

Abstract
Purpose
We have previously demonstrated a hyperplastic phenotype when Rb expression was disrupted within the intestinal epithelium. These findings mimic resection-induced adaptation suggesting a possible mechanistic role for Rb during adaptation. The purpose of the present study was to elucidate a mechanism for how Rb deficiency induces intestinal hyperplasia.
Methods
Enterocytes isolated from intestine-specific Rb knockout mice (Rb-IKO) underwent a microarray to elucidate their gene expression profile. IGF2 expression was significantly elevated, which was subsequently confirmed by RT-PCR and in situ mRNA hybridization. Mice with deficient expression of IGF2 or its receptor IGF1R were therefore crossed with Rb-IKO mice to determine the significance of IGF2 in mediating the Rb-IKO intestinal phenotype.
Results
Expression of IGF2 was significantly elevated in villus enterocytes of Rb-IKO mice. The mucosal hyperplasia in Rb-IKO mice was reversed when either IGF2 or IGF1R expression was genetically disrupted in Rb-IKO mice.
Conclusion
IGF-2 expression is significantly elevated in villus enterocytes and is required for the hyperplastic intestinal mucosal phenotype of Rb-IKO mice. The trophic effects of IGF2 require intact IGF1R signaling within the intestinal epithelium. These findings reveal novel regulatory roles for Rb in expanding intestinal mucosal surface area.
Keywords: Small bowel adaptation, Retinoblastoma protein, Insulin-like growth factor-2, Insulin-like growth factor 1 receptor
Adaptation is an endogenous compensatory response by the remaining bowel following massive small bowel resection (SBR). It is characterized by enhanced structural morphology consisting of taller villi and deeper crypts, likely driven by enhanced rates of enterocyte proliferation. While the mechanism for adaptation remains unknown, it is an important area of research as improved understanding may offer therapeutic options for patients suffering from short gut syndrome who are dependent upon parenteral nutrition.
The retinoblastoma protein (Rb) is a prototype tumor suppressor and cell cycle regulator [1–3]. Rb is expressed in virtually all tissues and controls cell cycle progression via interactions with the E2F family of transcription factors [4,5]. The contribution of Rb to normal intestinal homeostasis has been revealed using mutant mice in which Rb expression has been genetically disrupted within the intestinal epithelium. In these mice, enhanced rates of enterocyte proliferation, taller villi, and deeper crypts mirror what is observed during adaptation after massive SBR [6–8]. These findings therefore offer the possibility that Rb expression may be involved in resection-induced adaptation responses.
The purpose of the present study therefore was to determine a specific mechanism for Rb-directed intestinal mucosal expansion. We specifically sought to gain molecular insight into the pathogenesis of adaptation.
1. Methods
1.1. Animals
The protocol for this study was approved by the Washington University Institutional Animal Care and Use Committee (Protocol No. 20100103). Mice in which Cre recombinase expression is driven by the villin promoter within the intestinal epithelium were intercrossed with mice in which the exon 19 of Rb gene is flanked by loxP sites (Rb(flox/flox)). The offspring of this breeding strategy resulted in mice with constitutive intestine-specific disruption of Rb expression, referred to as intestinal Rb knockout (Rb-IKO). These mice were then bred with IGF2 knockout mice (a generous gift from Dr. Carla Kim, Department of Genetics; Harvard Medical School) to generate IGF2-null/Rb-IKO mice. In additional experiments, we bred mice with floxed IGF1R (Jackson Laboratory, Bar Harbor, ME) with Rb-IKO mice to generate IGF1R/Rb-IKO offspring. Mice were kept on a 12-h light–dark schedule and were harvested at aged 6–8 weeks.
1.2. Real-time PCR-IGF2
RNA was prepared from harvested ileal crypts and villi as previously described [8] and were homogenized in lysis buffer (RNAqueous kit, Ambion, Austin, TX). The RNA was extracted according to kit instructions and stored at −80 °C. Total RNA concentration was determined using a NanoDrop Spectrophotometer (ND-1000, NanoDrop Technologies, Wilmington, DE) and RNA quality evaluated using an Experion System with an RNA StdSens Chip and reagents (Bio-Rad Laboratories, Richmond, CA). β-Actin was used as the endogenous control. IGF2 gene expression was determined using primers and reagents from Applied Biosystems (Foster City, CA) and using an Applied Biosystems 7500 Fast Real-Time PCR system (Foster City, CA).
1.3. Microarray and in situ staining
RNA was prepared as above. Three separate WT or Rb-IKO samples were used for gene expression analysis by Agilent microarray in our Washington University DDRDC core facility. For in situ RNA hybridization assay, formalin-fixed and paraffin-embedded slides were used. The assay was performed by a commercial company (Affymetrix) based on a branched DNA amplification technology (QuantiGene ViewRNA) [9].
1.4. Rates of enterocyte proliferation and immunostaining
Ninety minutes before sacrifice, mice were given an intraperitoneal injection of 5-bromodeoxyuridine (BrdU; 0.1 ml/10 g body weight; Zymed Laboratories Inc., San Francisco, CA). Formalin-fixed tissue sections were embedded in paraffin, sectioned, deparaffinized, and blocked with 3% hydrogen peroxide in methanol. Antigen retrieval was performed using Diva Decloaking solution (Biocare Medical, Concord, CA) (120 °C for 5 min followed by 100 °C for 10 min). Slides were blocked with avidin-pink and biotin-blue (Biocare Medical), treated with anti-BrdU antibody (1:500, Accurate Chemical, Westbury, NY) in DaVinci Green (Biocare Medical), stored overnight at 4 °C, and visualized with biotinylated goat anti-rat IgG (Accurate Chemical) followed by streptavidin–horseradish peroxidase (HRP; Invitrogen, Camarillo, CA) and diaminobenzidine (DAB; Sigma-Aldrich, St Louis, MO), and hematoxylin counterstaining. The number of positively staining cells and the total number of cells per crypt were counted from at least 20 well-oriented crypts by blinded scoring. A proliferative index was calculated from the ratio of these measurements.
1.5. Small bowel harvest and enterocyte isolation
A 2-cm segment of tissue was removed 12 cm proximal to the terminal ileum and fixed for histology in 10% neutral-buffered formalin. The next 10-cm segment of ileum was cut open and transferred into tubes containing 5 ml of ice-cold PBS with protease inhibitors for Western blot analysis. Crypts and villi were separated from the resected intestine using our previously described technique of enterocyte isolation involving calcium chelation and mechanical dissociation [8].
1.6. Histology
All histologic measurements were performed by one investigator who was blinded with regard to mouse strain. Five-micrometer-thick longitudinal sections of paraffin-embedded tissue sections were mounted on glass slides. Hematoxylin-and-eosin-stained sections were used to measure villus height and crypt death with a video-assisted computer program (Metamorph, UIC; Dowington, PA). At least 20 well-oriented crypts and villi were counted per slide. Crypts were counted only if the crypt–villus junctions on both sides of the crypt were intact and if Paneth cells were present at the base of the crypt. Villi were counted only if the central lymphatic channel extended from the villus base to the tip and if the mucosal surface was in continuity with an intact crypt. Apoptotic rate was determined by counting the number of apoptotic bodies, defined by the presence of pyknotic, fragmented nuclei, in 50 well-oriented crypts.
1.7. Western blot
Frozen isolated crypt samples were thawed, reconstituted with Tris buffer, and sonicated for 10 s. The samples were lysed with sodium dodecyl sulfate sample buffer. The lysate was then heated for 5 min at 100 °C and the protein concentration was determined by using the RC DC kit (Bio-Rad; Hercules, CA). Proteins were loaded in equal amounts for Western blotting. Total Rb (5541436, BD Pharmingen, San Diego, CA), IGF-1R (Cell Signaling Technology, Danvers, MA) and tubulin (Cell Signaling Technology, Danvers, MA) antibodies were used.
2. Results
In order to investigate the mechanism of Rb deficiency-induced mucosal hyperplasia, a microarray was done to identify differences in enterocyte gene expression patterns compared with WT mice. The results of this microarray showed that the expression of IGF2 was increased nearly ninefold in villus enterocytes of Rb-IKO mice. This result was confirmed separately by RT-PCR and robust IGF2 expression within the villus epithelium was verified by in situ staining (Fig. 1).
Fig. 1.
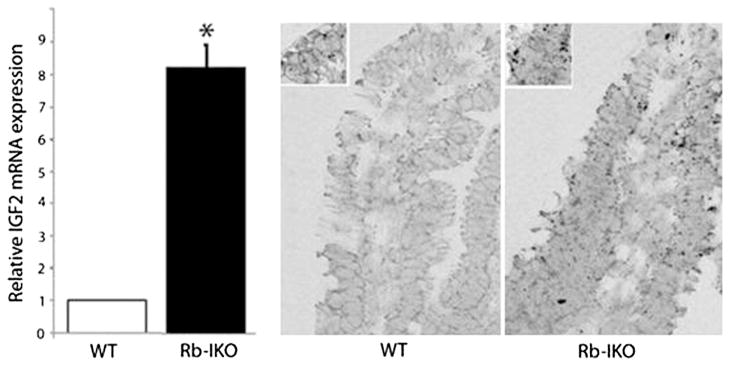
IGF2 mRNA expression measured by microarray in villus epithelium harvested from either wild-type (WT) or in mice which retinoblastoma protein expression was knocked out in the intestinal epithelium (Rb-IKO). The cellular location of IGF2 mRNA expression was verified within villus enterocytes using a QuantiGene ViewRNA assay.
To determine whether elevated intestinal epithelial IGF2 expression was required for mucosal hyperplasia in Rb-IKO mice, we bred IGF2-null mice with our Rb-IKO animals to generate IGF2-null/Rb-IKO offspring. Disrupted Rb expression was confirmed by Western blot while IGF2 deletion was confirmed with RT-PCR (data not shown). Disrupting IGF2 expression in Rb-IKO mice prevented the development of intestinal mucosal hyperplasia (Fig. 2A and B). Despite having shorter villi, crypt cell proliferation rate was no different between the IGF2-null/Rb-IKO and the Rb-IKO strains (Fig. 2C). Similar to rates of proliferation, there was no difference in the rate of crypt cell apoptosis between IGF2-null/Rb-IKO mice (data not shown).
Fig. 2.

Disrupting insulin-like growth factor-2 (IGF2) expression prevents intestinal mucosal hyperplasia in Rb-deficient mice. Villus height (A), crypt depth (B), and rates of enterocyte proliferation (C; % crypt cells staining for BrdU) recorded in wild-type (WT; n = 8), intestinal epithelial Rb-knockout (Rb-IKO; n = 4), and IGF2-null/Rb-IKO double-knockout (IGF2-null/Rb-IKO; n = 7) mice (*p ≤ 0.05, **p ≤ 0.001, Student’s t-test).
The most significant receptor responsive to IGF2 is insulin-like growth factor receptor-1 (IGF1R), which is expressed in both intestinal epithelium and subepithelial myofibroblasts [10–12]. We therefore questioned whether increased intestinal epithelial expression of IGF2 generates taller villi due to stimulation of IGF1R within the epithelial or subepithelial mesenchymal cell compartment. To test this, we crossed floxed IGF1R mice with Villin Cre mice to generate intestinal-specific IGF1R-deficient mice. As previously reported, ablation of IGF1R expression in enterocytes did not affect normal small intestine development or morphology [13]. This mouse line was further bred with our Rb-IKO mice to produce a double knockout of IGF1R and Rb specifically within the intestinal epithelium. The efficiency of Rb and IGF1R deletions were confirmed via Western blot (data not shown).
Disrupted expression of IGF1R within intestinal epithelial cells greatly reduced the hyperplastic intestinal phenotype observed in Rb-IKO mice (Fig. 3A and B). Crypt cell proliferative rates were lower in IGF1R/Rb-IKO double-knockout mice when compared to Rb-IKO mice (Fig. 3C). These data suggest the increased levels of IGF2 in the setting of Rb deficiency functions via an epithelial IGF1R to enhance intestinal morphology and that IGF1R are involved in Rb regulated crypt cell proliferation.
Fig. 3.

Disrupting intestinal epithelial insulin-like growth factor-1 receptor (IGF1R) expression prevents intestinal mucosal hyperplasia in Rb-deficient mice. Villus height (A), crypt depth (B), and rates of enterocyte proliferation (C; % crypt cells staining for BrdU) were recorded in (WT; n = 8), intestinal epithelial Rb-knockout (Rb-IKO; n = 4), and IGF1R/Rb-IKO double-knockout (IGF1R/Rb-IKO; n = 7) mice (*p ≤ 0.05, **p ≤ 0.001, Student’s t-test).
3. Discussion
In the present study, we first identified elevated expression of a known enterotrophic growth factor (IGF2) within the intestinal epithelium in association with the expanded mucosal surface area characteristic of Rb-deficient mice. Next, we confirmed the significance of IGF2 as a critical factor by genetically disrupting IGF2 expression in Rb-IKO mice—a manipulation that blocked the development of mucosal hyperplasia. Finally, by perturbing intestinal epithelial IGF1R expression within Rb-IKO mice, we have revealed that the intestinal epithelium is the necessary cell compartment responsive to IGF2 and villus growth. These results allow us to propose a novel mechanism for villus growth that involves attenuated intestinal epithelial Rb expression leading to increased IGF2 expression, which signals in an autocrine/paracrine manner via the epithelial IGF1R. These observations may provide molecular insight into mechanisms for regulation of mucosal surface area under conditions of normal growth and development as well as adaptation to massive intestinal loss.
The most obvious mechanism for villus growth and accelerated rates of enterocyte proliferation observed in Rb-IKO mice is the simple fact that the expression of a cell cycle inhibitor has been disrupted. As such, rates of enterocyte proliferation would be expected to be heightened on this basis alone. On the other hand, besides direct cell cycle regulation [1–5], Rb has been demonstrated to be involved in a broad range of cellular functions including Ras signaling, metabolic pathways, mitochondrial function, cell fate decisions, stem cells, and chromatin remodeling, to name a few [14]. This was the rational for seeking to identify expression profile differences between Rb-IKO and WT mice.
Within the 44,000 genes analyzed in the array, there were several genes overexpressed in addition to IGF2, including cyclinE1, proliferating cell nuclear antigen, and p107. However, IGF2 was the only factor that was found to be elevated which is known to be an intestinotrophic factor. Although IGF1 has been studied in the context of intestinal growth and adaptation after SBR [10,15–17], less is known regarding the role of IGF2. IGF2 is considered to be the most important IGF in adult humans [18] and inappropriate IGF2 expression has been implicated in a growing number of diseases [19]. Loss of imprinting (LOI) of the IGF2 gene is an epigenetic alteration resulting in a modest increase in IGF2 expression and is associated with increased cellular proliferation and many common cancers [20]. Specific to the gut, the allelic dosage of IGF2 directly correlates with intestinal mucosal surface area and crypt cell number [21]. In the context of IGF2 LOI, the intestinal expression of proliferation-related genes is elevated [22]. Further, ectopic overexpression of IGF2 in smooth muscle cells results in a significantly longer intestine [23]. Conversely, administration of an IGF receptor inhibitor [22] and transgenic expression of a decoy IGF receptor [20] reverses these enterotrophic effects. Taken together, these findings support an important role for IGF2 in normal homeostasis of the intestinal mucosa. The results of the microarray therefore provided the rationale for further experiments.
Our observation of elevated IGF2 expression in Rb-deficient enterocytes is novel. The hyperplastic phenotype seen in Rb-IKO mice is negated when IGF2 is also deleted, which confirms that elevated IGF2 expression is required for the hyperplastic villus growth in Rb-IKO mice. Since IGF2 was deleted in all cells of the body in our specific knockout mouse line, we next sought to investigate the cell compartment responsible for the trophic mucosal effects. Available evidence suggests that mesenchymal cells within the lamina propria are the major sites for IGF2 production within the normal small bowel [10,24]. Thus, mesenchymal signaling to the overlying epithelial cells may be one mechanism for villus growth. Cultured intestinal myofibroblasts express endogenous IGF2, but not IGF1 and conditioned media from these cells stimulates proliferation of intestinal epithelial cells, an effect that is attenuated by IGF2 neutralizing antibody [10]. In marked contrast, our discovery of elevated IGF2 expression in differentiated enterocytes of Rb-IKO mice would suggest that epithelial-produced IGF2 may signal to the underlying mesenchyme to induce villus growth. In a similar vein, epithelial Hedgehog signaling to the underlying intestinal mesenchyme has previously been demonstrated [25–27].
IGF1R is a transmembrane tyrosine kinase receptor with structural homology to insulin receptors. Both IGF1 and IGF2 bind to the IFG1R with high affinity. Insulin is also capable of binding to the IGF1R, but to a lesser extent. IGF1R mediates all known physiologic actions of IGF1 and IGF2 [10–12]. Activation of IGF1R leads to autophosphorylation and activation of signaling cascades such as IRS-1/PI3K/AKT and GRB2/Ras/ERK pathways [28]. Although the IGF2 receptor binds to IGF2 exclusively, it does not activate any signaling pathways itself but rather functions as a clearance receptor and acts to sequester IGF2 to prevent it from binding to IGF1R [29]. Since the IGF1R is the main receptor for IGF2 signaling, we therefore chose to delete the expression of this receptor in the intestinal epithelium employing the villin promoter to drive the production of Cre recombinase. An alternative method would be to disrupt IGF1R expression within the intestinal mesenchyme; however, there are no intestine-specific mesenchyme promoters presently available. Since IFG1R expression within the intestinal epithelium appears to be required for the hyperplastic mucosa of Rb-IKO mice, we would conclude that IGF1R signaling within subepithelial mesenchymal cells is not important for stimulating villus growth. This observation would support the notion that villus growth can be driven solely by growth factor signaling within the overlying intestinal epithelium. In contrast with this finding is a recent study from our laboratory in which disruption of epidermal growth factor (EGF) receptor expression within the intestinal epithelium had no apparent phenotype and did not affect resection-induced villus growth [30]. It is possible that the disparate findings are due to various growth factors (IGF2 versus EGF) having a differential effect on the epithelium. Further, the roles for other growth factors in the regulation of villus growth must be considered involving either epithelial or mesenchymal signaling.
While there was no difference in crypt cell proliferation between IGF2-null/Rb-double IKO and Rb-IKO lines, rates of proliferation were significantly decreased in the IGF1R/Rb-double IKO mice. As mentioned previously, IGF1R binds to multiple ligands, including IGF1, a known enterotrophic factor that also stimulates crypt cell proliferation [31–34]. IGF1 is also a key mediator of other enterotrophic factors such as growth hormone and glucagon-like peptide-2 (GLP-2). Inducible deletion of IGF-1R has also shown to abrogate any trophic effects of GLP-2 [35]. Thus, deleting IGF-1R may also have the effect of blunting the activity of these other trophic factors.
The effect of disrupting IGF2 and IGF1R expression on normal adaptation after resection has yet to be explored. Such experiments may outline an important role for the Rb/IGF axis as a major contributor of resection-induced villus growth.
Acknowledgments
This work was supported by National Institutes of Health Grants R01 DK 059288 (Warner), T32 GM008795 (Choi), and P30DK52574—Morphology and Murine Models Cores of the Digestive Diseases Research Core Center of the Washington University School of Medicine, and the Children’s Surgical Sciences Institute of the St. Louis Children’s Hospital Foundation. Dr. Choi is also supported by The Marion and Van Black Endowed Pediatric Surgical Research Fellowship.
References
- 1.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–26. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 3.Zheng L, Lee WH. The retinoblastoma gene: a prototypic and multifunctional tumor suppressor. Exp Cell Res. 2001;264:2–18. doi: 10.1006/excr.2000.5129. [DOI] [PubMed] [Google Scholar]
- 4.Attwooll C, Lazzerini DE, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–16. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 6.Haigis K, Sage J, Glickman J, et al. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. 2006;281:638–47. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 7.Leinicke JA, Longshore S, Wakeman D, et al. Regulation of retinoblastoma protein (Rb) by p21 is critical for small bowel adaptation. J Gastrointest Surg. 2012;16:148–55. doi: 10.1007/s11605-011-1747-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo J, Longshore S, Nair R, et al. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem. 2009;284(1):134–40. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ting DT, Lipson D, Paul S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011 Feb 4;331(6017):593–6. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lund PK. Molecular basis of intestinal adaptation: the role of the insulin-like growth factor system. Ann N Y Acad Sci. 1998;859:18–36. doi: 10.1111/j.1749-6632.1998.tb11108.x. [DOI] [PubMed] [Google Scholar]
- 11.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999 May;19(5):3278–88. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heidegger I, Pircher A, Klocker H, et al. Targeting the insulin-like growth factor network in cancer therapy. Cancer Biol Ther. 2011;11(8):701–7. doi: 10.4161/cbt.11.8.14689. [DOI] [PubMed] [Google Scholar]
- 13.Wolf E, Kramer R, Blum WF, et al. Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: endocrine changes and effects on body and organ growth. Endocrinology. 1994 Nov;135(5):1877–86. doi: 10.1210/endo.135.5.7525257. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi C, Sasaki N, Kitajima S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 2012;103:1182–8. doi: 10.1111/j.1349-7006.2012.02284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeigler TR, Mantel MP, Chow JC, et al. Gut adaptation and the insulin-like growth factor system: regulation by glutamine and IGF-1 administration. Am J Physiol Gastrointest Liver Physiol. 1996;271:G866–75. doi: 10.1152/ajpgi.1996.271.5.G866. [DOI] [PubMed] [Google Scholar]
- 16.Bortvedt SF, Lund PK. Insulin-like growth factor 1: common mediator of multiple enterotrophic hormones and growth factors. Curr Opin Gastroenterol. 2012;28(2):89–98. doi: 10.1097/MOG.0b013e32835004c6. DS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemmey AB, Martin AA, Read LC, et al. IGF-I and the truncated analogue des-(1–3)IGF-I enhance growth in rats after gut resection. Am J Physiol. 1991;260(2 Pt 1):E213–9. doi: 10.1152/ajpendo.1991.260.2.E213. [DOI] [PubMed] [Google Scholar]
- 18.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–37. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 19.Reik W, Constancia M, Dean W, et al. Igf2 imprinting in development and disease. Int J Dev Biol. 2000;44(1):145–50. [PubMed] [Google Scholar]
- 20.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004 Feb;4(2):143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 21.Harper J, Burns JL, Foulstone EJ, et al. Soluble IGF2 receptor rescues Apc(Min/+) intestinal adenoma progression induced by Igf2 loss of imprinting. Cancer Res. 2006;66(4):1940–8. doi: 10.1158/0008-5472.CAN-05-2036. [DOI] [PubMed] [Google Scholar]
- 22.Kaneda A, Wang CJ, Cheong R, et al. Enhanced sensitivity to IGF-II signaling links loss of imprinting of IGF2 to increased cell proliferation and tumor risk. Proc Natl Acad Sci USA. 2007;104(52):20926–31. doi: 10.1073/pnas.0710359105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaina S, Pettersson L, Thomsen AB, et al. Shortened life span, bradycardia, and hypotension in mice with targeted expression of an Igf2 transgene in smooth muscle cells. Endocrinology. 2003;144(6):2695–703. doi: 10.1210/en.2002-220944. [DOI] [PubMed] [Google Scholar]
- 24.Winesett DE, Ulshen MH, Hoyt EC, et al. Regulation and localization of the insulin-like growth factor system in small bowel during altered nutrient status. Am J Physiol. 1995;268(4 Pt 1):G631–40. doi: 10.1152/ajpgi.1995.268.4.G631. [DOI] [PubMed] [Google Scholar]
- 25.Madison BB, Braunstein K, Kuizon E, et al. Epithelial hedgehog signals pattern the intestinal crypt–villus axis. Development. 2005 Jan;132(2):279–89. doi: 10.1242/dev.01576. [DOI] [PubMed] [Google Scholar]
- 26.Zacharias WJ, Li X, Madison BB, et al. Hedgehog is an anti-inflammatory epithelial signal for the intestinal lamina propria. Gastroenterology. 2010 Jun;138(7):2368–77. doi: 10.1053/j.gastro.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zacharias WJ, Madison BB, Kretovich KE, et al. Hedgehog signaling controls homeostasis of adult intestinal smooth muscle. Dev Biol. 2011 Jul 1;355(1):152–62. doi: 10.1016/j.ydbio.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foulstone E, Prince S, Zaccheo O, et al. Insulin-like growth factor ligands, receptors, and binding proteins in cancer. J Pathol. 2005;205(2):145–53. doi: 10.1002/path.1712. [DOI] [PubMed] [Google Scholar]
- 29.Kornfeld S. Structure and function of the mannose 6-phosphate/insulin-like growth factor II receptors. Annu Rev Biochem. 1992;61:307–30. doi: 10.1146/annurev.bi.61.070192.001515. [DOI] [PubMed] [Google Scholar]
- 30.Rowland KJ, McMellen ME, Wakeman D, et al. Enterocyte expression of epidermal growth factor receptor is not required for intestinal adaptation in response to massive small bowel resection. J Pediatr Surg. 2012 Sep;47(9):1748–53. doi: 10.1016/j.jpedsurg.2012.03.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conteas CN, McMorrow B, Luk GD. Modulation of epidermal growth factor-induced cell proliferation and receptor binding by insulin in cultured intestinal epithelial cells. Biochem Biophys Res Commun. 1989;161:414–9. doi: 10.1016/0006-291x(89)92614-4. [DOI] [PubMed] [Google Scholar]
- 32.Kurokowa M, Lynch K, Podolsky DK. Effects of growth factors on an intestinal epithelial cell line: transforming growth factor beta inhibits proliferation and stimulates differentiation. Biochem Biophys Res Commun. 1987;142:775–82. doi: 10.1016/0006-291x(87)91481-1. [DOI] [PubMed] [Google Scholar]
- 33.Ohneda K, Ulshen MH, Fuller CR, et al. Enhanced growth of small bowel in transgenic mice expressing human insulin-like growth factor I. Gastroenterology. 1997;112:444–54. doi: 10.1053/gast.1997.v112.pm9024298. [DOI] [PubMed] [Google Scholar]
- 34.Duncan MD, Korman LY, Bass BL. Epidermal growth factor primes intestinal epithelial cells for proliferative effect of insulin-like growth factor I. Dig Dis Sci. 1994;39:2197–201. doi: 10.1007/BF02090371. [DOI] [PubMed] [Google Scholar]
- 35.Rowland KJ, Trivedi S, Lee D, et al. Loss of glucagon-like peptide-2-induced proliferation following intestinal epithelial insulin-like growth factor-1-receptor deletion. Gastroenterology. 2011;141:2166–75. doi: 10.1053/j.gastro.2011.09.014. [DOI] [PubMed] [Google Scholar]