared via the sub-monomer route indeed display the anticipated conformational constraints, we constructed N-methyl-(S)-alanine oligomers of various lengths via the sub-monomer route on Rink amide resin. As shown in Fig 3., an increased circular dichroism signal was observed as the length of the oligomer increased from a dimer to a tetramer to a hexamer, suggesting the build-up of specific conformers as the length of the molecule increases. The CD spectrum of the hexamer shown in Fig. 3 is essentially identical with the published spectrum of the same compound made by traditional amino acid couplings (Zhang et al., 2006). Thus, this chemistry provides a route to highly structured oligomers.
Figure 3.

CD spectrum of the N-methyl-alanine dimer, tetramer and hexamer synthesized via the sub-monomer route. A) Spectrum obtained in an aqueous solution (PBS buffer pH=7). B) Spectrum obtained in a trifluoroethanol solution. All spectra were recorded at 0.3 μM compound, path length 10 mm, at 298°K.
Synthesis and characterization of a combinatorial library
With the appropriate conditions in hand, we proceeded to construct a combinatorial library of trimeric PTAs following an invariant linker. The synthesis was conducted on TentaGel beads, which is the preferred resin for subsequent screening (Alluri et al., 2003). 2-Bromoacetic acid, (S)-2-bromopropanoic acid-d4 and (R)-2-bromopropanoic acid) were employed as sub-monomers along with 16 amines (see Fig. 4). The theoretical diversity of the library was 110,592 compounds. The amines were chosen carefully such that any combination of amine and acid sub-monomers, could be identified uniquely by tandem mass spectrometry upon gas phase cleavage of the amide bond. Methionine was added to the beads first using standard peptide coupling conditions (HBTU, Fmoc-Met-OH, DIEA) to facilitate selective release of the compounds from the beads after the screen by treatment with CNBr. Following the methionine, several invariant peptoid residues were added. The two Nlys residues facilitate display of the molecule in aqueous solution while the alkyne and furan side chains facilitate subsequent labeling of the compound (Hintersteiner et al., 2009) and immobilization on a microarray (Astle et al., 2010) if desired. Moreover, the additional mass serves to move the molecular ion and important fragment peaks out of the range of the matrix peaks, which facilitates compound characterization.
Figure 4.

Structure of the library constructed. The invariant linker is shown in blue. The acid sub-monomers employed (X) are shown in red. The amines used are shown in black. Protecting groups employed on the acid and alcohols are not shown.
Following linker synthesis, the library was split in three, treated with DIC and (R)-2-bromopropanoic acid, (S)-2-bromopropanoic acid-d4 or bromoacetic acid respectively. The beads were then mixed together and then split in 16 equal parts. One of the amines was added to each tube. The same procedures were then repeated two more times in order to form the trimer. The acylation step was monitored by the chloranil test and the amine substitution was monitored by the silver acetate test (details are described in supplemental material). In addition, individual beads were sampled at each step. After releasing the compound using CNBr, the progress of the reaction was checked by MALDI mass spectrometry. This monitoring is important to create a high quality library. The acylation step in PTA synthesis, in particular, can be significantly slower than is the case for peptoid synthesis. Thus, while the conditions reported above for DIC-mediated coupling work well much of the time, this depends to some extent on the N-substituent. The blind use of a single set of reaction conditions without monitoring the reaction can result in a significant fraction of incomplete coupling.
In tubes where the reaction had not proceeded to >90% completion using the standard procedure, a second round of coupling was done by adding a 2:1 mixture of the bromopropanoic acid and DIC (after a pre-activation time of 10 minutes, final concentration of activated bromopropanoic acid at 1M). Prolonged reaction time (24 hours) provided a satisfactory yield in these difficult reactions. With respect to assessing the progress of the reactions by mass spectrometry, it is noteworthy that the amide bond on the C-terminal side of the PTA fragments easily and is much more acid labile than in standard peptoids (see Supplementary Figure S5). This results in the production of an abundant b ion on the MALDI mass spectrum even without high energy MS/MS fragmentation. The same effect has been noticed previously in mass spectrometric analysis of N-methyl peptides. (Vaisar and Urban, 1998)
After the final step, the entire library was washed thoroughly and 40 beads were picked randomly. The beads were treated with CNBr to release the compounds from the beads, which were then analyzed by tandem MALDI mass spectrometry. 37 out of 40 beads were sequenced successfully (see supplemental Figure S4 for some examples). All 16 of the amines and all three of the acids were found in these spectra, suggesting that all of the sub-monomers had incorporated in this synthesis. However, we observed no trimers in which a methyl group was present at all three positions and some dimers were also found. This is clear evidence that in the demanding library synthesis format, the reaction conditions employed resulted in some level of failure when attempting to string together three chiral centers in a row, despite all of our efforts. As will be described below, a modified coupling protocol has solved this problem. Nonetheless, since most compounds in the library could be sequenced, we decided to proceed with a screening experiment to probe the idea that the greater conformational constraints in the PTA relative to peptoids might result in higher affinity ligands.
Isolation and characterization of protein ligands from the PTA library
The library shown in Fig. 4 was screened for ligands to a single chain variable fragment (scFv) antibody called PX4-4, which was derived from a patient with the skin blistering disease Pemphigus vulgaris (Yamagami et al., 2010). Approximately 250,000 beads were incubated with 10 μM of purified scFv PX4-4 in the presence of 1 mg/ml E. coli lysate as a diverse source of competitor proteins. To isolate beads that display ligands that captured significant amounts of the target antibody, magnetic beads displaying protein L were added to the mixture. Protein L will bind tightly to the scFv antibody through the kappa light chains. After a brief incubation, a powerful magnet was employed to segregate the beads that had also affixed to the magnetic protein L-displaying beads (Astle et al., 2010). 33 beads were isolated as possible hits.
To confirm that these beads were true hits, they were stripped of protein by incubation in a trypsin solution at 37°C for one hour, then washed thoroughly. The beads were then incubated with 10 μM of purified scFv PX4-4 again for 30 minutes and washed gently. A 1:200 dilution of red quantum dots conjugated to protein L was then added. After 30 minutes of incubation, brightly glowing beads were picked manually using a low power fluorescence microscope to visualize them. 17 of the hits picked up magnetically displayed a red glow (not shown), consistent with their being PX4-4 ligands. These validated hits were released from the beads with CNBr and sequenced by tandem mass spectrometry.
The most promising hit from the bead screen (as judged by the intensity of the red halo after quantum dot addition), compound 1, was re-synthesized on Rink amide resin with a fluorescein tag, cleaved and purified by HPLC. As shown in Fig. 5A, this compound proved to have chiral centers at the first and third variable positions of the library, whereas there was no Cα substituent at the second position.
Figure 5.

Structure and characterization of a ligand for the scFv PX4-4. A. Structure of a fluorescein-labeled derivative of one of the screening hits, compound 1, and the peptoid analogue 2, which lacks the methyl groups at the chiral centers. The invariant linker is shown in blue. B. Fluorescence polarization assay employing the fluorescein-tagged compounds and the indicated proteins. PTA 1 binds the PX4-4 scFv with a KD of approximately 3 μM, but binds much less well to a collection of IgG antibodies, indicating selectivity for PX4-4. The peptoid 2 binds poorly to PX4-4.
The affinity of compound 1 for PX4-4 was determined by fluorescence anisotropy. As shown in Fig. 5B, PTA fluorescein-conjugated 1 exhibited a saturable binding curve when titrated with increasing concentrations of PX4-4, indicating a KD of approximately 5 μM. In contrast, little binding was observed when fluorescein-tagged 1 was mixed with bulk IgG antibodies, indicating selectivity for PX4-4.
The peptoid analogue of 1, compound 2, which lacks any chiral centers, was also made. As shown in Fig. 5B, this compound had a much lower affinity for PX4-4. The large difference in affinity of PTA 1 and peptoid 2, which differ only in the presence or absence of the methyl groups at the chiral centers, is striking. While we cannot rule out the possibility that the Cα methyl groups in PTA 1 contact the scFv antibody directly, these data strongly support the idea that the conformational constraints afforded by the presence of the chiral centers strongly stabilizes binding of the small molecule to the antibody fragment.
To more thoroughly probe the influence of the stereochemistry at each position of PTA 1, a small library was synthesized in which the amines were held invariant as found in 1, but all possible combinations of the three acid sub-monomers were employed at each alpha position. In order to ensure the efficient synthesis of PTA trimers, we performed an extensive optimization of acylation conditions as well as conditions for amination. Of the eight coupling reagents examined, the triphosgene reagent bis(trichloromethyl) carbonate (BTC) developed by Jung and colleagues (Thern et al., 2002) was found to be the best by far, providing superior yields with no racemization (see supplemental material Figure S6).
Several beads were chosen randomly from the library, and the compounds were released and analyzed by mass spectrometry. Gratifyingly, in this case, several of the compounds were trimers with chiral centers at each position. Indeed, close to the expected ratio of compounds was observed (see supplemental material Figure S6), demonstrating that the use of the BTC-based protocol is appropriate for the synthesis of high quality PTA libraries.
96 beads were chosen randomly from the library and placed individually into the wells of a 96-well filter plate. Each well was treated with fluorescein azide and a copper catalyst to attach fluorescein to the alkyne handle (Hintersteiner et al., 2009) and then the compounds were cleaved from beads. The beads were filtered out and the soluble compounds were used for the fluorescence polarization (FP) assay. All 27 possible structures were found in this collection of molecules by tandem MS sequencing. Semi-quantitative binding constants for the PX4-4 antibody for all 27 of the compounds were determined by titrating the compounds with the protein in the microwell format and monitoring the increase in FP (see Supplementary Table S2). The six highest affinity compounds, which included A48, identical to hit 1 in the variable region, were re-synthesized and purified to allow more accurate binding data to be acquired. All six compounds showed an increase in FP when titrated with PX4-4 (Figure 6). Most of the curves approached saturation, allowing for an accurate determination of the KD. In contrast, compound A41, corresponding to peptoid 2, was not saturated in the protein concentration range tested (100 nM to 400 μM) (Figure 6).
Figure 6.

Binding isotherms for A48 (the labeled analogue of 3) and the five other highest affinity members of the library in which the stereochemistry at each alpha-carbon of 3 was altered. A) General structure of the tested compounds and the KD determined by FP assay after re-synthesis as shown in B. The KD values are given in μM. B) Fluorescent polarization assay of a titration of fluorescein-labeled compound with PX4-4 C) Titration of fluorescein-labeled compound with a collection of human IgG antibodies (C). Note the different scales of the y-axes in B and C. The binding of the peptoid A41 to PX4-4 and whole human IgG is similar.
These data confirmed that compound 1 is the highest affinity PX4-4 ligand of the six and that the chirality of the stereogenic centers can have a profound effect on binding affinity. It is interesting that the compounds with the S,S,R or S,R,R configurations (the same as compound 1 in the first and third positions, but with a chiral center at position 2) were not high affinity ligands for PX4-4 (see supplementary material Table S2). This indicates that a stereocenter at the second position does not allow the molecule to access the bound conformation.
To address the selectivity of compounds A14, A7, A68, A41 and A24 for PX4-4, the FP experiment was repeated using a mixture of human IgG as we did previously for compound 1. A14, A7, A68 and A24 showed no binding with whole human IgG while A41, the analogue of peptoid 2, had a similar affinity for these random antibodies as it did for PX4-4 (Figure 6). This implies that greater conformational flexibility is associated with binding promiscuity, which has been noted previously in studies of N-methylated peptides (Doedens et al., 2010; Fiacco and Roberts, 2008).
Beyond N-substituted oligo-N-substituted alanines
The synthetic route employed to access the chiral bromides works well with other amino acids besides alanine (Izumiya and Nagamatsu, 1952; Tanasova et al., 2009). Thus, we conducted a small, preliminary study of whether bulkier substituents at the α-carbons in 1 might increase the affinity of the PTA ligand for PX4-4. Four fluorescein-labeled compounds were synthesized in which a benzyl or an isobutyl group was substituted for the chiral methyl groups of 1. The crude HPLCs of three of these compounds were quite clean while the fourth had some level of impurities (see Supplementary Material Figures S7 and S8). Their affinities for PX4-4 were measured by fluorescence polarization spectroscopy. As shown in Supplementary Figure S7, none of the compounds bound PX4-4 as tightly as the parent compound. The affinities were reduced between 2- to 20-fold for the four compounds. While improved PX4-4 ligands were not discovered in this small experiment, it shows that PTAs other than oligo-alanines can be accessed via this chemistry.
Summary and Conclusion
We report the synthesis of diverse libraries of peptide tertiary amides (PTAs) using the sub-monomer approach first developed for peptoid synthesis (Figliozzi et al., 1996; Zuckermann, 1992). This involves the use of chiral, 2-bromo acids as sub-monomers in place of 2-bromoacetic acid, several of which can be derived easily from amino acids in high yield and optical purity (Izumiya and Nagamatsu, 1952). Moreover, at least in the case of the alanine-derived 2-bromoacid employed in this study, a simple transaminase-mediated deuteration protocol provides isotopically labeled material to allow the absolute stereochemistry at the chiral center of the PTA to be determined by mass spectrometry. This, in turn, allows both the R and S acids to be used as sub-monomer diversity elements at any given position in the chain. As anticipated from studies of N-methyl peptide synthesis, the peptide bond couplings are more difficult than is the case for simple peptoids due to greater steric crowding, but the use of highly potent carboxylate-activating agents such as Jung’s BTC reagent (Thern et al., 2002; Videnov et al., 1996) allows the synthesis of high quality libraries by spit and pool synthesis. In this initial report, we have mostly limited the substitution at the α-carbon to methyl (i.e., the synthesis of N-alkylated oligoalanines), but, as shown in Supplementary Figures S7 and S8, other groups can be accommodated at this position.
The impetus behind the development of this chemistry was the need to create libraries of oligomers with greater conformational constraints than simple peptoids. Peptoid libraries are a useful source of protein ligands, but the primary hits from these libraries tend to be of modest affinity. This is likely due, in part, to the fact that peptoids are quite floppy molecules that must sacrifice a good deal of entropy in order to assume their bound conformation. Although various clever strategies have been reported to conformationally constrain peptoids (Yoo and Kirshenbaum, 2008), none of these have been applied to the synthesis of large combinatorial libraries, probably because of the extremely high efficiency demanded of the chemistry for this application (stepwise yields in excess of 95%).
N-methyl peptides are know to have significant conformational constraints (Chatterjee et al., 2009; Goodman and Fried, 1967; Zhang et al., 2006) resulting from both a strong preference for the trans amide bond geometry as well as potent allylic 1,3 strain effects that bias the conformations of the other two types of bonds in the molecule. Although chemistry exists to access Fmoc-protected, N-methyl amino acid monomers (Biron and Kessler, 2005), only small libraries of N-methyl peptides have been created by parallel synthesis (Ovadia et al., 2011). To the best of our knowledge, large libraries of these molecules have never been created synthetically and screened for protein ligands, though there has been considerable activity in the creation of such libraries using biological approaches (Kawakami et al., 2008; Subtelny et al., 2008). Thus, the PTA library described here is, to the best of our knowledge, the first large, chemically synthesized, conformationally constrained library of N-alkylated peptides.
To determine if this type of library would indeed be superior to a collection of simple peptoids, we created a library with three variable positions and screened it against an autoantibody of interest. 16 amines and three bromoacids (R- and S-2-bromopropionic acid and bromoacetic acid) were employed as sub-monomers. Thus, the library contained 3375 peptoids (163) amongst the 110,592 PTAs, yet no simple peptoids were isolated as hits from the screen. All of the hits had at least one chiral center. Moreover, characterization of the best hit, compound 1, demonstrated that it evinced a high affinity for the target antibody (KD = 5 μM), whereas the peptoid analogue, 2, had a much lower affinity for the autoantibody and also demonstrated more promiscuous binding behavior. Finally, an analysis of all possible stereoisomers of hit 1 revealed that the compound isolated was indeed the highest affinity ligand amongst this groups and that the vast majority of stereoisomers evinced much lower affinity for the target. Only two other compounds bound to PX4-4 with an affinity within ten-fold of A48. While it is impossible to understand exactly what this means in the absence of a structure of the PTA-antibody complexes, it may be that compounds A14 and A7 (Table 1) can place the side chains in something close to the three-dimensional arrangement achieved by A48 in it bound conformation, despite their different backbone stereochemistry. Efforts to obtain crystal structures are ongoing.
In conclusion, this work makes accessible libraries of conformationally constrained N-alkylated peptide oligomers that promise to be exceptional ligands for biological targets of interest.
Significance
Peptide tertiary amides (PTAs) (also called N-alkylated peptides) have long been known to be conformationally constrained. This suggests that libraries of these compounds might be a superior source of protein ligands than the corresponding libraries of relatively “floppy” peptoids or peptides. However, chemistry to create high quality PTA libraries was lacking. This report demonstrates that large one bead one compound libraries of N-alkylated alanines can be constructed by a peptoid-like sub-monomer synthesis using either enantiomer of 2-bromopropionic acid as a building block. A model screening experiment suggests that these molecules will indeed provide higher affinity binding to protein targets than unconstrained peptoids.
Materials and Methods
Synthesis of chiral bromoacid
D-alanine (8.9 g, 0.1 mol) and and KBr (11.9 g, 0.1 mol) was dissolved in 100ml, of a 30% HBr water solution and kept in −15°C dry ice/ethylene glycol bath. NaNO2 (10.35 g, 0.15 mmol) were dissolved in 15 ml water, and slowly dripped in the above solution under argon atmosphere. The reaction was allowed to proceed for 3 hours and to warm from −15°C to room temperature. It was then put under vacuum for 30 min. Product was extracted by diethyl ether (25ml × 3). Organic phase were combined and dried over Na2SO4. Then the solvent was evaporated under vacuum and the crude product were further purified by distillation at 115°C, high vacuum. The pure product was obtained as colorless oil in 82% yield.
Isotopic labeling of alanine
L-alanine (300 mg, 3.36 mmol) was dissolved in 10 ml of D2O, α-ketoglutarate (10 mg, 0.068 mmol) was then added as co-substrate, the whole system was then warmed up to 37°C and adjusted pD to 8.5~8.7 with NaOD. Alanine transaminase (0.1 mg, EC 2.6.1.2 from pig heart, Roche Diagnostics, Indianapolis, IN) was added, and incubated at 37°C overnight with mild shaking. 90% of D2O was recovered by distillation and L-alanine-d4 was obtained by lyophilization.
Library synthesis with DIC
Tentagel beads (1 g, 160 μm, ~500,000 beads, 0.52 mmol/g, cat# HL 12 162, Rapp-Polymere GmbH, Germany) were swelled in DMF for 2 hours before use. DMF was used as solvent unless otherwise mentioned. Fmoc-Met-OH (0.77 g, 2.08 mmol) was coupled on the beads using HBTU (0.77 g, 2.08 mmol), DIPEA (0.45 ml, 2.6 mmol) for 3 hours. Fmoc was deprotected by 20% piperidine for 30 min. Beads were washed thoroughly with DMF after each step. The beads were split in three portions after deprotection. 2ml 1M DIC solution and 2ml 1M corresponding bromoacid (2-Bromoacetic acid, (S)-2-bromopropanoic acid-d4 or (R)-2-bromopropanoic acid) solution were added together for preactivation for 5 min. The 4 ml combined solution was then added to one portion of beads and gently shook till completion. The reaction was monitored by chloranil test, a clear negative result after 5 min indicates the amine was acylated by the corresponding bromoacid. The beads was then thoroughly washed and pooled together, then split in 16 portions. Each portion was incubated with one of the amines listed in supplemental S13. 2M solution of the amine was used with an incubation time of 12 hours at 50°C. Silver acetate test and chloranil test were used to monitor the completion of the reaction. When reaction is complete, the beads were washed and pooled together. The acylation and amination steps were then repeated two more times for forming the trimer library.
Peptoid linker synthesis with DIC
Peptoid linker was synthesized by standard peptoid synthesis methods using microwave condition and 2M solutions of DIC, bromoacetic acid, and the corresponding amines in DMF.
Library synthesis with BTC
Tentagel beads with Rink linker (1 g, ~100,000 beads, 0.27 mmol/g, cat. # MB 250 230, Rapp-Polymere GmbH, Germany) was used in order to avoid long exposure to acid during cleavage. Beads were first treated by 20% piperidine for 30 min in DMF in order to remove Fmoc and then the linker part is synthesized as described above. The beads was then split in three equal portions, coupled with 2-Bromoacetic acid, (S)-2-bromopropanoic acid-d4 and (R)-2-bromopropanoic acid respectively. For bromoacetic acid, 2M solution of DIC and 2M solution of bromoacetic acid were used as described above. For bromopropanoic acids, BTC was used as coupling reagent. BTC (92.1 mg, 0.31 mmol) was dissolved in 5 ml anhydrous THF in a glass vial. Bromopropionic acid (89 μl, 0.95 mmol) was then added to the vial and the whole vial was kept in −20 °C freezer for 15 min. Beads were washed using DCM, DMF and then THF respectively for 5 times each, then 2:1 THF/DIPEA (750 μl THF, 375 μl DIPEA, 2.2 mmol) was added to the beads and shook gently. 2,4,6-Trimethylpyridine (356 μl, 2.7 mmol), was added to the cold solution of bromopropionic acid with BTC, white precipitation was formed following the addition. The white suspension was then applied to the beads and the reaction vessel was put on shaker for 2 hours at room temperature. The solution in the vessel should be a pale yellowish suspension during the whole course of the reaction. A darker color is an indication of excessive heat released during the initial addition of the acid chloride solution. It can be solved by further cooling down the acid chloride solution and the beads. The beads were washed with DCM for five times when reaction was done and then DMF for 5 times. Chloranil test was used to monitor the completion of the reaction. All three portions of beads were then pooled together and the beads were incubated with 2M solution of the corresponding amine in DMF at 60°C overnight. The completion of the reaction was monitored by chloranil and silver acetate test.
Supplementary Material
Acknowledgments
This work supported by a contract from the NHLBI (NO1-HV-00242). We thank Prof. John Stanley (U. of Pennsylvania) for providing the expression vector for PX4-4.
References
- Aina OH, Marik J, Liu R, Lau DH, Lam KS. Identification of novel targeting peptides for human ovarian cancer cells using “one-bead one-compound” combinatorial libraries. Mol Cancer Ther. 2005;4:806–813. doi: 10.1158/1535-7163.MCT-05-0029. [DOI] [PubMed] [Google Scholar]
- Alfredson TV, Bruins PW, Maki AH, Excoffier JL. Conformer interconversion in the LC analysis of triostin A and its under-N-methylated synthetic analogue. J Chromatogr Sci. 1994;32:132–138. doi: 10.1093/chromsci/32.4.132. [DOI] [PubMed] [Google Scholar]
- Alluri PG, Reddy MM, Bacchawat-Sikder K, Olivos HJ, Kodadek T. Isolation of protein ligands from large peptoid libraries. J Amer Chem Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- Aquino C, Sarkar M, Halmers CMJ, Mendes K, Kodadek T, Micalizio G. A biomimetic polyketide-inspired approach to small molecule ligand discovery. Nature Chem. 2011;4:99–104. doi: 10.1038/nchem.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle JM, Simpson LS, Huang Y, Reddy MM, Wilson R, Connell S, Wilson J, Kodadek T. Seamless bead to microarray screening: Rapid identification of the highest affinity protein ligands from large combinatorial libraries. Chem & Biol. 2010;17:38–45. doi: 10.1016/j.chembiol.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron E, Kessler H. Convenient synthesis of N-methylamino acids compatible with Fmoc solid-phase peptide synthesis. J Org Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- Burkoth TS, Fafarman AT, Charych DH, Connolly MD, Zuckermann RN. Incorporation of unprotected heterocyclic side chains into peptoid oligomers via solid-phase submonomer synthesis. J Amer Chem Soc. 2003;125:8841–8845. doi: 10.1021/ja0352101. [DOI] [PubMed] [Google Scholar]
- Chatterjee J, Ovadia O, Gilon C, Hoffman A, Mierke D, Kessler H. N-methylated cyclic pentapeptides as template structures. Adv Exp Med Biol. 2009;611:109–110. doi: 10.1007/978-0-387-73657-0_48. [DOI] [PubMed] [Google Scholar]
- Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens L, Opperer F, Cai M, Beck JG, Dedek M, Palmer E, Hruby VJ, Kessler H. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. Journal of the American Chemical Society. 2010;132:8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco SV, Roberts RW. N-Methyl scanning mutagenesis generates protease-resistant G protein ligands with improved affinity and selectivity. Chembiochem. 2008;9:2200–2203. doi: 10.1002/cbic.200800208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
- Goodman M, Fried M. Conformational aspects of polypeptide structure. XX. Helical poly-N-methyl-L-alanine. Experimental results. Journal of the American Chemical Society. 1967;89:1264–1267. doi: 10.1021/ja00981a042. [DOI] [PubMed] [Google Scholar]
- Goodman M, Levine L. Peptide synthesis via active esters. IV. Racemization and ring-opening reactions of opticlly active oxazolones. J Amer Chem Soc. 1964;86:2918–2922. [Google Scholar]
- Gorske BC, Blackwell HE. Tuning peptoid secondary structure with pentafluoroaromatic functionality: A new design paradigm for the construction of discretely folded peptoid structures. J Amer Chem Soc. 2006;128:14378–14387. doi: 10.1021/ja065248o. [DOI] [PubMed] [Google Scholar]
- Hintersteiner M, Kimmerlin T, Kalthoff F, Stoeckli M, Garavel G, Seifert JM, Meisner NC, Uhl V, Buehler C, Weidemann T, et al. Single bead labeling method for combining confocal fluorescence on-bead screening and solution validation of tagged one-bead one-compound libraries. Chem Biol. 2009;16:724–735. doi: 10.1016/j.chembiol.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Huang K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. A threaded loop conformation adopted by a family of peptoid nonamers. Journal of the American Chemical Society. 2006;128:1733–1738. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya N, Nagamatsu A. Walden invesrion of amino acids. VI. The synthesis of D-Surinamine (N-methyl-D-tyrosine) Bull Chem Soc Japan. 1952;25:265–267. [Google Scholar]
- Kawakami T, Murakami H, Suga H. Messenger RNA-programmed incorporation of multiple N-methyl-amino acids into linear and cyclic peptides. Chem Biol. 2008;15:32–42. doi: 10.1016/j.chembiol.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Kodadek T. Synthetic receptors with antibody-like binding affinities. Current opinion in chemical biology. 2011;14:713–720. doi: 10.1016/j.cbpa.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodadek T, Reddy MM, Olivos HJ, Bachhawat-Sikder K, Alluri PG. Synthetic molecules as antibody replacements. Acc Chem Res. 2004;37:711–718. doi: 10.1021/ar030145l. [DOI] [PubMed] [Google Scholar]
- Kwon YU, Kodadek T. Quantitative evaluation of the relative cell permeability of peptoids and peptides. J Amer Chem Soc. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YU, Kodadek T. Chemical communications. Cambridge, England: 2008. Encoded combinatorial libraries for the construction of cyclic peptoid microarrays; pp. 5704–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- Lee J, Reddy MM, Kodadek T. Discovery of a Positive Allosteric Potentiator of the Orexin Receptors. Chem Sci. 2010a;1:48–54. doi: 10.1039/C0SC00197J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Kim HS, Lim HS. Design and Facile Solid-Phase Synthesis of Conformationally Constrained Bicyclic Peptoids. Organic letters. 2011;13:5012–5015. doi: 10.1021/ol201773f. [DOI] [PubMed] [Google Scholar]
- Lee JH, Meyer AM, Lim HS. A simple strategy for the construction of combinatorial cyclic peptoid libraries. Chemical communications (Cambridge, England) 2010b;46:8615–8617. doi: 10.1039/c0cc03272g. [DOI] [PubMed] [Google Scholar]
- Lim HS, Archer CT, Kodadek T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J Amer Chem Soc. 2007;129:7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg Med Chem Lett. 1994;4:2657–2662. [Google Scholar]
- Oshima T, Tamiya N. Mechanism of transaminase action. Biochem J. 1961;78:116–119. doi: 10.1042/bj0780116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia O, Greenberg S, Chatterjee J, Laufer B, Opperer F, Kessler H, Gilon C, Hoffman A. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol Pharm. 2011;8:479–487. doi: 10.1021/mp1003306. [DOI] [PubMed] [Google Scholar]
- Paulick MG, Hart KM, Brinner KM, Tjandra M, Charych DH, Zuckermann RN. Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J Comb Chem. 2006;8:417–426. doi: 10.1021/cc0501460. [DOI] [PubMed] [Google Scholar]
- Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. Conformational flexibility, internal hydrogen bonding and passive membrane diffusion permeability: successful in silico prediction of the relative permeabilites of cyclic peptides. J Amer Chem Soc. 2006a;128:14073–14080. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J Amer Chem Soc. 2006b;128:2510–2511. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- Sanchez-Perez AM, Montoliu C, Felipo V. Trialkylglycines: a new family of compounds with in vivo neuroprotective activity. CNS Drug Rev. 2003;9:263–274. doi: 10.1111/j.1527-3458.2003.tb00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma BK, Kodadek T. Acylhydrazides as peptoid sub-monomers. Chem Comm. 2011;47:10590–10592. doi: 10.1039/c1cc12750k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. Oligo(N-aryl glycines): A New Twist on Structured Peptoids. Journal of the American Chemical Society. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]
- Shin SB, Yoo B, Todaro LJ, Kirshenbaum K. Cyclic peptoids. Journal of the American Chemical Society. 2007;129:3218–3225. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, et al. Peptoids: a modular approach to drug discovery. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer JR, Crapster JA, Guzei IA, Blackwell HE. Extraordinarily Robust Polyproline Type I Peptoid Helices Generated via the Incorporation of alpha-Chiral Aromatic N-1-Naphthylethyl Side Chains. Journal of the American Chemical Society. 2011;39:15559–15567. doi: 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Hartman MC, Szostak JW. Ribosomal synthesis of N-methyl peptides. Journal of the American Chemical Society. 2008;130:6131–6136. doi: 10.1021/ja710016v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanasova M, Yang TQ, Olmsted CC, Vasileiou C, Li X, Anyika M, Borhan B. An unusual conformation of alpha-haloamides due to cooperative binding with zincated porphyrins. Eur J Org Chem. 2009;2009:4242–4253. [Google Scholar]
- Teixido M, Albericio F, Giralt E. Solid-phase synthesis and characterization of N-methyl-rich peptides. J Pept Res. 2005;65:153–166. doi: 10.1111/j.1399-3011.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- Thakkar A, Cohen AS, Connolly MD, Zuckermann RN, Pei D. High-Throughput Sequencing of Peptoids and Peptide-Peptoid Hybrids by Partial Edman Degradation and Mass Spectrometry. Journal of combinatorial chemistry. 2009 doi: 10.1021/cc8001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thern B, Rudolph J, Jung G. Total synthesis of the nematicidal cyclododecapeptide omphalotin A by using racemization-free triphosgene-mediated couplings in the solid phase. Angewandte Chemie (International ed. 2002;41:2307–2309. doi: 10.1002/1521-3773(20020703)41:13<2307::AID-ANIE2307>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. A peptoid “antibody surrogate” that antagonizes VEGF receptor 2 activity. J Amer Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- Uno T, Beausoleil E, Glodsmith RA, Levine BH, Zuckermann RN. New submonomers for poly N-substituted glycines (peptoids) Tet Lett. 1999;40:1475–1478. [Google Scholar]
- Vaisar T, Urban J. Gas phase fragmentation of protonated mono-N-methylated peptides. Analogy with solution phase acid-catalyzed hydrolysis. J Mass Spectrom. 1998;33:505–524. [Google Scholar]
- Videnov GI, Kaiser D, Kempter MC, Jung G. Synthesis of naturally occurring, conformationally restricted oxazole- and thiazole-containing di- and tripeptide mimetics. Angew Chem Intl Ed. 1996;35:1503–1506. [Google Scholar]
- White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nature chemical biology. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenn SJ, Weisinger RM, Halpin DR, Harbury PB. Synthetic ligands discovered by in vitro selection. Journal of the American Chemical Society. 2007;129:13137–13143. doi: 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Design and synthesis of a cell permeable synthetic transcription factor mimic. J Comb Chem. 2007;9:592–600. doi: 10.1021/cc070023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. The Journal of clinical investigation. 2010;120:4111–4117. doi: 10.1172/JCI44425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo B, Kirshenbaum K. Peptoid architectures: elaboration, actuation, and application. Current opinion in chemical biology. 2008 doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Yu P, Liu B, Kodadek T. A high-throughput assay for assessing the cell permeability of combinatorial libraries. Nature Biotech. 2005;23:746–751. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Blomberg D, Sethson I, Brickmann K, Ekholm K, Johansson B, Nilsson A, Kihlberg J. Synthesis and pharmacological evaluation of an analogue of the peptide hormone oxytocin that contains a mimetic of an inverse gamma-turn. Journal of medicinal chemistry. 2002;45:2512–2519. doi: 10.1021/jm0110744. [DOI] [PubMed] [Google Scholar]
- Zhang S, Prabpai S, Kongsaeree P, Arvidsson PI. Chemical communications. Cambridge, England: 2006. Poly-N-methylated alpha-peptides: synthesis and X-ray structure determination of beta-strand forming foldamers; pp. 497–499. [DOI] [PubMed] [Google Scholar]
- Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient method for the preparation of peptoids (oligo N-substituted glycines) by submonomer solid-phase synthesis. J Amer Chem Soc. 1992;114:10646–10647. [Google Scholar]
- Zuckermann RN, Kerr JM, Kent SBH, Moos WH. Efficient method for the preparation of peptoids [Oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- Zuckermann RN, Kodadek T. Peptoids as potential therapeutics. Curr Opin Mol Ther. 2009;11:299–307. [PubMed] [Google Scholar]
- Zuckermann RN, Martin EJ, Spellmeyer DC, Stauber GB, Shoemaker KR, Kerr JM, Figliozzi GM, Goff DA, Siani MA, Simon RJ, et al. Discovery of nanomolar ligands for 7-transmembrane G-protein-coupled receptors from a diverse N-(substituted)glycine peptoid library. J Med Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.