

Abstract
Transforming growth factor-β (TGFβ) is an immunosuppressive cytokine produced by tumour cells and immune cells that can polarize many arms of the immune system. This Review covers the effects of TGFβ on NK cells, dendritic cells, macrophages, neutrophils, CD8+ and CD4+ effector and regulatory cells, and NKT cells in preclinical animal tumour models and in patients with cancer. Collectively, many recent studies favour the idea that blocking TGFβ signalling in the tumour microenvironment enhances antitumour immunity and may be beneficial for cancer therapy. An overview of the current drugs and reagents for inhibiting TGFβ signalling and their phase in clinical development is also provided.
Introduction
As most tumours present self antigens, peripheral tolerance [G] has an important role in contributing to immune evasion by tumours. In addition, the overproduction of immunosuppressive cytokines, including transforming growth factor-β (TGFβ), by tumour cells and tumour-infiltrating lymphocytes also contributes to an immunosuppressive microenvironment. Many studies indicate that TGFβ can promote cancer metastasis through effects on the tumour microenvironment, by enhancing tumour cell invasion and by inhibiting the function of immune cells 1, 2. These findings have promoted interest in targeting TGFβ and its signalling pathway in patients with cancer. However, such targeting of TGFβ could result in adverse effects in normal tissues, as this pathway is also involved in multiple homeostatic processes (Figure 1). For example, TGFβ can function as a tumour suppressor to prevent tumorogenesis; however, overproduction of TGFβ is frequently associated with tumour metastasis and poor prognosis in patients with cancer (Figure 1). Although the molecular mechanisms behind this dichotomy of TGFβ functions are not fully elucidated, progress has been made in understanding the role of TGFβ in different stages of cancer. This topic has recently been reviewed and is not discussed here 1, 3–5.
Figure 1. The yin and yang of TGFβ in tumour development maintenance, and metastasis formation.
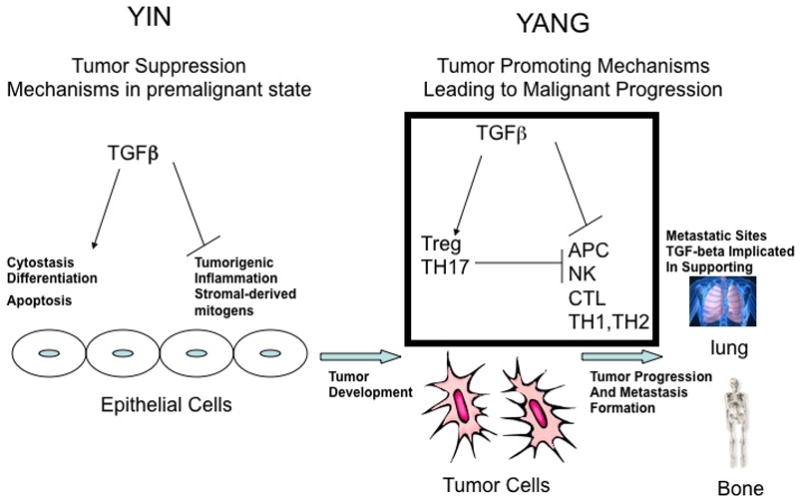
Before epithelial cells transform into a malignant tumour, transforming growth factor-β (TGFβ) functions as a tumour-suppressor, by blocking expression of stromal-derived mitogens and suppressing pro-tumourogenic inflammation. Furthermore, TGFβ supports the cytostasis, terminal differentiation and apoptosis of premalignant cells which harbor either an overexpressed oncogene or suppressed tumour suppressor gene. Once the epithelial cells become fully malignant, TGFβ has the opposite effect by blocking the antitumour immune response through support for the activity of regulatory cells and through direct inhibition of effector cell mechanisms from clearing the established tumour, as described in the main text and summarised in Figure 2. Once tumours are established, TGFβ further supports the formation of metastases to several sites, including bone and lung tissues. Additional nonimmune mechanisms outside the scope of this Review that support tumorigenesis and metastasis formation are addressed in References 1 and 2.
This Review focuses on the tumour-promoting properties of TGFβ, which prevent effective antitumour immune responses once cancer has been established in the host. A successful immune response requires the proper activation and maturation of antigen-presenting cells (APCs) of the innate immune system that present antigen to adaptive immune cells. TGFβ can suppress or alter the activation, maturation and differentiation of both innate and adaptive immune cells, including natural killer (NK) cells, dendritic cells (DCs), macrophages, neutrophils, and CD4+ and CD8+ T cells 6. A dampened innate immune response leads to poor adaptive immunity, resulting in persistence of the tumour. In addition, TGFβ has an important role in the differentiation and induction of natural and induced regulatory T (TReg) cells, which also contribute to the tolerizing environment. Furthermore, in the presence of IL-6, TGFβ induces the differentiation of IL-17–producing CD4+ T helper 17 (TH17) cells and CD8+ cytotoxic T cells; although the role of IL-17-producing cells still remains controversial in tumour biology, given that these cells can exhibit both tumour-promoting and antitumour activities 7. As we discuss, many recent discoveries have been made towards understanding the biological effects of TGFβ on different immune cells, although multiple areas require further investigation. Finally, there is compelling evidence to support targeting TGFβ with inhibitors to enhance antitumour immunity in patients with cancer.
Effects of TGFβ on innate immune cells
NK cells
NK cells are innate lymphoid cells that have an important role in the antitumour response by recognizing and directly killing tumours and by rapidly producing chemokines and cytokines crucial for this function. For example, interferon-γ (IFNγ) production by NK cells is important for stimulating effector CD4+ TH1 cells that are required for clearing tumours. TGFβ attenuates IFNγ production by and the lytic activity of NK cells 8, 9. These might be direct effects of TGFβ or might result indirectly from cell-cell contact between NK cells and regulatory T cells producing this cytokine 10. In support of a direct effect, TGFβ signalling can suppress IFNγ production through SMAD3, a transcription factor downstream from this pathway, resulting in suppression of T-bet, a transcription factor required for IFNγ production 11.
Targeted killing by NK cells requires stimulating the NK activating receptors NKG2D, NKp46, NKp44 and NKp30 12. It has been shown that exogenously administered TGFβ inhibits NKp30 and NKG2D expression, leading to decreased ability of NK cells to kill target cells 13. TGFβ also decreases expression of NKG2D by NK cells and CD8+ T cells from glioma patients with a high tumour burden 14. In patients with lung and colorectal cancer, the downmodulation of NKG2D has been associated with increased serum levels of TGFβ 15. Furthermore, recent studies of isolated NK cells from healthy donors have shown that platelet-derived TGFβ results in downmodulation of NKG2D, causing a decrease in IFNγ production and degranulation functions essential for tumour destruction by these cells 16. Finally, surface-bound TGFβ on myeloid-derived suppressor cells [G] can inhibit NK cell cytolytic activity in an orthotopic liver cancer model 17. These observations indicate that TGFβ has immunosuppressive effects on NK cell killing functions in patients with cancer, and therefore might be a target for enhancing NK cell- mediated antitumour immune responses.
Dendritic cells
DCs are APCs that have a major role in the initial activation and subsequent regulation of immune responses 18. In addition to activating adaptive immunity mediated by T cells, DCs can also activate NK cells 19. DCs can present antigen in an immunogenic or tolerogenic manner, and so they have an important role in determining the host response to tumours 18, 20. DC activation involves the upregulation of MHC and costimulatory molecule expression, alteration in motility and the formation of dendrites to increase the surface area for antigen presentation and interaction with lymphocytes 21. Non-activated or immature DCs can still present antigen, but in the absence of proper costimulation this results in T cell tolerance 22, 23. In the presence of immune-inhibitory signals such as IL-10 and/or corticosteroids, DCs can induce tolerance by T cell deletion and/or the activation and induction of TReg cells 24, 25. Hence, DCs can induce either immunity or peripheral tolerance and are an essential component of tumour immunity. TGFβ affects DC biology in several ways. TGFβ can immobilize DCs, thereby interfering with their migration and the transport of antigen to draining lymph nodes for presentation to adaptive immune cells, and might also directly induce DC apoptosis 26–28. Tumour-infiltrating DCs secrete TGFβ and respond to TGFβ, either in an autocrine or paracrine manner, by down-regulating expression of MHC class II molecules, the co-stimulatory molecules CD40, CD80 and CD86, and tumour necrosis factor (TNF), IFNα, IL-12 and CC-chemokine ligand 5 (CCL5) 6, 29. These immature or tolerogenic DCs promote the formation of TReg cells that potently inhibit the function of other T cells 30, 31. Activated DCs are also able to activate both natural and induced TReg cells 32–35; interestingly, the capacity of DCs to induce both types of TReg cell is greatly increased by TGFβ and IL-10 36, 37. So, in the context of these immunosuppressive cytokines in the tumour microenvironment, DCs take up tumour cells, become tolerogenic TGFβ-secreting cells, and promote the induction of tumour-specific TReg cells in both mice and humans 38–41. Therefore, a growing body of evidence points to the induction of TReg cells by TGFβ produced by DCs; as TReg cells are a major obstacle to tumour immunity, this indicates that the TGFβ pathway might be targeted to augment antitumour immunity in patients with cancer.
Macrophages
The mass of most solid tumours is made up of ~50% macrophages, and high levels of tumour- associated macrophages (TAMs) [G] are correlated with poor cancer prognosis 42, 43. Macrophages are a heterogeneous population and are typically defined as being of an M1 (classically activated) [G] or M2 (alternatively activated) [G] phenotype, similar to the CD4+ TH1 versus TH2 cell paradigm. M1 macrophages are induced by IFNγ and other proinflammatory stimuli and are efficient at presenting antigens, producing proinflammatory cytokines, activating TH1 responses, and in general mediating anti-tumor responses. In contrast, there are a variety of M2 macrophages induced by IL-10, immune complexes, glucocorticoids, IL-4 and IL-13 with various phenotypes that are involved in remodeling and repair of damaged tissue, parasite resistance, immune regulation and/or tumour promotion 44. Both TAMs and myeloid-derived suppressor cells (MDSCs), which are a heterogeneous population of immature DCs, macrophages, granulocytes, and other myeloid cells in early stages of their differentiation and have properties similar to those that have been described for M2 cells, are involved in cancer progression and metastasis. In skin cancer, TGFβ-mediated recruitment of macrophages into tumours has an important role in immune escape, as it converts a regressing tumour into a progressing tumour 45. It is suggested that TGFβ-recruited TAMs are highly phagocytic and can compete with DC function, thereby markedly decreasing the ability of DCs to present tumour antigens to the adaptive immune system 45. TAMs acquire their phenotype by expressing high levels of TGFβ, IL-10, macrophage galactose N-acetyl-galactosamine-specific lectin 1 (MGL1), Dectin-1, CXC-chemokine ligand 10 (CXCL10), CXCL9 and other IFN-responsive genes. By contrast, they produce low levels of IL-12, TNF and nitric oxide synthase 2 (NOS2). Interestingly, this cytokine phenotype is mediated by the inhibitory NF-κB subunit p50 and it is thought that the cytokine milieu of the tumour microenvironment is necessary to maintain the phenotype46–48. During tumour progression, TAMs can switch from an M1 to M2 phenotype, which is paralleled by a gradual inhibition of NF-κB activity 49.
Peritoneal macrophages from tumour-bearing hosts produce increased levels of TGFβ, are less differentiated and have deficiencies in inflammatory cytokine production owing to decreased expression of NF-κB and C/EBP transcription factors 50. In this model, tumour-derived factors TGFβ and prostaglandin E2, individually and additively downregulate NF-κB and C/EBP. There is also evidence that tumour-infiltrating MDSCs secrete high levels of TGFβ, which upregulates CD206 (a deactivation marker characteristic of M2 cells) expression in an autocrine manner 51. It is unclear how TGFβ production is induced in MDSCs and macrophages, but the mechanism might involve IL-13 and glucocorticoids 52, 53; the induced TGFβ might then contribute to the alternative activation of M2 macrophages by downregulating NF-κB expression.
Neutrophils
Neutrophils are short-lived polymorphonuclear leukocytes with potent antimicrobial and inflammatory capacities. Despite their known function as professional phagocytes, the role of neutrophils in tumour progression has been controversial and has received little attention compared with that of macrophages. Initial studies characterizing the effect of TGFβ on the control of inflammatory responses showed that this factor was a potent chemotactic factor for neutrophils, promoting their migration but not degranulation or activation 6. Subsequent studies showed that neutrophil migration could also be indirectly affected by TGFβ through regulating the expression of adhesion molecules in the endothelium 54, 55, and that TGFβ could inhibit neutrophil cytotoxicity, suggesting that TGFβ might influence human neutrophil activity in vivo56. Recently, the contradictory role of neutrophils in both tumour suppression and tumour promotion either by directly or indirectly controlling tumour growth, angiogenesis and metastasis (reviewed in 57) was reevaluated in terms of the characterization of different types of tumour associated neutrophil (TAN) with polarized N1 or N2 phenotypes 58. These polarized populations are similar to those that have been described for macrophages; they are influenced by the microenvironment and seem to be controlled by TGFβ in the tumour proximity. N2 cells are characterized by an expression profile that promotes tumour angiogenesis and metastasis 59–61, and inhibits the antitumour immune response by the secretion of reactive oxygen species (ROS). ROS normally act as potent microbicidal agents but in the context of the tumour microenvironment, ROS could lead to oxidative damage and inhibition of T cell function 62. Depletion of this neutrophil subpopulation in untreated tumour-bearing mice was sufficient to inhibit tumour growth, even when CD8+ T cells were absent; highlighting the immunosuppressive potential of N2 cells 58–60. Under TGFβ-inhibiting conditions, as well as in response to certain activation signals, neutrophils acquire an antitumour N1 phenotype that promotes tumour death and inhibits tumour growth 57, 58, 63, 64. The lack of systemic effects on neutrophil polarization during TGFβ neutralization experiments indicated that the effect is mainly intratumoral, characterized by increased numbers of N1 TANS that express activating chemokines and cytokines as well as by changes in endothelial adhesion molecules expression 58. Interestingly N1 and N2 neutrophils were shown to control the activation status of CD8+ T cells. This interplay seemed to be reciprocal as activated CD8+ T cells were controlling the activation and migration of neutrophils to the tumour microenvironment as well65. Clearly a reevaluation of the role of neutrophils in tumour immunology as well as characterization of this polarized subpopulation by TGFβ may be required to design more effective immunotherapies.
Effects of TGFβ on effector T cells
CD8+ T cells
CD8+ T cells are a crucial component of antitumour immunity, as tumour antigen-specific cytotoxic T lymphocytes (CTLs) have an important role in the cytolytic killing of tumour cells. Several studies have shown a direct correlation between the frequency of CTLs in tumour infiltrating lymphocytes (TILs) and the overall survival of cohorts of patients with cancer; in particular, the high ratio of intratumoral activated cytotoxic CD8+ T cells to Tregs leads to improved prognosis 66–69. TGFβ signalling in tumour-specific CTLs dampens their function and frequency in the tumour 70, and blocking TGFβ signalling on CD8+ T cells results in more rapid tumour surveillance and the presence of many more CTLs at the tumour site71. Several experimental protocols have been used to render CD8+ T cells unresponsive to TGFβ. In a model where a dominant-negative form of TGFβ receptor II (TGFβRII) is expressed by both CD4+ and CD8+ T cells, a strong antitumour immune response was associated with the proliferation and increased activity of tumour-specific CTLs 72, 73. Similarly, when tumour-specific CD8+ T cells are rendered unresponsive to TGFβ signalling by transduction with a similar TGFβRII dominant-negative construct before adoptive transfer, these cells infiltrate into tumours, secrete cytokines such as IFNγ and can successfully kill tumour cells 74, 75.
In some tumour models, systemic blockade of TGFβ using a monoclonal antibody or kinase inhibitors to block downstream signalling prevents tumour recurrence by impacting the activity of various cell types, including an increase in the cytotoxic activity of CTLs76–79. However, inhibition of TGFβ by using the monoclonal antibody alone is not always sufficient to promote tumour rejections in all animal tumour models. In such models, the combination of a TGFβ-specific antibody with a vaccine resulted in a synergistic improvement in the inhibition of tumour growth that is mediated by increased number and activity of CD8+ T cells 80–82. It is speculated that in these models, the source of inhibitory TGFβ is the immune system itself and not the tumour, because the antibody-mediated blockade is effective at enhancing antitumour immune responses even when antibody is administered with a prophylactic vaccine before injection of tumour cells80. This effect of TGFβ is consistent with the recent finding where TGFβ was shown to be responsible for the apoptosis of short-lived effector T cells that comprise more than 90% of the effector pool after immunization with Listeria monocytogenes 83. TGFβ promotes the apoptosis of these effector T cells by downregulating the expression of BCL-2, which opposes the survival function of IL-15 on the short-lived effector population 83. It is possible that blocking TGFβ signalling with the neutralizing antibody during administration of the tumour vaccine inhibits the apoptosis of tumour-specific short-lived effector CD8+ T cells and therefore prevents the termination of expanding CTLs.
TGFβ-mediated inhibition of CTL function during tumour immunity might be through several mechanisms. TGFβ directly inhibits CTL function by suppressing the expression of several cytolytic genes, including the genes encoding granzyme A, granzyme B, IFNγ and Fas ligand 70. TGFβ also attenuates the effector function of antigen-specific memory CD8+ T cells by inhibiting T-bet expression resulting in inhibition of IFNγ production 84. TGFβ might also block T cell receptor (TCR) signalling of TILs by upregulating the expression of SPRED1 (sprouty-related, EVH1 domain containing 1), which is an inhibitor of the Ras/MAPK pathway 85. Interestingly, TGFβ can also influence CD8+ T cell-mediated tumour immunity by inducing IL-17 production by CD8+ T cells, although the effect of IL-17 on tumour growth versus immune surveillance remains controversial 86–89.
CD4+ T cells
CD4+ T cells are central orchestrators of adaptive immunity; however, their role in antitumour immune responses has largely been overlooked, mainly owing to the lack of MHC class II expression by tumour cells. TGFβ has been shown to have effects on all subsets of CD4+ effector T cells controlling the expression of master transcriptional regulators in these cells. TGFβ inhibits T-bet and GATA-3 expression (which determine CTL, TH1 and TH2 cell differentiation) while it promotes FOXP3 and Rorγt expression (which determine TReg and TH17 cell differentiation) (reviewed in 90). The role of CD4+ T cells in tumour biology has been classically studied in the context of TReg cells, which are covered in a separate section of this Review. This section focuses on the specific role of TH cell subpopulations in the control of antitumour immune responses and how TGFβ within the tumour microenvironment could influence the polarization of each subset.
Early studies trying to understand the mechanism of tumor-induced immunosuppression 91 identified TGFβ as one of the major inhibitors of immune responses in the tumour microenvironment. Tumour-derived TGFβ was shown to inhibit TH1 responses by shifting infiltrating T cells towards a TH2 phenotype 92, and hence promoting a less efficient anti-tumor immune response. However, later studies comparing the efficacy of TH1 and TH2 effector cell subsets in mediating anticancer responses showed that both TH1 and TH2 cells increased the CTL-mediated antitumour response, although TH1 cells secreting IFNγ seemed to be more effective by promoting APC activation 93, 94. Studies using TCR-transgenic mice further support the requirement for CD4+ T cells to activate memory CTLs in vivo 95, and interestingly show tumour eradication by CD4+ T cells even in cases where tumours were resistant to CD8+ T cell- mediated rejection 96. These findings suggested a potential benefit of TH cells in cancer immunotherapy and started a search for the most effective anticancer CD4+ effector T cell population.
Contradictory reports regarding the role of IL-17 in cancer have made it difficult to conclude whether or not T cells expressing this cytokine would be beneficial against tumours 97, 98. Accumulation of TH17 cells in the tumour microenvironment has been reported in several types of cancer, as well as the expression of IL-6, IL-1β and TGFβ by tumour cells, which are key cytokines controlling TH17 cell differentiation and proliferation 99, 100. TH17 cell-polarized tumour specific CD4+ T cells were shown to be more efficient than TH1 -polarized cells in tumour rejection after adoptive transfer and this efficiency was probably dependent on IFNγ rather than IL-17 production 88. Similar observations, transferring CD8+IL-17+ cells which then become IFNγ+ producing cells were reported; however, some discrepancies have been found regarding the role of IFNγ in these models as the use of lymphopenic hosts promotes loss of a TH17 cell phenotype and acquisition of a TH1 cell phenotype in the transferred cells, potentially masking the real effects of IL-17 in controlling antitumour immunity 87, 89, 101.
Recent findings indicate that the differentiation state of T cells, naïve versus effector/memory, might also be important for mounting more efficient antitumour responses as single transfers of naïve CD4+ T cells were able to eradicate established tumours independent of CD8+ T cells, NK cells and NKT cells 102, 103.
Growing evidence suggests that control of the cytokines expressed in the tumour microenvironment can promote tumour eradication by controlling TReg and TH17 cell polarization in the tumour. Exogenous administration of IL-2 in tumour-bearing mice increased TReg cell and decreased TH17 cell frequencies in the tumour 104, whereas antagonizing the effects of TGFβ by administering IL-7 has been shown to be useful in the promotion of TH17 cells 105. A complete understanding of the dynamic cytokine network, including the role of TGFβ in controlling T cell polarization in the tumour, as well as characterization of the molecular signals mediating TH cell differentiation will be crucial for dissecting the beneficial use of TH cells in future immunotherapies against cancer.
Effects of TGFβ on regulatory cells
CD4+ TReg cells
TReg cells are an immunosuppressive T cell population that express the forkhead family transcription factor, FoxP3, and can suppress antitumour immune responses 106. These cells are a heterogeneous population containing at least two distinct subsets known as natural TReg (nTReg) cells and adaptive or induced TReg (iTReg) cells 106. nTReg cells develop in the thymus, express the IL-2 receptor α chain (CD25) and maintain self tolerance in an antigen-independent manner. iTReg cells, by contrast, develop in the periphery in response to self- or tumour- antigens and express variable levels of CD25 106. Although nTReg cells and iTReg cells have been identified as separate subsets of regulatory T cells, their phenotype and function have not been fully established in preclinical tumour-bearing animal models and patients with cancer. TGFβ could be involved in generating TReg cells in vivo and this cytokine may assist subsets of TReg cells in suppressing effector cell function in the tumour microenvironment (reviewed in 2 and covered in more detail in the section detailing effects of TGFβ on effector cells). High levels of TReg cells in patients with cancer can be inversely correlated with survival 107, 108. Although the precise mechanism(s) behind increased TReg cells in malignancies are unknown, TGFβ, as well as other tumour-produced chemical mediators working in concert with this cytokine, PGE2 and H-Ferritin have been implicated in inducing TReg cells 109, 110. In addition, the production of CCL22 by TAMs surrounding tumours might mediate TReg cell trafficking to the tumour bed through CCR4 111. Recently, IL-23 production in the tumour microenvironment has been implicated in promoting the proliferation of intratumoural TReg cells as these cells express the IL-23 receptor, have evidence of STAT3 signalling (downstream of IL-23 receptor) and are decreased in number in tumour-bearing animals treated with a blocking antibody specific for the IL-23 receptor 25. This cytokine may complement the effects of TGFβ, which also seems to increase the number of intratumoural TReg cells.
Recently a new regulatory T cell subtype has been identified that can be induced and expanded in mice bearing orthotopic liver, lung and melanoma tumours 112. Unlike the conventional TReg cells described above, this regulatory subtype lacks expression of CD25 and FOXP3. Instead, the cells express the IL-2 receptor β chain (CD122), IL-10, TGFβ1 and the early activation marker, CD69 112. Activation of CD69 by the agonistic Ab against CD69 (H1.2F3) results in high levels of membrane-bound TGFβ expression by these cells through the activation of ERK and this might contribute to the ability of this regulatory T cell subset to suppress CD4+ T cell proliferation and promote the growth of established tumours 112. Although these results suggest that another subset of regulatory T cells associated with TGFβ production can suppress the antitumour response, the role of these cells in patients with cancer remains to be determined. TGFβ, in combination with IL-2, is required for the conversion of naïve T cells to iTReg cells in vitro 113, 114. Furthermore, the induction of TReg cells by TGFβ might be a mechanism by which tumours escape the antitumour immune response as several tumours can produce TGFβ2. Blockade of TGFβ with antibodies or genetic manipulation leads to decreased numbers of induced TReg cells in some models of tumour-bearing animals 39, 115. Therefore, targeting TGFβ signalling in the tumour microenvironment could attenuate the immunosuppressive effects of iTReg cells, resulting in increased antitumour immunity.
CD8+ regulatory T cells
CD8+ T cells can become suppressor cells similar to CD4+ TReg cells, and TGFβ can induce CD8+ T cells to express FOXP3 116, 117. CD8+ regulatory T cells are induced under immunosuppressive conditions such as the tumour microenvironment 118–121. The role of tumour-infiltrating CD8+ regulatory T cells is less well understood than that of CD4+ TReg cells. It was recently shown that infiltration of CD8+ T cells into the immunosuppressive microenvironment of prostate tumours can convert tumour-specific CD8+ effector T cells into regulatory cells, and that this regulatory activity could be blocked by a TGFβ-specific antibody119. In another recent study, CD8+CD25+FOXP3+ regulatory T cells were isolated from colorectal cancer tissue and shown to have suppressive activity ex vivo. In this study, TGFβ and IL-6 induced the generation of CD8+ regulatory T cells in a synergistic manner 120. However, as these CD8+ regulatory T cells represent only a small number of CD8+ T cells in vivo, more investigation is needed to fully understand how they are induced and what is their clinical relevance in patients with cancer.
NKT cells
NKT cells are a heterogenous subset of T cells that also have properties of NK cells, and thus bridge the innate and adaptive immune responses. Unlike other T cells that recognize MHC class I-presented peptides, NKT cells recognize self and foreign glycolipids presented by the nonclassical MHC class I molecule CD1d 122. There are two main subtypes of NKT cells that have opposing roles in the antitumour immune response: Type I NKT cells (invariant NKT or iNKT cells) and Type II NKT cells.
iNKT cells are defined by use of a semi-invariant TCR involving Vα14Jα18 in mice and Vα24Jα18 in humans, and respond to α-galactosylceramide, resulting in increased antitumour responses through IFNγ production that activates CD8+ T cells and NK cells 123, 122. Defects in iNKT cells have been identified in patients with cancer in later stages of the disease and increased numbers of circulating and intratumoural iNKT cells have been associated with improved prognosis 124. Targeting iNKT cells with activating agents is being evaluated in clinical trials (reviewed in 123). TGFβ has been implicated in suppressing this cell subset in patients with cancer and a recent evaluation of iNKT cells from patients with metastatic melanoma and renal cell carcinoma suggested that blocking TGFβ in vitro could enhance iNKT cell activation ex vivo 125.
Type II NKT cells have diverse repertoires of TCRs and, in contrast to iNKT cells, suppress the antitumour response through several mechanisms, including TGFβ production 122. In a murine fibroblast tumor model this subset of NKT cells can express high levels of IL-13, leading to the production of TGFβ by MDSCs, which in turn results in attenuated antitumour responses by CD8+ effector T cells 76. However, in a different tumour model, evaluating antibody-mediated blockade of TGFβ combined with a peptide vaccine against a lung cancer tumor line (TC1) suggested that the IL-13 pathway augmented by type II NKT cells might not be the mechanism behind the enhanced antitumour activity observed in vaccinated mice 81. These results suggest a more complex interplay, yet to be determined, between TGFβ, type II NKT cells and effector immune cells responsible for the antitumour response.
Summary
Successful cancer immunotherapy depends on overcoming the immunosuppressive milieu in the tumour microenvironment in patients with cancer. TGFβ has a crucial immunosuppressive role in both the innate and the adaptive arms of the immune response (Figures 2 and 3). In terms of the innate immune response, TGFβ inhibits IFNγ production by NK cells causing dampened CD4+ TH1 cell responses. It downregulates expression of the activating receptor NKG2D on NK cells resulting in decreased cytolytic activity and overall poor antitumour responses. TGFβ also influences tumour antigen presentation by decreasing DC migration and promoting DC apoptosis in some tumour models. In general, TGFβ inhibits DC maturation and cytokine production, thereby promoting a tolerogenic environment. In addition, TGFβ produced by tolerogenic DCs contributes to TReg cell differentiation. TGFβ can also favour the differentiation of M2 versus M1 macrophages by inhibiting NF-κB activation. TAMs are a subtype of M2 cells that are recruited to the tumour by TGFβ and also produce high levels of TGFβ. TAMs in the tumour microenvironment compete with DCs for antigen uptake but cannot properly present antigen. TGFβ also promotes the differentiation of N1 to N2 neutrophils, which similar to M2 macrophages, are less cytotoxic. So, blocking TGFβ can induce an expression profile in the tumour microenvironment that promotes better antigen uptake and presentation, resulting in more robust priming and activation of the adaptive anti-tumour immune response.
Figure 2. Effects of TGFβ on innate immune cells.
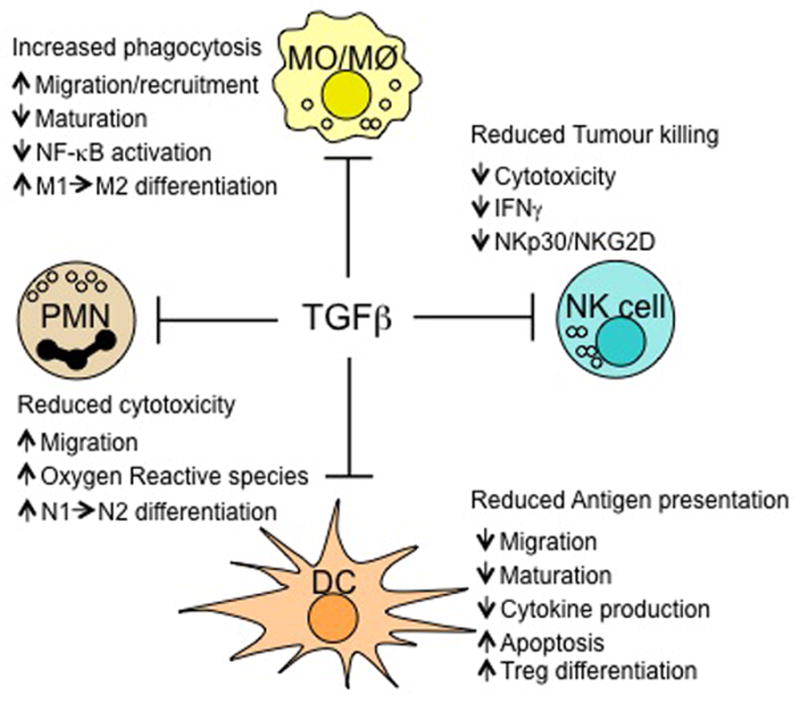
Transforming growth factor-β (TGFβ) has an inhibitory effect on innate immunity in the tumour microenvironment through several pathways. It inhibits natural killer (NK) cell function by downregulating interferon-γ (IFNγ) production and expression of the activating receptors NKp30 and NKG2D, thereby decreasing NK cell killing activity. In the presence of TGFβ, dendritic cells (DCs) acquire a tolerogenic phenotype involving decreased migration, maturation and cytokine production and increased apoptosis; they gain the ability to induce regulatory T (TReg) cell differentiation. TGFβ can also convert N1 neutrophils to a N2 phenotype, which is less cytotoxic. Similarly, TGFβ can promote the recruitment of M2 over M1 macrophages and of tumour-associated macrophages (TAMs), and can decrease cytokine production by these macrophages by inhibiting nuclear factor-κB (NF-κB) activity.
Figure 3. Effects of TGFβ on effector T cells.
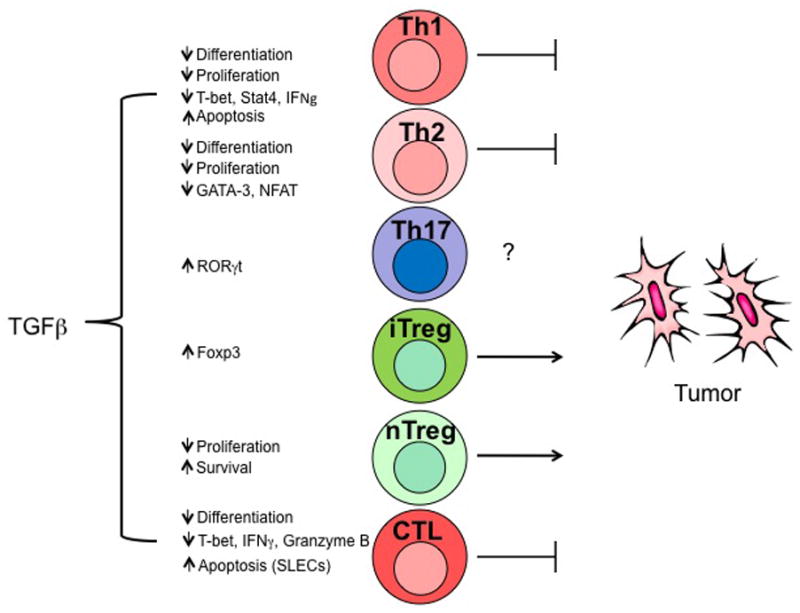
Transforming growth factor-β (TGFβ) differentially regulates the survival, differentiation, proliferation and apoptosis of T cell subsets. Among TH subpopulations, both TH1 and TH2 can provide antitumour responses, however TH1 seem to be more efficient. Both nTreg and iTreg populations inhibit antitumour immune responses. Within the tumour microenvironment, TGFβ can promote tumour growth by the maintenance of TReg cell and differentiation of iTreg subpopulations. TGFβ can also inhibit TH1 cell and CTL functions by downregulating T-bet and IFNγ expression and probably promoting a shift towards TH2 differentiation. CTLs are potent antitumour effector cells. TGFβ could also inhibit tumour immune surveillance by the induction of apoptosis in short-lived effector CTLs. The role of TH17 cells in tumour biology is still controversial and requires further characterization.
In terms of the adaptive immune response, TGFβ can also directly dampen the function of CD8+ and CD4+ T cells while promoting the recruitment and differentiation of regulatory T cells at the tumour bed (Figure 3). This cytokine inhibits the cytotoxic function of tumour specific CTLs and promotes apoptosis of the short-lived effector CD8+ T cells. TGFβ also controls the differentiation of several key CD4+ T cell subsets in tumour immunology, including TH1, TH17 and TReg cell subpopulations. Importantly, the effect of TGFβ on the differentiation of CD4+ T cells is influenced by the cytokine milieu in the tumour microenvironment, suggesting that modulating the relative abundance of such factors could probably promote antitumour responses in vivo. It is well documented that both TGFβ and regulatory T cells have key roles in suppressing the antitumour immune response; however, the precise contributions of TGFβ and different regulatory T cell subsets in suppressing effector cell function are still being evaluated (Figure 4). For example, although TGFβ can induce CD8+ T cells to become regulatory cells expressing FOXP3, the precise role of CD8+FOXP3+ T cells in tumour immunity remains unclear. TGFβ is also implicated in suppressing antitumour iNKT cell function; however, the interplay between TGFβ and immunosuppressive Type II NKT cells is less clear. Given that TGFβ can actively modulate inflammation and tolerance induction in the many ways described above, TGFβ blockade might enhance antitumour immunity through effects on numerous components of the immune response.
Figure 4. Effects of TGFβ on regulatory cells.
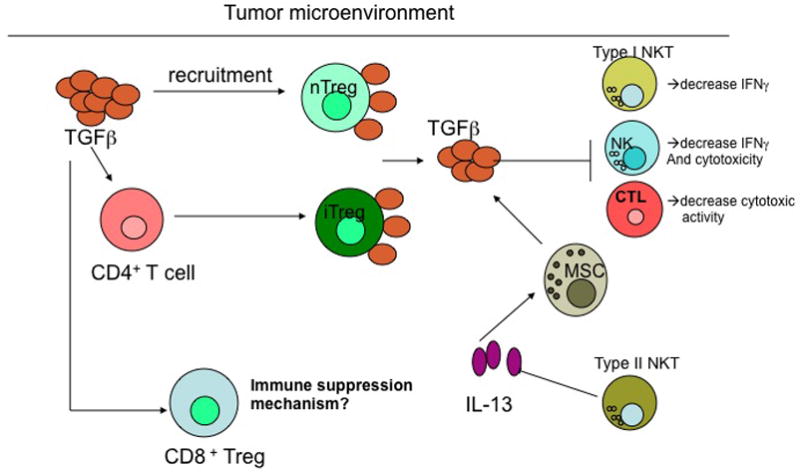
Within the tumour microenvironment, transforming growth factor-β (TGFβ) has been implicated in recruiting natural regulatory T (nTReg) cells as well as converting CD4+ effector T cells to induced TReg (iTReg) cells. These TReg cells can express cell surface-bound TGFβ and can inhibit effector cells, including natural killer (NK) cells and CD8+ T cells, in the tumour microenvironment by cell–cell contact to dampen the antitumour response. Type I NKT cells, which are responsible for recruiting effector immune cells to the tumour through the production of large amounts of IFNγ, can be suppressed by intratumoural TGFβ, whereas Type II NKT cells support increased TGFβ production by myeloid-derived suppressor cells (MDSCs) through the generation of interleukin-13 (IL-13). CD8+ regulatory T cells have been observed in lung tumours and these might result from the production of IL-10 by antigen-presenting cells, leading to increased TGFβ production in the tumour microenvironment. The precise immunosuppressive mechanisms of CD8+ regulatory T cells in regulating the antitumour immune response have yet to be identified.
Targeting TGFβ signalling for immunotherapy of cancer
The immunosuppressive effects of TGFβ on immune cell subsets leading to dampened antitumour immune responses as described above strongly support the development of TGFβ inhibitors to treat patients with cancer. Several inhibitors of TGFβ signalling, summarized in Table 1, are in various stages of development, targeting several steps in the TGFβ signalling cascade (Figure 5). Although most of these approaches are in preclinical studies, several clinical trials have evaluated TGFβ inhibition in patients with cancer using an antibody (GC1008), blocking oligonucleotides (AP12009), small molecule inhibitors (LY373636 and LY2157299) and a vaccine approach 126–133. The results from these trials evaluating TGFβ blockade alone indicate that there is limited clinical benefit; however, trials evaluating the small molecule inhibitors and antibody-mediated blockade are ongoing (Table 1).
Table 1.
Summary of TGFβ signalling inhibitors being evaluated as immunotherapies.
| Drug | Mechanism of Action | Stage of Development | Referenced Summary of Results |
|---|---|---|---|
| AP12009 | Antisense Oligonucleotide against TGFβ2 | Phase I/II and Phase III (enrolling) | AP 12009 was safe and well-tolerated in high grade glioma patients. 7/24 patients with stable disease and 2/24 patients with complete response 25 months after initiating therapy 130, 132. |
| AP-11014 | Antisense Oligonucleotide Against TGFβ1 | Preclinical | Decreases TGFb1 secretion by and subsequent proliferation of lung, colon and prostate cancer cell lines 140. |
| Lerdelimumab (CAT 152) | Antibody specific for TGFβ2 | Phase III | Evaluated for eye surgeries to prevent scarring after primary traveculectomies. Deemed safe but ineffective at improving scarring following eye surgery when compared to placebo 141 |
| Metelimumab (CAT 192) | Antibody specific for TGFβ1 | Phase II | May prevent excessive post operative scarring for glaucoma surgery; also when added to ACE-I drug can arrest diabetic nephropathy in rats 142 |
| GC-1008 | Antibody specific for all isoforms of TGFβ | Phase I | Safe and well-tolerated in advanced melanoma and kidney cancer patients 143. |
| ID11 | Antibody specific for all isoforms of TGFβ | Preclinical | One study indicates that pretreatment of mice engrafted with syngeneic breast cancer cell line with this antibody suppresses breast cancer metastases to lungs 144. |
| TR1 and MT1 | Antibodies specific for TGFβRII | Preclinical | Enhances antitumor responses against murine breast and colon cell lines by increasing CTL and NK activity while decreasing Tregs and myeloid derived suppressor cells in mice treated with these antibodies 145. |
|
Ly550410 Ly580276 Ly364947 Ly2109761 |
Small Molecule Inhibitors of TGFβRI kinase | Preclinical | While majority of these inhibitors have not been assessed in animal models, nanoparticle delivery Ly364947 has resulted in antitumor activity against human pancreatic and gastric xenografts in immunodeficient mice.146, 147. |
| Ly573636 | Small Molecule Inhibitor of TGFβRI kinase | Phase II | Results unavailable as Phase II trial evaluating this drug in advanced stage melanoma patients is ongoing 133. |
| Ly2157299 | Small Molecule Inhibitor of TGFβRI kinase | Phase I | 40mg and 80mg daily doses of this drug were safe and well-tolerated in colon cancer, prostate cancer, adrenal gland cancer, breast cancer and melanoma patients enrolled in this study126. |
| SB-505124 SB-431542 |
Small Molecule Inhibitor of TGFβRI kinase | Preclinical | No in vivo data available at this time. Inhibition of TGF-beta signaling has been established in vitro148, 149. |
| SD-208 SD-093 |
Small Molecule Inhibitor of TGFβRI kinase | Preclinical | Inhibits multiple myeloma (SD-093) and glioma (SD208) growth in vivo150, 151. |
| Ki26894 | Small Molecule Inhibitor of TGFβRI kinase | Preclinical | Inhibits breast cancer metastasis and enhanced survival in preclinical murine model treated with this drug 152. |
| Sm16 | Small Molecule Inhibitor of TGFβRI kinase | Preclinical | In vivo effects include inhibiting murine mesothelioma tumor growth and recurrence following resection of this tumor in a mouse model153,154. |
| Trx-xFoxH1b Trx-Lef1 |
Interacting peptide aptamers against smads | Preclinical | Bind to smads to inhibit TGF-beta mediated gene expression using in vitro luciferase reporter assays 155. |
| P144, P17 | 14mer peptide blocking TGFβ binding to TGFβRI and TGFRβRII | Preclinical | Administration of both peptides with adjuvant molecules poly(I:C) and agonistic anti-CD40 antibody increased antitumor activity against the ova-expressing lymphoma cell line E.G7 ova in mice by enhancing NK, tumor- specific CTL, DC activity while suppressing MDSCs and inhibition of TGFb production by Tregs156. |
| Belagenpumatucel-L: Antisense- transfected tumour cells | Vaccine against TGFβRII | Phase I and II | No significant toxicities observed in cancer patients enrolled in the studies. When stratified for circulating tumor cells patients with less than 2 circulating tumor cells per 7.5mL of blood demonstrated median survival of 660 days vs 150 days in patients with greater circulating tumor cells 127–129. |
| Soluble TBR2-FC | Stabilized soluble protein against TGFβRs | Preclinical | Lifetime exposure was tolerated in mice and decreased incidence of metastasis formation in a metastatic melanoma model and inducible transgenic breast cancer model157. |
| Plasmid | Plasmid DNA encoding TGFβRII fused to human IgG heavy chain | Preclinical | Administration of this plasmid to tumor draining lymph nodes from implanted E.G7 lymphomas and B16 melanomas in mice inhibited lymphoma tumor growth and primary melanoma growth and lung metastases 158. |
Figure 5. Targets for inhibiting TGFβ and downstream signalling events.
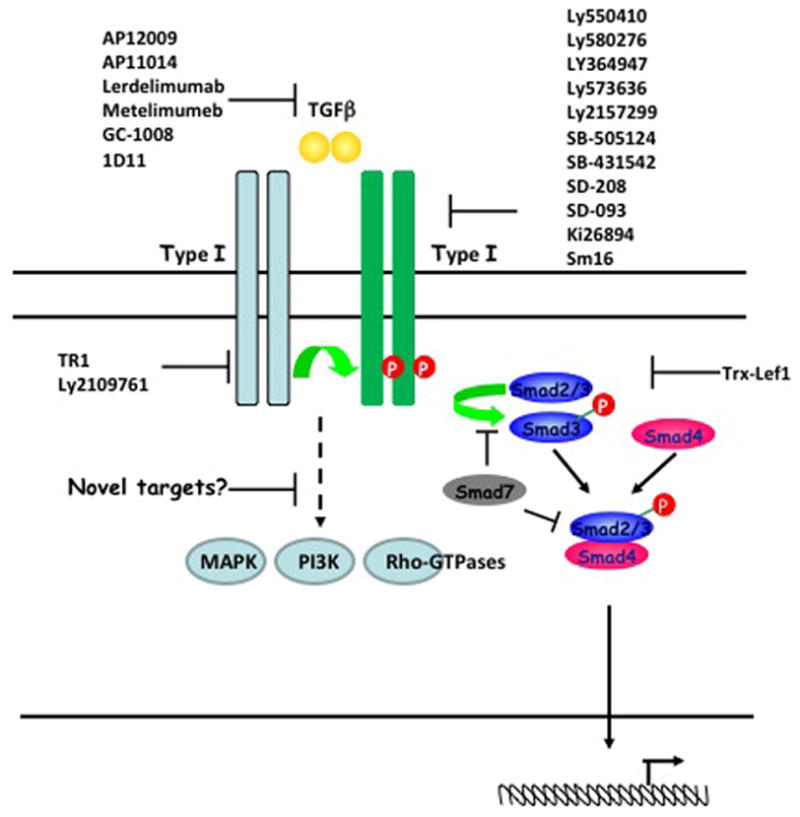
The transforming growth factor-β (TGFβ)-dependent signalling pathway depends on Type I and Type II serine-threonine kinase receptors and transcription factors known as SMADs. The dimeric bioactive ligand binds to a Type II receptor, which in turn phosphorylates and activates a Type I receptor. Once the Type I receptor is activated, it phosphorylates the receptor SMADs (R-SMADs: SMAD2 and SMAD3), which promotes their interaction with the common mediator SMAD (SMAD4) and translocation to the nucleus. The inhibitory SMAD7 negatively regulates TGFβ signalling by competing with R-SMADs for interaction with the Type I receptor or SMAD4. Current TGFβ signalling inhibitors (listed in Table I ans shown in scheme) include ligand, receptor and SMAD antagonists. However, additional SMAD-independent pathways have been reported to be induced in response to TGFβ, including the activation of MAPK, Rho-like GTPase and phosphatidylinositol 3-Kinase (PI3K) signalling pathways; a complete understanding of these alternative pathways could potentially offer additional downstream molecules that could be targeted in future therapeutic approaches. (Figure adapted from 138)
Although systemic blockade of TGFβ has been well-tolerated in preclinical studies, given the pleiotropic effects of this cytokine, one potential concern of this type of therapy is the development of significant autoimmune toxicities in humans. This could be particularly problematic if TGFβ blockade is used in combination with other immune-activating agents, such as CTLA-4- or PD1-specific antibodies (which are also being evaluated as single agents in clinical trials and have shown several autoimmune toxicities) 134. Other potential toxicities of blocking TGFβ might result from the cytokine’s homeostatic functions in other tissues outside of the immune system, including angiogenesis and the development of musculoskeletal tissues.
The manipulation of local TGFβ sources in the tumour should be considered in the future as a strategy to inhibit the dominant immunosuppressive intratumoural environment while promoting antitumour immunity. Challenges to this approach will include being able to target the tumour microenvironment with TGFβ inhibitors without affecting TGFβ function in the rest of the host to maintain systemic homeostatic processes. This might require novel delivery systems, as well as effective TGFβ-specific drugs that have minimum systemic toxicities to the recipient.
Future Directions
Given that TGFβ affects the activity and differentiation of numerous immune cell types, it is unclear which of the effects of TGFβ is most important in the tumour microenvironment. This is, in part, due to the pleiotropic nature of TGFβ and the contextual and combinatorial effects that this cytokine can have at a range of biological concentrations on different cell types at various stages of their development. Therefore, there remain several questions regarding the basic biology of this cytokine as well as regarding the best strategy to modulate this pathway alone and in combination with other pathways to enhance antitumour immunity in patients with cancer.
Although it is thought that tumour cells are an important source of TGFβ in the tumour microenvironment, immune cells themselves might in fact be a larger source of this cytokine, which is produced by effector T cells, regulatory T cells, APCs and MDSCs. Identifying the most relevant source of TGFβ will be important, as localized immunotherapy in the tumour microenvironment might be safer than systemic therapies that could interfere with the systemic homeostatic functions of TGFβ. In addition, TGFβ is expressed and synthesized as an inactive latent form unable to bind to its receptor. TGFβ becomes activated by interacting with molecules such as plasmin, matrix metalloproteinase, reactive oxygen species, thrombospondin-1, or integrins αvβ6 or αvβ8 6, 90. Notably, the cells that can activate TGFβ may be different than those that produce this potent cytokine, and thus this activation step provides a means for TGFβ to integrate signals from multiple cell types 90. We know little about how TGFβ is activated in a tumour-bearing environment and whether tolerogenic DCs, TAMs or MDSCs have a greater capacity to activate TGFβ than their immunogenic counterparts. Therefore, a precise understanding of the mechanisms by which immunosuppressive cell subsets work alone and together, and their specific involvement in producing and/or activating TGFβ, may improve cancer immunotherapies.
Further studies are also warranted to evaluate the effect of increasing innate and adaptive immune responses in a tumour-bearing host. For example, inhibition of TGFβ offers a means to manipulate T cell polarization in vivo that can change an immunosuppressive environment into a more antitumour environment once a tumour has established in the host, as is the case in cancer patients when these types of therapies are generally being considered. Once the exact role of IL-17 and TH17 cells in antitumour immunity has been defined, modulation of TGFβ levels might also be used to alter the ratio between TReg cells and TH17 cells in the tumour microenvironment. In addition, blocking TGFβ signalling in combination with tumour vaccines promotes antitumour immunity that is mediated, in part, by CD8+ T cells80–82, 135, which could lead to a long-term response with immunological memory.
Additional areas for future research related to the development of agents that efficiently block TGFβ and its activity include pharmacodynamic profiling of tissue TGFβ concentrations and the optimization of strategies to block the most appropriate TGFβ-dependent signalling pathways. For example, the blockade of SMAD-independent pathways of TGFβ-dependent signalling, including MAPKs, Rho GTPases and PI3K that are involved in tumour progression and metastasis, could lead to new strategies to enhance antitumour immunity (Figure 5). Mutations in SMAD2, SMAD3 and SMAD4 lead to cancer progression 136, 137, indicating that the tumour-suppressor properties of TGFβ involve SMAD-dependent signalling and therefore SMAD-dependent pathways may not be ideal therapeutic targets. Understanding the intricate signalling pathways controlled by TGFβ, as well as the mechanism(s) leading to its opposing effects in tumour biology, could lead to new strategies against cancer 138.
Furthermore, identifying the ideal timing of TGFβ blockade in the host, if used in combination with vaccines, cytokine therapies (IL-2 and IL-15) or other immune-activating antibodies (such as CTLA4- or PD1/PD1L-specific antibodies), would be very informative 139. Finally, designing optimal methods to deliver the most effective TGFβ inhibitor to the tumour microenvironment and the evaluation of exposing expanded T cell subsets to these drugs ex vivo to enhance adoptive T-cell based immunotherapies are all areas requiring further investigation.
So far, the clinical trials evaluating blockade of TGFβ in patients with cancer do not show a clear clinical benefit. Therefore, larger studies are warranted to clarify the toxicity and efficacy of these strategies. In addition, the optimal dose and timing of TGFβ blockade, as well as the ideal combination of this approach with other immunotherapies, remain unknown. These questions are currently being addressed in ongoing preclinical studies and are likely to be the focus of future clinical trials.
Text box 1. Non-immune effects of TGFβ in tumours.
In addition to the effects of TGFβ on various immune cell subsets to promote tumour progression, this pleiotropic cytokine enhances tumourigenesis in a number of pathways not directly involving the antitumor response. For example, TGFβ promotes autocrine mitogen production to enhance tumor cell proliferation. In addition, TGFβ augments a number of biologic process supporting metastasis formation, including: tumor cell motility, cancer cell priming for metastasis development, extravasation of tumor cells from the primary site, osteoclast mobilization which can support osseus metastasis deposits and angiogenesis to nourish primary tumor and secondary metastases. These effects are beyond the scope of this review and are discussed in detail in References 1 and 2.
Acknowledgments
R.A.F is an investigator of the Howard Hughes Medical Institute. This work is supported by a post-doctoral fellowship grant from the Cancer Research Institute (to S.S.), the Yale Skin SPORE through a Yale Skin SPORE Career Development Award (to S.H.W.) and a post-doctoral fellowship from PEW Charitable Trust: PEW Latin American Fellow Program in Biomedical Sciences (to P.L.L.). Additional support from NIH grants CA121974 and DK051665 (to R.A.F.) and JDRF grant 32-2008-352 (to R.A.F.).
Glossary
- Peripheral tolerance
The lack of responsiveness of mature lymphocytes in the periphery to specific self antigens. These mechanisms can control potentially self-reactive lymphocytes that have escaped central tolerance or prevent immune responses to specialized self proteins that were not present during establishment of central tolerance. Peripheral tolerance is associated with suppression of self-reactive antibody production by B cells and inhibition of self-reactive effector cells, such as T helper cells and cytotoxic T lymphocytes
- Myeloid-derived suppressor cells (MDSCs)
A group of immature CD11b+GR1+ cells (which include precursors of macrophages, granulocytes, dendritic cells and myeloid cells) that are produced in response to various tumour-derived cytokines. These cells have been shown to induce tumour-associated antigen-specific CD8+ T-cell tolerance
- Tumour-associated macrophages (TAMs)
An important component of the tumour microenvironment. These cells differentiate from circulating blood monocytes that have infiltrated tumours. These cells can have positive or negative effects on tumorigenesis (that is, tumour promotion or immunosurveillance, respectively)
- M1 (classically activated) macrophage
A macrophage that is activated by Toll-like receptor ligands (such as LPS) and interferon-gamma and that express, among others, inducible nitric-oxide synthase and nitric oxide
- M2 (alternatively activated) macrophage
A macrophage that is stimulated by interleukin-4 (IL-4) or IL-13 and that expresses arginase-1, the mannose receptor CD206 and the IL-4 receptor alpha-chain
Footnotes
The authors declare that they have no competing financial interests.
References
- 1.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wrzesinski SH, Wan YY, Flavell RA. Transforming Growth Factor-{beta} and the Immune Response: Implications for Anticancer Therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 3.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 4.Tian M, Schiemann WP. The TGF-beta paradox in human cancer: an update. Future Oncol. 2009;5:259–71. doi: 10.2217/14796694.5.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2009 doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 7.Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–56. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–7. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 9.Rook AH, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. 1986;136:3916–20. [PubMed] [Google Scholar]
- 10.Wahl SM, Wen J, Moutsopoulos NM. The kiss of death: interrupted by NK-cell close encounters of another kind. Trends Immunol. 2006;27:161–4. doi: 10.1016/j.it.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Trotta R, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181:3784–92. doi: 10.4049/jimmunol.181.6.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castriconi R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. 2003;100:4120–5. doi: 10.1073/pnas.0730640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crane CA, et al. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010;12:7–13. doi: 10.1093/neuonc/nop009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta 1 Secretion and Down- Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J Immunol. 2004;172:7335–7340. doi: 10.4049/jimmunol.172.12.7335. [DOI] [PubMed] [Google Scholar]
- 16.Kopp HG, Placke T, Salih HR. Platelet-Derived Transforming Growth Factor-{beta} Down-Regulates NKG2D Thereby Inhibiting Natural Killer Cell Antitumor Reactivity. 2009;69:7775–7783. doi: 10.1158/0008-5472.CAN-09-2123. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182:240–9. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 18.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 19.Ferlazzo G, Munz C. Dendritic cell interactions with NK cells from different tissues. J Clin Immunol. 2009;29:265–73. doi: 10.1007/s10875-009-9283-y. [DOI] [PubMed] [Google Scholar]
- 20.Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. 2008;15:39–50. doi: 10.1038/sj.cdd.4402247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–8. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 22.Hawiger D, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki S, Steinman RM. Dendritic cells as controllers of antigen-specific Foxp3+ regulatory T cells. J Dermatol Sci. 2009;54:69–75. doi: 10.1016/j.jdermsci.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kortylewski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito M, et al. Tumor-derived TGFbeta-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol. 2006;176:5637–43. doi: 10.4049/jimmunol.176.9.5637. [DOI] [PubMed] [Google Scholar]
- 27.Weber F, et al. Transforming growth factor-beta1 immobilises dendritic cells within skin tumours and facilitates tumour escape from the immune system. Cancer Immunol Immunother. 2005;54:898–906. doi: 10.1007/s00262-004-0652-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halliday GM, Le S. Transforming growth factor-beta produced by progressor tumors inhibits, while IL-10 produced by regressor tumors enhances, Langerhans cell migration from skin. Int Immunol. 2001;13:1147–54. doi: 10.1093/intimm/13.9.1147. [DOI] [PubMed] [Google Scholar]
- 29.Bekeredjian-Ding I, et al. Tumour-derived prostaglandin E and transforming growth factor-beta synergize to inhibit plasmacytoid dendritic cell-derived interferon-alpha. Immunology. 2009;128:439–50. doi: 10.1111/j.1365-2567.2009.03134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, et al. CD4-8- dendritic cells prime CD4+ T regulatory 1 cells to suppress antitumor immunity. J Immunol. 2005;175:2931–7. doi: 10.4049/jimmunol.175.5.2931. [DOI] [PubMed] [Google Scholar]
- 31.Roncarolo MG, Levings MK, Traversari C. Differentiation of T regulatory cells by immature dendritic cells. J Exp Med. 2001;193:F5–9. doi: 10.1084/jem.193.2.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamazaki S, et al. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen- processing dendritic cells. J Exp Med. 2003;198:235–47. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–77. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM. Expansion of FOXP3 high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood. 2006;108:2655–61. doi: 10.1182/blood-2006-03-011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung DJ, et al. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood. 2009;114:555–63. doi: 10.1182/blood-2008-11-191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo X, et al. Dendritic cells with TGF-beta1 differentiate naive CD4+CD25− T cells into islet- protective Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104:2821–6. doi: 10.1073/pnas.0611646104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levings MK, Bacchetta R, Schulz U, Roncarolo MG. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol. 2002;129:263–76. doi: 10.1159/000067596. [DOI] [PubMed] [Google Scholar]
- 38.Ghiringhelli F, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta- secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202:919–29. doi: 10.1084/jem.20050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu VC, et al. Tumor Evasion of the Immune System by Converting CD4+CD25− T Cells into CD4+CD25+ T Regulatory Cells: Role of Tumor-Derived TGF-beta. J Immunol. 2007;178:2883–2892. doi: 10.4049/jimmunol.178.5.2883. [DOI] [PubMed] [Google Scholar]
- 40.Yamazaki S, et al. CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol. 2008;181:6923–33. doi: 10.4049/jimmunol.181.10.6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD. Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2009;182:2795–807. doi: 10.4049/jimmunol.0712671. [DOI] [PubMed] [Google Scholar]
- 42.Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–30. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Steidl C, et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 45.Byrne SN, Knox MC, Halliday GM. TGFbeta is responsible for skin tumour infiltration by macrophages enabling the tumours to escape immune destruction. Immunol Cell Biol. 2008;86:92–7. doi: 10.1038/sj.icb.7100116. [DOI] [PubMed] [Google Scholar]
- 46.Biswas SK, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–22. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 47.Saccani A, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432–40. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 48.Porta C, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009;106:14978–83. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010 doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 50.Torroella-Kouri M, et al. Identification of a subpopulation of macrophages in mammary tumor- bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009;69:4800–9. doi: 10.1158/0008-5472.CAN-08-3427. TGFβ and prostaglandin E2, individually and additively downregulate NF-κB and C/EBP in a unique less differentiated macrophage subpopulation. [DOI] [PubMed] [Google Scholar]
- 51.Umemura N, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008;83:1136–44. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- 52.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 53.Gratchev A, et al. Activation of a TGF-beta-specific multistep gene expression program in mature macrophages requires glucocorticoid-mediated surface expression of TGF-beta receptor II. J Immunol. 2008;180:6553–65. doi: 10.4049/jimmunol.180.10.6553. [DOI] [PubMed] [Google Scholar]
- 54.Allen SS, et al. Altered inflammatory responses following transforming growth factor-beta neutralization in experimental guinea pig tuberculous pleurisy. Tuberculosis (Edinb) 2008;88:430–6. doi: 10.1016/j.tube.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Smith WB, et al. Transforming growth factor-beta 1 inhibits the production of IL-8 and the transmigration of neutrophils through activated endothelium. J Immunol. 1996;157:360–8. [PubMed] [Google Scholar]
- 56.Shen L, et al. Inhibition of human neutrophil degranulation by transforming growth factor- beta1. Clin Exp Immunol. 2007;149:155–61. doi: 10.1111/j.1365-2249.2007.03376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Di Carlo E, et al. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97:339–45. doi: 10.1182/blood.v97.2.339. [DOI] [PubMed] [Google Scholar]
- 58.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94. doi: 10.1016/j.ccr.2009.06.017. This study provides the first evidence that TGFβ controls the pro/anti-tumor responses by polarizing neutrophil subpopulations N1–N2 within the tumor microenvironment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–8. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181:435–40. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tazawa H, et al. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003;163:2221–32. doi: 10.1016/S0002-9440(10)63580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60. [PubMed] [Google Scholar]
- 63.Colombo MP, Modesti A, Parmiani G, Forni G. Local cytokine availability elicits tumor rejection and systemic immunity through granulocyte-T-lymphocyte cross-talk. Cancer Res. 1992;52:4853–7. [PubMed] [Google Scholar]
- 64.Hicks AM, et al. Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc Natl Acad Sci U S A. 2006;103:7753–8. doi: 10.1073/pnas.0602382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stoppacciaro A, et al. Regression of an established tumor genetically modified to release granulocyte colony-stimulating factor requires granulocyte-T cell cooperation and T cell- produced interferon gamma. J Exp Med. 1993;178:151–61. doi: 10.1084/jem.178.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher K, Haensch W, Roefzaad C, Schlag PM. Prognostic significance of activated CD8(+) T cell infiltrations within esophageal carcinomas. Cancer Res. 2001;61:3932–6. [PubMed] [Google Scholar]
- 67.Nakano O, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–6. [PubMed] [Google Scholar]
- 68.Zhang L, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 69.Gao Q, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25:2586–93. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 70.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Q, et al. Blockade of transforming growth factor-{beta} signaling in tumor-reactive CD8(+) T cells activates the antitumor immune response cycle. Mol Cancer Ther. 2006;5:1733–43. doi: 10.1158/1535-7163.MCT-06-0109. [DOI] [PubMed] [Google Scholar]
- 72.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–81. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 73.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Q, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+T cells: eradication of autologous mouse prostate cancer. Cancer Res. 2005;65:1761–9. doi: 10.1158/0008-5472.CAN-04-3169. [DOI] [PubMed] [Google Scholar]
- 75.Wang L, et al. Immunotherapy for human renal cell carcinoma by adoptive transfer of autologous transforming growth factor beta-insensitive CD8+ T cells. Clin Cancer Res. 2010;16:164–73. doi: 10.1158/1078-0432.CCR-09-1758. [DOI] [PubMed] [Google Scholar]
- 76.Terabe M, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol. 2000;1:515–20. doi: 10.1038/82771. [DOI] [PubMed] [Google Scholar]
- 77.Terabe M, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nam JS, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835–43. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallace A, et al. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. 2008;14:3966–74. doi: 10.1158/1078-0432.CCR-08-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takaku S, et al. Blockade of TGF-beta enhances tumor vaccine efficacy mediated by CD8(+) T cells. Int J Cancer. 2010;126:1666–74. doi: 10.1002/ijc.24961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Terabe M, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res. 2009;15:6560–9. doi: 10.1158/1078-0432.CCR-09-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ueda R, et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res. 2009;15:6551–9. doi: 10.1158/1078-0432.CCR-09-1067. These studies demonstrate that the combination of a TGFβ-specific antibody with a vaccine results in a synergistic improvement in the inhibition of tumour growth that is mediated by increased number and activity of CD8+ T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–44. doi: 10.1016/j.immuni.2009.04.020. This study demonstrates the ability of TGF-beta to promote apoptosis of effector CD8+ T cells under immunogenic conditions, such as vaccination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–23. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.di Bari MG, et al. TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol Immunother. 2009;58:1809–18. doi: 10.1007/s00262-009-0692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nam JS, et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008;68:3915–23. doi: 10.1158/0008-5472.CAN-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hinrichs CS, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114:596–9. doi: 10.1182/blood-2009-02-203935. This study demonstrate that IL-17 producing CD8+ mediate tumor regression and persist longer than normal CD8+ cells upon adoptive transfer in tumor-bearing mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Muranski P, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–73. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin-Orozco N, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–98. doi: 10.1016/j.immuni.2009.09.014. These studies provide compiling evidence comparing the therapeutic potential of adoptively transferred T helper subpopulations and show that Th17 polarized cells where the most effective in tumor eradication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li XF, et al. Transforming growth factor-beta (TGF-beta)-mediated immunosuppression in the tumor-bearing state: enhanced production of TGF-beta and a progressive increase in TGF-beta susceptibility of anti-tumor CD4+ T cell function. Jpn J Cancer Res. 1993;84:315–25. doi: 10.1111/j.1349-7006.1993.tb02873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maeda H, Shiraishi A. TGF-beta contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. J Immunol. 1996;156:73–8. [PubMed] [Google Scholar]
- 93.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–8. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muranski P, Restifo NP. Adoptive immunotherapy of cancer using CD4(+) T cells. Curr Opin Immunol. 2009;21:200–8. doi: 10.1016/j.coi.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gao FG, et al. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res. 2002;62:6438–41. [PubMed] [Google Scholar]
- 96.Perez-Diez A, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–54. doi: 10.1182/blood-2006-10-051318. This report shows the potential of CD4+ T cells in tumor elimination even in the absence of CD8+ T cells and independent on MHCII expression by tumor cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin-Orozco N, Dong C. The IL-17/IL-23 axis of inflammation in cancer: friend or foe? Curr Opin Investig Drugs. 2009;10:543–9. [PubMed] [Google Scholar]
- 98.Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol. 2009;183:4169–75. doi: 10.4049/jimmunol.0901017. [DOI] [PubMed] [Google Scholar]
- 99.Miyahara Y, et al. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:15505–10. doi: 10.1073/pnas.0710686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Su X, et al. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184:1630–41. doi: 10.4049/jimmunol.0902813. [DOI] [PubMed] [Google Scholar]
- 101.Nurieva R, Yang XO, Chung Y, Dong C. Cutting edge: in vitro generated Th17 cells maintain their cytokine expression program in normal but not lymphopenic hosts. J Immunol. 2009;182:2565–8. doi: 10.4049/jimmunol.0803931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xie Y, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–67. doi: 10.1084/jem.20091921. This report shows for the first time the potential use of naïve CD4+ T cells in transfer experiments to induce potent antitumor immunity in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Quezada SA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kryczek I, et al. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J Immunol. 2007;178:6730–3. doi: 10.4049/jimmunol.178.11.6730. [DOI] [PubMed] [Google Scholar]
- 105.Pellegrini M, et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med. 2009;15:528–36. doi: 10.1038/nm.1953. [DOI] [PubMed] [Google Scholar]
- 106.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626–35. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 107.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 108.Merlo A, et al. FOXP3 expression and overall survival in breast cancer. J Clin Oncol. 2009;27:1746–52. doi: 10.1200/JCO.2008.17.9036. [DOI] [PubMed] [Google Scholar]
- 109.Moo-Young TA, et al. Tumor-derived TGF-[beta] Mediates Conversion of CD4+Foxp3+ Regulatory T Cells in a Murine Model of Pancreas Cancer. J Immunother. 2009;32:12–21. doi: 10.1097/CJI.0b013e318189f13c. TGFβ derived from the tumor promotes in situ Treg differentiation demonstrating a mechanism of immune surveillance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006;108:804–811. doi: 10.1182/blood-2006-02-002774. [DOI] [PubMed] [Google Scholar]
- 111.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 112.Han Y, Guo Q, Zhang M, Chen Z, Cao X. CD69+ CD4+ CD25− T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol. 2009;182:111–20. doi: 10.4049/jimmunol.182.1.111. [DOI] [PubMed] [Google Scholar]
- 113.Chen W, et al. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-{beta} Induction of Transcription Factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fantini MC, et al. Cutting Edge: TGF-{beta} Induces a Regulatory Phenotype in CD4+CD25− T Cells through Foxp3 Induction and Down-Regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 115.Petrausch U, et al. Disruption of TGF-beta signaling prevents the generation of tumor-sensitized regulatory T cells and facilitates therapeutic antitumor immunity. J Immunol. 2009;183:3682–9. doi: 10.4049/jimmunol.0900560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Niederkorn JY. Emerging concepts in CD8(+) T regulatory cells. Curr Opin Immunol. 2008;20:327–31. doi: 10.1016/j.coi.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kapp JA, Bucy RP. CD8+ suppressor T cells resurrected. Hum Immunol. 2008;69:715–20. doi: 10.1016/j.humimm.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 118.Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. 2006;177:896–904. doi: 10.4049/jimmunol.177.2.896. [DOI] [PubMed] [Google Scholar]
- 119.Shafer-Weaver KA, et al. Cutting Edge: Tumor-specific CD8+ T cells infiltrating prostatic tumors are induced to become suppressor cells. J Immunol. 2009;183:4848–52. doi: 10.4049/jimmunol.0900848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chaput N, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58:520–9. doi: 10.1136/gut.2008.158824. [DOI] [PubMed] [Google Scholar]
- 121.Kiniwa Y, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13:6947–58. doi: 10.1158/1078-0432.CCR-07-0842. [DOI] [PubMed] [Google Scholar]
- 122.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Kaer LV. NKT cells: what’s in a name? Nat Rev Immunol. 2004;4:231–237. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- 123.Berzofsky J, Terabe M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunology, Immunotherapy. 2008;57:1679–1683. doi: 10.1007/s00262-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Berzofsky JA, Terabe M. The contrasting roles of NKT cells in tumor immunity. Curr Mol Med. 2009;9:667–72. doi: 10.2174/156652409788970706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van der Vliet HJ, et al. Circulating myeloid dendritic cells of advanced cancer patients result in reduced activation and a biased cytokine profile in invariant NKT cells. J Immunol. 2008;180:7287–93. doi: 10.4049/jimmunol.180.11.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Calvo-Aller E, BJ, Glatt S, et al. First human dose escalation study in patients with metastatic malignancies to determine safety and pharmacokinetics of LY2157299, a small molecule inhibitor of the transforming growth factor-beta receptor I kinase. ASCO Annual Meeting; 2008. [Google Scholar]
- 127.Fakhrai H, et al. Phase I clinical trial of a TGF-beta antisense-modified tumor cell vaccine in patients with advanced glioma. Cancer Gene Ther. 2006;13:1052–1060. doi: 10.1038/sj.cgt.7700975. [DOI] [PubMed] [Google Scholar]
- 128.Nemunaitis J, et al. Phase II Study of Belagenpumatucel-L, a Transforming Growth Factor Beta-2 Antisense Gene-Modified Allogeneic Tumor Cell Vaccine in Non-Small-Cell Lung Cancer. J Clin Oncol. 2006;24:4721–4730. doi: 10.1200/JCO.2005.05.5335. [DOI] [PubMed] [Google Scholar]
- 129.Nemunaitis J, et al. Phase II trial of Belagenpumatucel-L, a TGF-[beta]2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009;16:620–624. doi: 10.1038/cgt.2009.15. One of the few recent studies evaluating blocking TGFβ blockade in humans; there are multiple limitations regarding design of trial and true “efficacy” of this approach. [DOI] [PubMed] [Google Scholar]
- 130.Schlingensiepen KH, et al. Targeted tumor therapy with the TGF-beta2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006;17:129–139. doi: 10.1016/j.cytogfr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 131.Schlingensiepen R, et al. Intracerebral and Intrathecal Infusion of the TGF-beta2-Specific Antisense Phosphorothioate Oligonucleotide AP 12009 in Rabbits and Primates: Toxicology and Safety. Oligonucleotides. 2005;15:94–104. doi: 10.1089/oli.2005.15.94. [DOI] [PubMed] [Google Scholar]
- 132.http://clinicaltrials.gov/ct2/show/NCT00761280?term=AP12009&rank=3.
- 133.http://clinicaltrials.gov/show/NCT00383292.
- 134.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 135.Schreiber TH, Deyev VV, Rosenblatt JD, Podack ER. Tumor-induced suppression of CTL expansion and subjugation by gp96-lg vaccination. Cancer Res. 2009;69:2026–33. doi: 10.1158/0008-5472.CAN-08-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 137.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 138.Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19:77–91. doi: 10.1517/13543780903382609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wei S, et al. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008;68:5432–8. doi: 10.1158/0008-5472.CAN-07-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schlingensiepen KH, et al. The TGF-beta1 antisense oligonucleotide AP 11014 for the treatment of non-small cell lung, colorectal and prostate cancer: Preclinical studies. J Clin Immunol. 2004;22:3132. [Google Scholar]
- 141.Khaw P, et al. A phase III study of subconjunctival human anti-transforming growth factor beta(2) monoclonal antibody (CAT-152) to prevent scarring after first-time trabeculectomy. Ophthalmology. 2007;114:1822–30. doi: 10.1016/j.ophtha.2007.03.050. [DOI] [PubMed] [Google Scholar]
- 142.Benigni A, et al. Add-On Anti-TGF-beta Antibody to ACE Inhibitor Arrests Progressive Diabetic Nephropathy in the Rat. J Am Soc Nephrol. 2003;14:1816–1824. doi: 10.1097/01.asn.0000074238.61967.b7. [DOI] [PubMed] [Google Scholar]
- 143.Morris JC, GIS, Tan AR, Lawrence DP, Olencki TE, Dezube BJ, Hsu FJ, Reiss M, Berzofsky JA. Phase I/II study of GC1008: A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC) J Clin Oncol. 2008;26 doi: 10.1371/journal.pone.0090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nam JS, et al. Bone Sialoprotein Mediates the Tumor Cell-Targeted Prometastatic Activity of Transforming Growth Factor beta in a Mouse Model of Breast Cancer. Cancer Res. 2006;66:6327–6335. doi: 10.1158/0008-5472.CAN-06-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhong Z, et al. Anti-Transforming Growth Factor beta Receptor II Antibody Has Therapeutic Efficacy against Primary Tumor Growth and Metastasis through Multieffects on Cancer, Stroma, and Immune Cells. Clin Cancer Res. 2010;16:1191–1205. doi: 10.1158/1078-0432.CCR-09-1634. Newest study evaluating an antibody to TGFBRII which demonstrates antitumour responses against murine breast and colon cancer cell lines by increasing CTL and NK activity while decreasing Tregs and MSDC in mice treated with these antibodies. [DOI] [PubMed] [Google Scholar]
- 146.Kano MR, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. PNAS. 2007;104:3460–3465. doi: 10.1073/pnas.0611660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scott Sawyer J, et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorganic & Medicinal Chemistry Letters. 2004;14:3581–3584. doi: 10.1016/j.bmcl.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 148.Inman GJ, et al. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor- beta Superfamily Type I Activin Receptor-Like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 149.Saunier EF, Akhurst RJ. TGF Beta Inhibition for Cancer Therapy. Curr Cancer Drug Targets. 2006;6:565–578. doi: 10.2174/156800906778742460. [DOI] [PubMed] [Google Scholar]
- 150.Hayashi T, et al. Transforming Growth Factor beta Receptor I Kinase Inhibitor Down-Regulates Cytokine Secretion and Multiple Myeloma Cell Growth in the Bone Marrow Microenvironment. Clin Cancer Res. 2004;10:7540–7546. doi: 10.1158/1078-0432.CCR-04-0632. [DOI] [PubMed] [Google Scholar]
- 151.Uhl M, et al. SD-208, a Novel Transforming Growth Factor beta Receptor I Kinase Inhibitor, Inhibits Growth and Invasiveness and Enhances Immunogenicity of Murine and Human Glioma Cells In vitro and In vivo. Cancer Res. 2004;64:7954–7961. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- 152.Ehata S, et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 2007;98:127–133. doi: 10.1111/j.1349-7006.2006.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kim S, et al. Systemic Blockade of Transforming Growth Factor-{beta} Signaling Augments the Efficacy of Immunogene Therapy. Cancer Res. 2008;68:10247–10256. doi: 10.1158/0008-5472.CAN-08-1494. Recent evidence of successful combinatorial therapy using TGF-beta blocking reagents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Suzuki E, et al. A Novel Small-Molecule Inhibitor of Transforming Growth Factor beta Type I Receptor Kinase (SM16) Inhibits Murine Mesothelioma Tumor Growth In vivo and Prevents Tumor Recurrence after Surgical Resection. Cancer Res. 2007;67:2351–2359. doi: 10.1158/0008-5472.CAN-06-2389. [DOI] [PubMed] [Google Scholar]
- 155.Qiqi C, Sang Kyun L, Bryan Z, Michael Hoffmann F. Selective inhibition of TGF-beta responsive genes by Smad-interacting peptide aptamers from FoxH1 Lef1 and CBP. Oncogene. 2005;24:3864–3874. doi: 10.1038/sj.onc.1208556. [DOI] [PubMed] [Google Scholar]
- 156.Llopiz D, et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int J Cancer. 2009;125:2614–23. doi: 10.1002/ijc.24656. [DOI] [PubMed] [Google Scholar]
- 157.Yang Y-a, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fujita T, et al. Inhibition of Transforming Growth Factor-{beta}-Mediated Immunosuppression in Tumor-Draining Lymph Nodes Augments Antitumor Responses by Various Immunologic Cell Types. Cancer Res. 2009;69:5142–5150. doi: 10.1158/0008-5472.CAN-08-2499. [DOI] [PubMed] [Google Scholar]