

Abstract
Lipopolysaccharide (LPS) is the major cell surface molecule of gram-negative bacteria, deposited on the outer leaflet of the outer membrane bilayer. LPS can be subdivided into three domains: the distal O-polysaccharide, a core oligosaccharide, and the lipid A domain consisting of a lipid A molecular species and 3-deoxy-D-manno-oct-2-ulosonic acid residues (Kdo). The lipid A domain is the only component essential for bacterial cell survival. Following its synthesis, lipid A is chemically modified in response to environmental stresses such as pH or temperature, to promote resistance to antibiotic compounds, and to evade recognition by mediators of the host innate immune response. The following protocol details the small- and large-scale isolation of lipid A from gram-negative bacteria. Isolated material is then chemically characterized by thin layer chromatography (TLC) or mass-spectrometry (MS). In addition to matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS, we also describe tandem MS protocols for analyzing lipid A molecular species using electrospray ionization (ESI) coupled to collision induced dissociation (CID) and newly employed ultraviolet photodissociation (UVPD) methods. Our MS protocols allow for unequivocal determination of chemical structure, paramount to characterization of lipid A molecules that contain unique or novel chemical modifications. We also describe the radioisotopic labeling, and subsequent isolation, of lipid A from bacterial cells for analysis by TLC. Relative to MS-based protocols, TLC provides a more economical and rapid characterization method, but cannot be used to unambiguously assign lipid A chemical structures without the use of standards of known chemical structure. Over the last two decades isolation and characterization of lipid A has led to numerous exciting discoveries that have improved our understanding of the physiology of gram-negative bacteria, mechanisms of antibiotic resistance, the human innate immune response, and have provided many new targets in the development of antibacterial compounds.
Keywords: Chemistry, Issue 79, Membrane Lipids, Toll-Like Receptors, Endotoxins, Glycolipids, Lipopolysaccharides, Lipid A, Microbiology, Lipids, lipid A, Bligh-Dyer, thin layer chromatography (TLC), lipopolysaccharide, mass spectrometry, Collision Induced Dissociation (CID), Photodissociation (PD)
Introduction
Lipopolysaccharide (LPS) is the major outer surface molecule of nearly all gram-negative organisms and consists of three molecular domains: a distal O-antigen polysaccharide, a core oligosaccharide, and the membrane-associated lipid A domain deposited on the outer leaflet of the outer membrane bilayer1,2. The lipid A domain consists of 3-deoxy-D-manno-oct-2-ulosonic (Kdo) residues and a lipid A molecular species, where lipid A can be defined as the chloroform soluble portion of LPS upon mild-acid hydrolysis1,2. The standard lipid A molecule can be chemically defined as a diglucosamine backbone that is hexa-acylated and bis-phosphorylated; consistent with the major lipid A species observed in the model organism Escherichia coli (E. coli)1,2. Nine constitutively expressed genes, conserved throughout gram-negative bacteria, are responsible for the production of the lipid A domain (Figure 1)1,2. Most bacteria have an additional set of genes, which vary in degree of phylogenetic conservation, that participate in further chemical modification of lipid A3. Dephosphorylation, removal of acyl chains, and the addition of chemical moieties such as amino sugars (e.g. aminoarabinose) and/or phosphoethanolamine are the most commonly observed activities (Figure 1). Many of the enzymes responsible for lipid A modification are directly activated by environmental signals, such as divalent cations, or their expression is regulated by two component response-regulator systems3.
Recognition of lipid A species by the host innate immune system is mediated by the Toll-like receptor 4/myeloid differentiation factor 2 (TLR4/MD2) co-receptor4. Hydrophobic forces between MD2 and the lipid A acyl chains, as well as between TLR4 and the 1 and 4 'phosphate groups of lipid A promote the strong association of lipid A with TLR4/MD24,5. Modifications that alter acylation state or the negative charge of lipid A impact TLR4/MD2 based lipid A recognition and downstream stimulation of the innate immune response activators NF-κB and mediators of inflammation such as TNFα and IL1-β6,7. Modifications that mask the negative charge of lipid A also prevent bactericidal cationic antimicrobial peptides from binding to gram-negative cell surfaces3,8. Many lipid A modifications are hypothesized to increase bacterial fitness under specific environmental conditions, such as inside the human host or in an ecological niche. For this reason many modification enzymes are attractive targets in the rational development of antimicrobial compounds. The chemical diversity of lipid A structures, with respect to organism and/or environment, and the biological implications of these diverse structures make the structural characterization of lipid A an important endeavor in the study of gram-negative bacteria.
Isolation of lipid A molecules from whole bacteria involves the extraction of LPS from the bacterial cell surface, a hydrolytic step to liberate lipid A, followed by a final purification procedure9-11. The most frequently cited LPS extraction procedure is the hot-phenol water extraction procedure, first introduced by Westphal and Jann10. After extraction whole LPS is subjected to mild-acid hydrolysis, which chemically separates Kdo from the 6'-hydroxyl of the distal glucosamine sugar of lipid A (Figure 1). Numerous pitfalls exist for the hot-phenol water procedure including the use of a high hazard reagent, the need to degrade co-extracted nucleic acids and proteins, and several days are required to complete the protocol10.
Our lab has further developed the extraction and isolation of lipid A as first developed by Caroff and Raetz12,13. Compared to hot-phenol water procedures, the method presented here is more rapid and efficient and accommodates a wide range of culture volumes from 5 ml to multiple liters. Moreover, unlike hot-phenol water extractions, our method does not select for rough- or smooth-types of LPS, providing optimal recovery of lipid A species. In our protocol, chemical lysis of whole bacterial cells is performed using a mixture of chloroform, methanol and water, where LPS can be pelleted by centrifugation. A combination of mild-acid hydrolysis and solvent extractions (Bligh-Dyer) are used to liberate lipid A from covalently attached polysaccharide. The method of Bligh and Dyer was first applied to the extraction of lipid species from a variety of animal and plant tissues14, modified here to separate hydrolyzed polysaccharide from lipid A. In this final separation step, chloroform soluble lipids selectively partition into the lower organic phase. To further purify lipid A, reverse-phase or anionic exchange column chromatography can be used12.
After isolation of lipid A species from whole cells, a number of analytical methods can be used to characterize the chemical structure of the isolated material such as NMR, TLC, and MS-based analysis. NMR allows for non-destructive structural elucidation, and provides structural detail related to glycosidic linkages, unequivocal assignment of acyl chain positions, and assignment of attachment sites for lipid A modifications like aminoarabinose or phosphoethanolamine15-17. NMR analysis of lipid A is not discussed within our protocol, but has been described adequately elsewhere15,16. For rapid analysis TLC based methods are frequently used, but fail to provide direct information regarding fine chemical structure. MS based protocols are the most frequently employed method to characterize lipid A structures18,19. Matrix associated laser desorption ionization (MALDI)-MS is often used to initially survey intact lipid A species. Singly charged ions are generated from analyte prepared according to our extraction procedures. As more fine structural analysis is required, MS/MS based methods prove more informative than MALDI-MS. Coupled to electrospray ionization (ESI) singly or multiply charged lipid A precursor ions are further fragmented by collision induced dissociation (CID) or ultraviolet photodissociation (UVPD), to generate structurally informative product ions18,20,21. Neutral loss products from lipid A precursor ions are also frequently generated during ESI-MS providing an additional layer of structural information.
Tandem mass spectrometry (MS/MS) has proven to be an indispensable and versatile method for the elucidation of lipid A structures. During MS/MS, ions are activated to yield a diagnostic fragmentation pattern that can be used to elucidate the structure of the precursor ion. The most widely available MS/MS method is CID. This method produces fragment ions via collisions of the selected precursor ion with an inert target gas, resulting in energy deposition that leads to dissociation. CID has proven a critical tool in the assignment of lipid A structure for a wide range of bacterial species22-33.
Although CID is the most universally implemented MS/MS method, it generates a limited array of product ions. 193 nm UVPD is an alternative and complementary MS/MS method. This method uses a laser to irradiate ions, and the absorption of photons results in energization of the ions and subsequent dissociation. This higher energy MS/MS technique produces a more diverse array of product ions than CID and thus provides more informative fragmentation patterns. In particular, UVPD affords information about subtle changes in lipid A species based on cleavages at glycosidic, amine, acyl and C-C linked bonds18,21,34.
Protocol
All solutions should be prepared with ultrapure water and HPLC grade methanol and chloroform. Prepared solutions that contain organic solvents such as methanol, chloroform, or pyridine and concentrated acids or bases should be prepared and used under a chemical fume hood. All solutions can be stored at RT. Solvents should be measured in a graduated glass cylinder and stored in glass solvent bottles with PTFE lined caps. For long-term storage chloroform-containing solvents should be stored in tinted amber glass bottles to avoid the production of phosgene, a highly reactive acid chloride. PTFE centrifuge tubes and rotary evaporator flasks should be rinsed with methanol and chloroform before use. Follow necessary federal, state and/or institutional waste disposal regulations when disposing of solvents and/or radioactive waste.
1. Large Scale Lipid A Extraction (50 ml to 1.5 L)
Prepare a single-phase Bligh-Dyer mixture: chloroform-methanol-1X phosphate buffered saline (PBS), pH 7.4; 1:2:0.8 v/v). Combine 200 ml of chloroform, 400 ml of methanol and 160 ml of PBS in 1 L solvent bottle. Cap bottle and mix by inversion (>30x). Loosen the cap periodically during mixing to vent and store sealed.
Prepare a pre-equilibrated two-phase Bligh-Dyer mixture- chloroform:methanol:water (2:2:1.8 v/v). Combine 400 ml of chloroform, 400 ml of methanol and 180 ml of water in 1 L solvent bottle. Cap bottle and mix by inversion, making sure to loosen cap periodically during mixing to vent. Let equilibrate O/N and store sealed.
Prepare the mild acid hydrolysis buffer (50 mM sodium acetate, pH 4.5; 1% sodium dodecyl sulfate (SDS)). For 500 ml of mild acid hydrolysis buffer, weigh 2.05 g of sodium acetate and transfer to a 500 ml beaker. Add water to a volume of ~350 ml and stir, then add 50 ml of 10% SDS. Mix and adjust pH to 4.5, transfer to a graduated cylinder, and add water to 500 ml.
Prepare chloroform:methanol (4:1, v/v): Measure 100 ml of chloroform and transfer to solvent bottle. Measure 25 ml of methanol and mix with chloroform. Store in amber glass bottle.
Inoculate 5 ml of media (Luria broth or other) from a single bacterial colony. Grow O/N at 37 °C, or at required °C for growth. A diagram of the extraction procedure is shown in Figure 2.
The next day, measure the OD600 and use the 5 ml O/N culture to inoculate 200 ml of culture to a starting OD600 of 0.05. Grow cells until an OD600 of 0.8-1.0 is reached.
Harvest cells via centrifugation at 10,000 x g for 10 min. Longer spins may be required for strains that pellet poorly. Pour off media supernatant.
Wash cell pellet with 50 ml of 1x phosphate buffered saline (PBS). Repeat centrifugation to pellet cells. Pour off supernatant and store cell pellet at -20 °C, or proceed to step 1.9.
Resuspend cells in 40 ml of 1x PBS and divide between two 250 ml PTFE centrifuge tubes (yielding 20 ml of cell suspension/tube). Add 25 ml of chloroform and 50 ml of methanol to each tube, for a single phase Bligh-Dyer (chloroform: methanol:water; 1:2:0.8 v/v) (Figure 2). Mix by inversion and incubate at RT for >20 min to ensure complete cell lysis.
Centrifuge the mixture at 2,000 x g for 20 min. LPS will pellet along with proteins and nucleic acids (Figure 2); however, phospholipids, isoprenyl lipids, and small, hydrophobic peptides will remain in the supernatant. Discard the supernatant.
Wash the LPS pellet with ~100 ml single phase Bligh-Dyer mixture. Centrifuge at 2,000 x g for 20 min. Discard supernatant. When isolating lipid A from E. coli or Salmonella, only one wash is required; however, additional wash steps may be needed to reduce phospholipid contamination when isolating lipid A from some organisms.
Add 27 ml of mild acid hydrolysis buffer (50 mM sodium acetate, pH 4.5; 1% SDS; see step 1.3) to LPS pellet, and mix by pipetting up and down until only small particles remain. Sonicate to homogeneously resuspend LPS pellet in solution with probe tip sonicator at a constant duty cycle for 20 sec at 50% output. Repeat sonication of sample 2x (20 sec/burst, ~5 sec between bursts).
Boil samples in a water bath for 30 min. Caution: make sure the caps are tight, but not fully sealed. Some organisms, like Vibrio cholerae (V. cholerae) require longer incubation times (1 hr) to increase overall yield of lipid A. Remove bottles from water bath and allow sample to cool to RT before proceeding.
To extract lipids after hydrolysis, convert the SDS solution into a two-phase Bligh-Dyer (Figure 2) mixture by adding 30 ml of chloroform and 30 ml of methanol, for a chloroform:methanol:water (2:2:1.8, v/v) mixture. Mix by inversion and centrifuge the sample for 10 min at 2,000 x g. Extract the lower phase into a clean PTFE centrifuge tube using a glass pipette.
Perform a second extraction by adding 30 ml of lower phase from a pre-equilibrated two-phase Bligh-Dyer mixture (step 1.2), to the upper phase from step 1.14. Mix, then centrifuge at 2,000 x g for 10 min. Extract the lower phase, and pool with the lower phase extracted in step 1.14.
Wash the pooled lower phases (60 ml total) by adding 114 ml of pre-equilibrated two phase Bligh-Dyer upper phase (step 1.2) to create a two-phase Bligh-Dyer mixture (chloroform:methanol:water; 2:2:1.8, v/v). Mix. Centrifuge at 2,000 x g for 10 min.
Remove the lower phase to a clean glass rotary evaporator flask and dry sample using rotary evaporation (Figure 2).
Add 5 ml of chloroform:methanol (4:1, v/v) to the rotary flask, and bath-sonicate (>30 sec) to aid in suspension of lipid from sides of flask. Use a glass transfer pipette to transfer lipid to a clean glass tube (13 x 100 mm or larger) capped with PTFE lined phenolic screw caps. Dry sample under a stream of nitrogen using a nitrogen dryer.
Resuspend dried lipid in 1 ml of chloroform:methanol (4:1, v/v). Transfer to small glass sample vial (12 x 32 mm) with a tapered base for more quantitative resuspension using small volumes (see Table of Equipment/Reagents). Dry using a nitrogen dryer.
Dried sample can be stored at -20 °C until subsequent TLC (Protocol 2) or MS analysis (Protocol 3, 4, or 5). (Note: Amount of material isolated and used in subsequent protocols is suggested based on what's sufficient for most organisms. The same lipid sample can be used in Protocols 2-5, taking note of % material removed. See Discussion for more details.).
2. Visualization of Lipid A Species via Thin Layer Chromatography
Prepare the TLC mobile phase solvent system (chloroform:pyridine:88% formic acid:water; 50:50:16:5 v/v). Combine 200 ml of chloroform, 200 ml of pyridine, 64 ml of 88% formic acid, and 20 of ml water in a 1 L solvent bottle. Cap bottle, mix by inversion multiple times, and vent.
Use a TLC tank that will accommodate 20 x 20 cm plates. Line TLC tank with ~40 cm chromatography paper.
To TLC tank add 200 ml of chloroform:pyridine:88% formic acid:water (50:50:16:5, v/v) mixture. Allow tank to pre-equilibrate for >3 hr, often O/N is preferred and more convenient.
Remove dried lipid samples from freezer (step 1.20) and let warm to RT.
With a razor remove the silica from the top edge of a silica gel 60 TLC plate. Using a dull pencil, draw a line parallel to the bottom of the plate, 2 cm from the bottom. This line is the origin for spotting samples. Mark increments along this reference line to spot samples 1 cm apart.
Dissolve dried lipid from step 2.4 in 200 μl of chloroform:methanol (4:1, v/v), vortex and sonicate (3x, ~15 sec each), yields ~100-500 ng/μl lipid A if extracted from E. coli.
Using a microcapillary glass pipet, spot one-tenth of the volume (20 μl) onto the TLC plate as marked in step 2.5. Allow samples to air-dry on TLC plate for ~15 min. (Note: Samples in small glass vials can be dried using a nitrogen dryer and stored at -20 °C for subsequent use in protocols 3-5. Take note of % material removed).
Place TLC plate containing spotted samples into the pre-equilibrated tank. Once solvent front reaches the top of the TLC plate (~2.5-3 hr), remove the plate and air dry (>60 min). A cold-air gun can be used to ensure complete dryness.
While plate is drying, turn on hot plate at 250 °C for charring.
In a ventilated chemical fume hood, prepare 10% sulfuric acid-ethanol mixture for charring lipids resolved on the silica TLC plate (concentrated sulfuric acid:100% ethanol; 1:9 v/v). Measure 10 ml of sulfuric acid and add slowly to 90 ml of 100% ethanol. Carefully mix and transfer to a glass chromatographic reagent atomizer.
In the fume hood, use a glass chromatographic reagent atomizer to spray the dried TLC plate with 10% sulfuric acid-ethanol mixture. Spray mixture evenly across the plate.
Place the TLC plate on the 250 °C hot plate until charred lipid samples appear as black/brown spots (<1 min). Do not overexpose the plate, as this will cause the entire plate to turn brown and make visualization of lipid A species difficult.
3. Structural Characterization of Lipid A via MALDI-TOF Mass Spectrometry
From step 1.20, (or step 2.7) remove lipid A sample from freezer and let warm to RT. Resuspend dried lipid A sample in ~20 μl chloroform:methanol (4:1, v/v) and vortex to obtain a ~1-5 μg/μl lipid A solution if extracted from E. coli. (Note: 10% less concentrated if material used from step 2.7).
Prepare ATT matrix components. Saturated 6-aza-2-thiothymine in 50% acetonitrile: add 500 μl of water and 500 μl of acetonitrile to a 1.5 ml microcentrifuge tube, and then add >10 mg 6-aza-2-thiothymine so that the 50% acetonitrile is super-saturated. Saturated tribasic ammonium citrate solution: Add >1 mg to 500 μl water, precipitate should be readily apparent. Vortex and centrifuge solutions before use; only saturated supernatant is used.
Prepare ATT matrix mixture: saturated 6-aza-2-thiothymine in 50% acetonitrile, saturated tribasic ammonium citrate (20:1, v/v). Mix matrix components together by adding 20 μl of ATT solution to 1 μl saturated tribasic ammonium citrate solution in 500 μl microcentrifuge tube, vortex to mix, and centrifuge before applying to MALDI plate.
Prepare MALDI plate by adding 0.5 μl calibrant mixture to the MALDI plate on a spot near where the samples will be deposited, to provide the most accurate mass/charge ratio determination.
Deposit 0.5 μl of ATT matrix onto MALDI plate on each spot where lipid A sample will be deposited.
Deposit 0.5 μl of sample (2.5% of total sample) onto the spot of ATT matrix, to mix on plate, and acquire spectra by scanning sample for optimal ion signals (Figure 3). Note: Amounts for the most optimal MS signal can vary with organism and need to be determined empirically. To add more material one can spot 0.5 μl multiple times allowing spotted mixture to dry in between addition of more sample. Sample can also be concentrated (dry and resuspend in a smaller volume) or diluted as necessary. More starting material (volume of starting cell culture) can also be used (See Discussion).
4. Electrospray Ionization Mass Spectrometry and Collision Induced Dissociation of Lipid A
Prepare a chloroform:methanol solvent mixture (1:1, v/v) by mixing a 200 μl stock of HPLC grade methanol with 200 ml HPLC grade chloroform in a glass solvent bottle.
Transfer 200 μl of the chloroform:methanol solvent mixture to the vial with lipid A (e.g. from step 1.20, 2.7, or dried after Protocol 3) and sonicate the lipid a solution for 5 min or until all material is dissolved.
Set up the mass spectrometer for negative mode electrospray ionization.
Using a 250 μl syringe and a syringe pump, directly infuse the diluted lipid A sample at a flow rate of 2.0-3.5 μl/min.
Optimize and enhance the lipid A ion signal by tuning the ion optics. A full mass spectrum can now be collected of the lipid A species (Figure 4).
Isolate and activate the target lipid A by selecting CID as the MS/MS method.
Increase the CID voltage (or normalized collision energy, NCE) until the precursor lipid A species is about 10% relative abundance compared to the highest product ion.
Acquire and average spectra until sufficient signal-to-noise is achieved for the product ions. The number of scans needed is dependent on the signal intensity of the original precursor and can range from 3-300 scans (Figure 5A).
5. MS/MS on Lipid A by Ultraviolet Photodissociation
Prepare sample and mass spectrometer as in steps 4.1-4.5.
Turn on the laser interfaced to the mass spectrometer. The mass spectrometer was outfitted with a 193 nm excimer laser and modified to allow UV activation in the HCD cell of the instrument. Photodissociation was implemented in a manner similar to that described previously35. We use a modified vacuum manifold with a CaF2 optical window to transmit photons into the higher-energy C-trap dissociation (HCD) cell. The laser is triggered during MS/MS by a transistor-transistor logic (TTL) signal from mass spectrometer to a pulse/delay generator.
Set up the instrument software so that the laser will trigger the excimer laser when the ions enter the HCD cell. This requires a modest modification of the software.
Turn on the pulse generator such that the laser is pulsed every 2 msec (500 Hz).
Isolate the lipid A precursor ion by selecting HCD as MS/MS method and adjust the collisional energy to 1% NCE. This allows the isolation of precursor ions for ultraviolet dissociation within the HCD cell. Although the HCD cell is used, the software is modified to perform UVPD during this interval.
Activate the isolated lipid A ion by increasing the laser energy and adjusting the number of laser pulses. A typical UVPD experiment will be performed using ten 6 ml pulses.
As in step 4.8, acquire and average spectra until sufficient signal-to-noise is achieved for the UVPD product ions. The number of scans needed is dependent on the signal intensity of the original precursor and can range from 3-300 scans (Figure 5B).
6. 32P-Labeling of Lipid A and Subsequent Isolation
Inoculate 5 ml of media (Luria broth or other media) from a single colony. Grow O/N at 37 °C or at required temperature.
The next day, measure the OD600 and use the O/N culture to inoculate 7 ml of culture to a starting OD600 of ~0.05, grown in a standard 20 x 150 mm disposable glass culture tube. Add 2.5 μCi/ml of inorganic 32P. Grow cells until an OD600 of 0.8-1.0 is reached. Please follow federal, state, and/or institutional guidelines for the safe and proper handling of radioactive materials.
Harvest cells in 16 x 125 mm glass centrifuge tubes with PTFE lined cap using a fixed angle clinical centrifuge at 1,500 x g for 10 min. Remove supernatant into appropriate radioactive waste container. All radioactive waste (e.g. liquid, glass, biohazard) generated in subsequent steps should be disposed of in accordance with federal, state, and/or institutional guidelines.
Wash cell pellet with 5 ml of 1x phosphate buffered saline (PBS). Centrifuge for 10 min at 1,500 x g. Discard supernatant.
Resuspend cells in 5 ml of single phase Bligh-Dyer mixture (Figure 2) consisting of chloroform:methanol:water (1:2:0.8, v/v). Vortex to mix, and incubate at RT for >20 min to ensure complete cell lysis.
Centrifuge in clinical centrifuge at 1,500 x g for 20 min. Gently pour off supernatant, which contains phospholipids and isoprenyl lipids.
Resuspend the LPS pellet in 1.8 ml of 50 mM sodium acetate, pH 4.5; 1% SDS buffer by vortexing. Sonicate sample using a bath sonicator until pellet is evenly dispersed (~30 sec).
Incubate sample for 30 min in boiling water bath. Make sure caps are tight but not sealed.
Remove from water bath and allow sample to cool at RT for 5-10 min.
Convert the solution into a two-phase Bligh-Dyer mixture by adding 2 ml of chloroform and 2 ml of methanol, yielding a chloroform:methanol:aqueous (2:2:1.8 v/v) mixture. Vortex to mix, and centrifuge for 10 min in clinical centrifuge at 1,500 x g to separate phases.
Extract the lower phase into a clean glass centrifuge tube using a glass pipette (first extraction).
Perform a second extraction on the sample by adding 2 ml of pre-equilibrated two-phase Bligh-Dyer lower phase (prepared as in step 1.2) to the remaining upper phase from step 11. Vortex and centrifuge 10 min. Remove lower phase and combine with the lower phase from the first extraction.
To the pooled lower phase (~4 ml total volume), add 7.6 ml of pre-equilibrated upper phase (step 1.2), yielding a two-phase Bligh-Dyer solution (chloroform:methanol:water; 2:2:1.8, v/v). Vortex and centrifuge for 10 min.
Remove the lower phase to a clean glass tube and dry using a nitrogen dryer. Dried samples can be stored at -20 °C until further use.
7. Visualization of 32P-labeled Lipid A Species via Thin Layer Chromatography
Prepare 32P-labeled lipid A sample, TLC tank, and TLC plates as described in steps 2.1-2.8.
Dissolve 32P-labeled sample in 500 μl of 4:1 chloroform-methanol (v/v). Vortex and bath sonicate to completely dissolve lipid material. Add 5 μl of sample to scintillation vial containing 5 ml of scintillation cocktail. Count in scintillation counter and calculate total counts/min of sample.
When visualizing radiolabeled lipid species, 10 x 20 cm or 20 x 20 cm TLC plates can be used. For 10x20 cm plates, samples should be spotted along the origin marked in step 2.5 so that the plate's longest dimension is vertical (i.e. samples spotted at origin marked 2 cm above the 10 cm edge).
Using a microcapillary pipette, spot 10,000-20,000 cpm/sample on plate and allow spots to dry (>15 min). Samples might need to be concentrated by drying under nitrogen and resuspended in an appropriate volume, to achieve 10,000-20,000 counts/min/spot (2 μl or 2 μl at a time for <10 μl total).
Place TLC plate containing radiolabeled samples into the pre-equilibrated tank. Once solvent front reaches the top of the TLC plate (~3 hr), remove the plate and air dry (>60 min). Caution: TLC plates run in the presence of pyridine must be dried thoroughly in a chemical fume hood, as trace amounts of pyridine can damage phosphorimaging screens. A cold-air gun can be used to ensure complete dryness.
Wrap the plate in plastic wrap and expose to phosphorImager screen in an autoradiography cassette O/N. The next morning, scan the screen to obtain the image (Figure 6). Using image densitometry analysis software, this method allows for accurate, relative quantification of lipid A species.
Representative Results
Canonical lipid A of E. coli and Salmonella enterica serovar Typhimurium is a hexa-acylated disaccharide of glucosamine with phosphate groups at the 1- and 4 '-positions. During growth in rich media (e.g. Luria Broth) a portion of the lipid A contains a pyrophosphate group at the 1-position yielding a tris-phosphorylated species36 (Figure 1). Kdo (3-deoxy-D-manno-octulosonic acid), is attached at the 6'-hydroxyl and serves as a bridge to link lipid A to the remaining carbohydrate domains (i.e. core oligosaccharide and O-antigen domains) of LPS. Although gram-negative bacteria share a conserved pathway for lipid A biosynthesis similar to that of E. coli K-12, there is a large amount of diversity in lipid A structures. This diversity arises from the action of latent enzymes that modify the lipid A structure, which are activated in response to environmental stimuli. For example, in S. enterica the phosphate groups of lipid A can be modified with the cationic sugar L-4-aminoarabinose (Figure 1; blue) or with a phosphoethanolamine residue (Figure 1; magenta). In Salmonella these modifications are regulated by two-component response regulator systems, added in response to low [Mg2+], mild-acidic pH, and the presence of cationic antimicrobial peptides. Additionally in Salmonella, palmitate can be added to form a hepta-acylated lipid A, which promotes resistance to cationic antimicrobial peptides (Figure 1; green)8. Other modifications include, but are not limited to, removal of phosphate groups and acyl chains, or dioxygenase catalyzed addition of a hydroxyl group to the 3'-linked secondary acyl chain observed in a number of organisms1,3.
Isolation of intact lipid A from whole cells by our modification of the method of Caroff and Raetz is described in protocol 1 (Figure 2). This isolation method has been used to estimate that 106 molecules of lipid A exist per bacterial cell of E. coli37. Following protocols 1 and 3, the negative ion MALDI-TOF mass spectrum of lipid A from E. coli K-12 (W3110) yields the singly deprotonated ion ([M-H]-) at m/z 1,796.20 as the majorly observed species (Figure 3). MALDI-TOF MS was performed in negative-reflectron mode to improve spectral resolution. Alternatively, the same lipid sample subjected to negative mode nano-ESI (Protocol 4) yields predominately doubly deprotonated lipid A ions at m/z 898.1 denoted by [M-2H]2- (Figure 4). Singly deprotonated lipid A ions [M-H]- at m/z 1,796.20 are observable, but are of lower relative abundance to [M-2H]2- ions (Figure 4). Multiply charged species predominate when using ESI. Fragmentation by CID (Figure 5A) or 193 nm UVPD (Figure 5B) was performed on the [M-H]- lipid A ion m/z 1,796.20. These techniques can be used to better assign chemical structures, particularly of lipid A species containing complex combinations of modifications, or previously uncharacterized chemical modifications. Fragmentation profiles are shown with dashed lines representing cleavage sites and are matched with the m/z values below each provided structure (Figure 5). The m/z values and cleavage sites highlighted in red font represent unique product ions associated with UVPD.
As described in protocols 6 and 7, 32P-labeled lipid A isolated from two different E. coli K-12 strains was analyzed by TLC (Figure 6). W3110 contains mostly bis- and tris- phosphorylated lipid A (Figure 6; left lane), whereas a more complex TLC pattern is observed with 32P-lipid A isolated from E. coli strain WD101 (Figure 6; right lane)38. WD101 produces lipid A heavily modified with aminoarabinose (L-Ara4N) and phosphoethanolamine (pEtN). Since both 1- and 4'-phosphates are available for modification, lipid A from WD101 can be described as singly modified, containing only one L-Ara4N or pEtN at either phosphate, or doubly modified where both phosphates are modified in a combinatorial manner. In addition to modification at the 1- and 4'- phosphates, palmitate addition is also observed (see Figure 1) and increases the Rf value of lipid A species in this solvent system (Figure 6). If desired, densitometry analysis using a phosphorimager, can be used to quantifiably estimate relative amounts of lipid A species within the same sample .
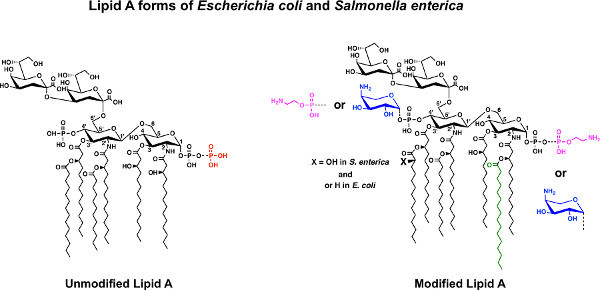 Figure 1. Representative lipid A domain structures from E. coli K-12 and S. enterica serovar Typhimurium. Modifications to the conserved lipid A structure are shown (right), as described in Representative Results. Click here to view larger figure.
Figure 1. Representative lipid A domain structures from E. coli K-12 and S. enterica serovar Typhimurium. Modifications to the conserved lipid A structure are shown (right), as described in Representative Results. Click here to view larger figure.
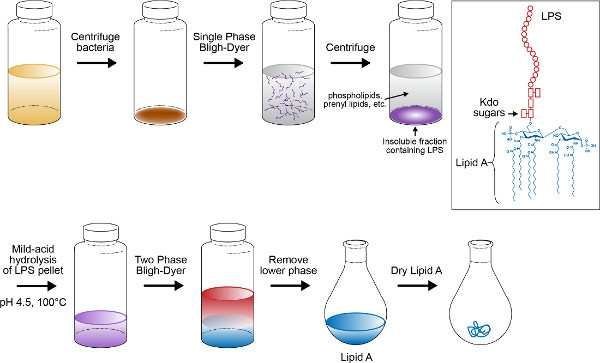 Figure 2. Schematic of lipid A isolation procedure. Outline depicts chemical lysis of bacterial cell pellet using single phase Bligh-Dyer mixture, centrifugation of lysate to pellet LPS, mild-acid hydrolysis to liberate lipid A from attached polysaccharide, and final purification of lipid A using two phase Bligh-Dyer extraction. Click here to view larger figure.
Figure 2. Schematic of lipid A isolation procedure. Outline depicts chemical lysis of bacterial cell pellet using single phase Bligh-Dyer mixture, centrifugation of lysate to pellet LPS, mild-acid hydrolysis to liberate lipid A from attached polysaccharide, and final purification of lipid A using two phase Bligh-Dyer extraction. Click here to view larger figure.
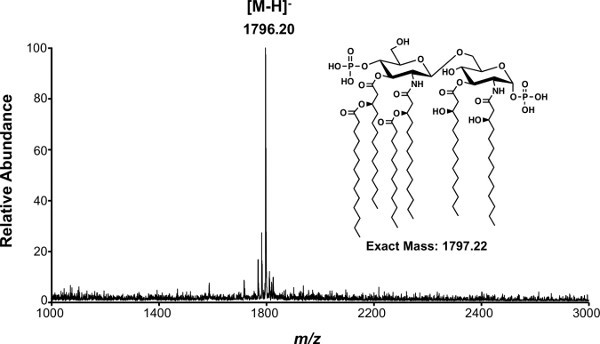 Figure 3. MALDI-MS analysis of lipid A isolated from E. coli K-12 (W3110). Spectra obtained from the average of >300 shots. Singly charged [M-H]- lipid A is observed as the molecular ion at m/z 1,796.2, which corresponds to a deprotonated species of the structure shown at right.
Figure 3. MALDI-MS analysis of lipid A isolated from E. coli K-12 (W3110). Spectra obtained from the average of >300 shots. Singly charged [M-H]- lipid A is observed as the molecular ion at m/z 1,796.2, which corresponds to a deprotonated species of the structure shown at right.
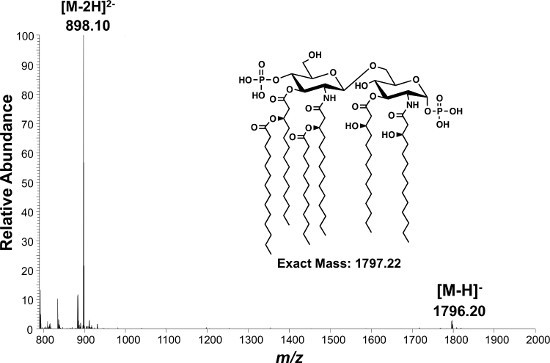 Figure 4. ESI-MS analysis of lipid A isolated from E. coli K-12 (W3110). Singly [M-H] and doubly-charged [M-2H]2- lipid A species are observed as molecular ions of m/z 1,796.2 and m/z 898.1, respectively.
Figure 4. ESI-MS analysis of lipid A isolated from E. coli K-12 (W3110). Singly [M-H] and doubly-charged [M-2H]2- lipid A species are observed as molecular ions of m/z 1,796.2 and m/z 898.1, respectively.
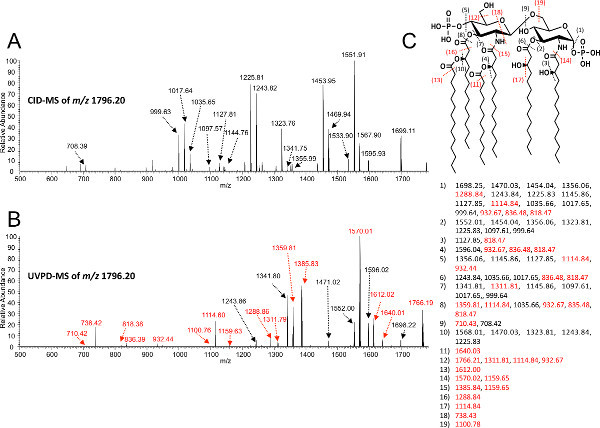 Figure 5. MS/MS analysis of lipid A isolated from E. coli K-12 (W3110). Collision induced dissociation (A) or ultraviolet photodissociation (B) were used to fragment the precursor ion m/z 1,796.2. UVPD specific product ions are indicated in red. A fragmentation map is provided (C), where black dashed lines indicate fragmentation associated with both CID and UVPD, and red dashed lines indicate UVPD specific fragmentation. Values listed beneath the fragmentation map correspond to exact masses of [M-H]- product ions. Click here to view larger figure.
Figure 5. MS/MS analysis of lipid A isolated from E. coli K-12 (W3110). Collision induced dissociation (A) or ultraviolet photodissociation (B) were used to fragment the precursor ion m/z 1,796.2. UVPD specific product ions are indicated in red. A fragmentation map is provided (C), where black dashed lines indicate fragmentation associated with both CID and UVPD, and red dashed lines indicate UVPD specific fragmentation. Values listed beneath the fragmentation map correspond to exact masses of [M-H]- product ions. Click here to view larger figure.
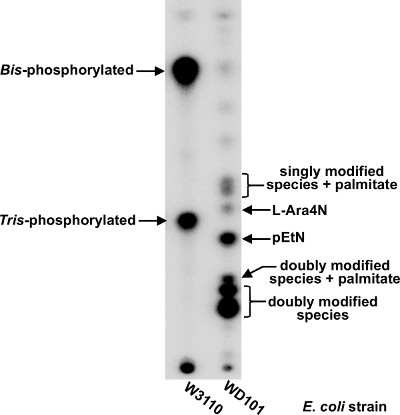 Figure 6. TLC-based separation of lipid A species isolated from E. coli K-12.32P-labeled lipid A was isolated from W3110 (left lane) or WD101 (right lane) and separated in a TLC tank solvent system containing chloroform:pyridine:88% formic acid:water (50:50:16:5 v/v).
Figure 6. TLC-based separation of lipid A species isolated from E. coli K-12.32P-labeled lipid A was isolated from W3110 (left lane) or WD101 (right lane) and separated in a TLC tank solvent system containing chloroform:pyridine:88% formic acid:water (50:50:16:5 v/v).
Discussion
In this protocol we have detailed the isolation of lipid A species from whole cells of bacteria, and described TLC or MS based analytical methods to chemically characterize this isolated material. Tandem mass spectrometry is a powerful strategy for de novo structural characterization of biological compounds, and is invaluable for the chemical characterization of the panoply of lipid A molecules observed in nature. CID and UVPD are two complementary activation methods that create different types of product ions that provide key fingerprints for lipid A molecules. MS/MS fragmentation using both CID and UVPD allows elucidation of subtle differences of lipid A structures, providing finer detail for chemical structure assignments. Data of this type are necessary to establish precise correlate structure/function relationships in the biology of lipid A molecular species. We have also described the procedure for 32P-radiolabeling lipid A species in small-scale bacterial cultures, where the chemical pattern of 32P-lipid A species can be visualized using TLC and autoradiography.
There are a number of features to this protocol that any user should be aware of. For large-scale lipid A preparations (protocol 1), the proportion of starting solvent to amount of bacterial pellet can be optimized to improve overall yield. However this proportion can only be determined empirically. The amounts described in this protocol represent a good starting point for most bacterial strains. Culture volumes for lipid A extraction and isolation can also be adjusted depending on the bacterial strain you are working with. For instance, V. cholerae requires at least 200 ml of culture in order to obtain high quality mass spectra; however, the culture volume for E. coli can be scaled down to 5 ml. More starting material (larger culture volumes) is required for some bacterial species because the hydrolysis step that releases lipid A from whole LPS is less efficient. For example organisms containing a functional Kdo dioxygenase or Kdo kinase exhibit decreased lipid A yields after mild-acid hydrolysis39. Often, mutants with altered LPS/lipid A structures or a particular bacterial species are often difficult to pellet during cell harvest. For these strains, the length of centrifugation can be extended to increase yield. Also of note, after cell lysis in a single-phase Bligh-Dyer mixture and LPS is pelleted (see step 1.9), additional wash steps may be required to reduce phospholipid contamination. When isolating lipid A from E. coli or Salmonella, only one wash is required. However, if isolating lipid A from other organisms (e.g. V. cholerae or Helicobacter pylori), additional wash steps are required.
After mild acid hydrolysis in SDS, the yield of lipid A species with reduced hydrophobicity (i.e. more phosphate groups >2 or fewer acyl chains <5) can be improved by using an acidic Bligh-Dyer extraction. For the volumes used in Protocol section 1, add 225 μl of concentrated HCl to the SDS solution containing hydrolyzed lipid A followed by 30 ml of chloroform and 30 ml of methanol for a two phase Bligh-Dyer mixture of chloroform:methanol: 0.1 M HCl (2:2:1.8, v/v). For the volumes used in Protocol section 6 add 15 μl of concentrated HCl to the SDS solution containing hydrolyzed lipid A followed by 2 ml of chloroform and 2 ml of methanol yielding a chloroform: methanol: 0.1 M HCl (2:2:1.8, v/v) mixture. After an acidic Bligh-Dyer extraction, pyridine can be added to the pooled lower phases to neutralize the acid (1 drop of pyridine/2 ml of final sample volume). This additional step should be performed before drying the sample, in a chemical fume hood. Be careful not to use excess pyridine, which can lead to the removal of ester-linked fatty acids. Additionally, if the lipid sample is difficult to dry under nitrogen, add a few milliliters of chloroform:methanol (4:1, v/v) to the flask and continue to dry the sample to completion.
The complex chemical heterogeneity of lipid A species observed in some organisms (e.g. V. cholerae, Yersinia pseudotuberculosis) can sometimes make TLC- or MS-based analysis difficult. Column chromatography can be employed upstream of these analytical techniques to pre-fractionate isolated lipid A species into more simple mixtures. Anion exchange with diethylaminoethyl (DEAE)-cellulose is most commonly used40. As a general guideline lipid A modified at either phosphate position, with non-acidic groups such as aminoarabinose or phosphoethanolamine, elutes before unmodified lipid A species40. Similarly tris-phosphorylated lipid A elutes well after unmodified bisphosphorylated species36,40. Reverse phase chromatography can be used to fractionate lipid A species of varying degrees of hydrophobicity41. Column chromatography is also useful in the removal of residual SDS after mild acid hydrolysis and subsequent Bligh-Dyer lipid A extraction steps. High levels of residual SDS can contribute to signal suppression in spectra obtained by our sensitive ESI-MS protocols.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by Grants AI064184 and AI76322 from the National Institutes of Health (NIH) and by Grant 61789-MA-MUR from the Army Research Office to M.S.T. Research was also supported by Welch Foundation Grant F1155 and NIH grant R01GM103655 to J.S. B.
References
- Trent MS, Stead CM, Tran AX, Hankins JV. Diversity of endotoxin and its impact on pathogenesis. Journal of endotoxin research. 2006;12:205–223. doi: 10.1179/096805106X118825. [DOI] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A Modification Systems in Gram-Negative Bacteria. Annu. Rev. Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Rietschel ET, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway C. Innate immunity. The New England journal of medicine. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- Guo L, et al. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- Yi EC, Hackett M. Rapid isolation method for lipopolysaccharide and lipid A from gram-negative bacteria. The Analyst. 2000;125:651–656. doi: 10.1039/b000368i. [DOI] [PubMed] [Google Scholar]
- Westphal OaJ, K Bacterial Lipopolysaccharide. Extraction with phenol-water and further application of the procedure. Methods Carbohydr. Chem. 1965;5:83–91. [Google Scholar]
- Galanos C, Luderitz O, Westphal O. A new method for the extraction of R lipopolysaccharides. European journal of biochemistry / FEBS. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- Raetz CR, Purcell S, Meyer MV, Qureshi N, Takayama K. Isolation and characterization of eight lipid A precursors from a 3-deoxy-D-manno-octylosonic acid-deficient mutant of Salmonella typhimurium. The Journal of biological chemistry. 1985;260:16080–16088. [PubMed] [Google Scholar]
- Caroff M, Deprun C, Karibian D, Szabo L. Analysis of unmodified endotoxin preparations by 252Cf plasma desorption mass spectrometry. Determination of molecular masses of the constituent native lipopolysaccharides. The Journal of biological chemistry. 1991;266:18543–18549. [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Ribeiro AA, Raetz CR. High-resolution NMR spectroscopy of lipid A molecules containing 4-amino-4-deoxy-L-arabinose and phosphoethanolamine substituents. Different attachment sites on lipid A molecules from NH4VO3-treated Escherichia coli versus kdsA mutants of Salmonella typhimurium. The Journal of biological chemistry. 2000;275:13542–13551. doi: 10.1074/jbc.275.18.13542. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Structure and biosynthesis of free lipid A molecules that replace lipopolysaccharide in Francisella tularensis subsp. novicida. Biochemistry. 2006;45:14427–14440. doi: 10.1021/bi061767s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain SM, et al. Location of polar substituents and fatty acyl chains on lipid A precursors from a 3-deoxy-D-manno-octulosonic acid-deficient mutant of Salmonella typhimurium. Studies by 1H, 13C, and 31P nuclear magnetic resonance. The Journal of biological chemistry. 1985;260:16089–16098. [PubMed] [Google Scholar]
- Madsen JA, Cullen TW, Trent MS, Brodbelt JS. IR and UV Photodissociation as Analytical Tools for Characterizing Lipid A Structures. Analytical Chemistry (Washington, DC, United States. 2011;83:5107–5113. doi: 10.1021/ac103271w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoub JH, El Aneed A, Cohen AM, Joly N. Structural investigation of bacterial lipopolysaccharides by mass spectrometry and tandem mass spectrometry. Mass Spectrom. Rev. 2010;29:606–650. doi: 10.1002/mas.20258. [DOI] [PubMed] [Google Scholar]
- Hankins JV, Trent MS. Secondary acylation of Vibrio cholerae lipopolysaccharide requires phosphorylation of Kdo. The Journal of biological chemistry. 2009;284:25804–25812. doi: 10.1074/jbc.M109.022772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins JV, et al. Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Molecular Microbiology. 2011;81:1313–1329. doi: 10.1111/j.1365-2958.2011.07765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Reinhold VN. Detailed structural characterization of lipid A: electrospray ionization coupled with tandem mass spectrometry. Anal. Biochem. 1994;218:63–73. doi: 10.1006/abio.1994.1141. [DOI] [PubMed] [Google Scholar]
- Boue SM, Cole RB. Confirmation of the structure of lipid A from Enterobacter agglomerans by electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2000;35:361–368. doi: 10.1002/(SICI)1096-9888(200003)35:3<361::AID-JMS943>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Kussak A, Weintraub A. Quadrupole ion-trap mass spectrometry to locate fatty acids on lipid A from Gram-negative bacteria. Anal. Biochem. 2002;307:131–137. doi: 10.1016/s0003-2697(02)00004-0. [DOI] [PubMed] [Google Scholar]
- El-Aneed A, Banoub J. Elucidation of the molecular structure of lipid A isolated from both a rough mutant and a wild strain of Aeromonas salmonicida lipopolysaccharides using electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005;19:1683–1695. doi: 10.1002/rcm.1971. [DOI] [PubMed] [Google Scholar]
- Murphy RC, Raetz CRH, Reynolds CM, Barkley RM. Mass spectrometry advances in lipidomica: collision-induced decomposition of Kdo2-lipid A. Prostaglandins Other Lipid Mediators. 2005;77:131–140. doi: 10.1016/j.prostaglandins.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-S, Kim Y-G, Joo H-S, Kim B-G. Structural analysis of lipid A from Escherichia coli O157:H7:K- using thin-layer chromatography and ion-trap mass spectrometry. J. Mass Spectrom. 2004;39:514–525. doi: 10.1002/jms.614. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li J, Altman E. Structural characterization of the lipid A region of Aeromonas salmonicida subsp. salmonicida lipopolysaccharide. Carbohydr. Res. 2006;341:2816–2825. doi: 10.1016/j.carres.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Mikhail I, Yildirim HH, Lindahl ECH, Schweda EKH. Structural characterization of lipid A from nontypeable and type f Haemophilus influenzae: variability of fatty acid substitution. Anal. Biochem. 2005;340:303–316. doi: 10.1016/j.ab.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Madalinski G, Fournier F, Wind F-L, Afonso C, Tabet J-C. Gram-negative bacterial lipid A analysis by negative electrospray ion trap mass spectrometry: Stepwise dissociations of deprotonated species under low energy CID conditions. Int. J. Mass Spectrom. 2006;249:77–92. [Google Scholar]
- Silipo A, et al. Structural characterizations of lipids A by MS/MS of doubly charged ions on a hybrid linear ion trap/orbitrap mass spectrometer. J. Mass Spectrom. 2008;43:478–484. doi: 10.1002/jms.1333. [DOI] [PubMed] [Google Scholar]
- Jones JW, Shaffer SA, Ernst RK, Goodlett DR, Turecek F. Determination of pyrophosphorylated forms of lipid A in gram-negative bacteria using a multivaried mass spectrometric approach. Proc. Natl. Acad. Sci. U.S. A. 2008;105:12742–12747. doi: 10.1073/pnas.0800445105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JW, Cohen ie, Turecek F, Goodlett DR, Ernst RK. Comprehensive structure characterization of lipid A extracted from Yersinia pestis for determination of its phosphorylation configuration. J. Am. Soc. Mass Spectrom. 2010;21:785–799. doi: 10.1016/j.jasms.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in gram-positive and gram-negative bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8722–8727. doi: 10.1073/pnas.1201313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SW, et al. Tyrosine sulfation in a Gram-negative bacterium. Nature. 2012;3:1153–1110. doi: 10.1038/ncomms2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touze T, Tran AX, Hankins JV, Mengin-Lecreulx D, Trent MS. Periplasmic phosphorylation of lipid A is linked to the synthesis of undecaprenyl phosphate. Mol. Microbiol. 2008;67:264–277. doi: 10.1111/j.1365-2958.2007.06044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway SM, Raetz CR. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. The Journal of biological chemistry. 1990. pp. 265–6394. [PubMed]
- Trent MS, et al. Accumulation of a polyisoprene-linked amino sugar in polymyxin-resistant Salmonella typhimurium and Escherichia coli: structural characterization and transfer to lipid A in the periplasm. The Journal of biological chemistry. 2001;276:43132–43144. doi: 10.1074/jbc.M106962200. [DOI] [PubMed] [Google Scholar]
- Chung HS, Raetz CR. Dioxygenases in Burkholderia ambifaria and Yersinia pestis that hydroxylate the outer Kdo unit of lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 2011;108:510–515. doi: 10.1073/pnas.1016462108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Lin S, Cotter RJ, Raetz CR. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-L-arabinose, phosphoethanolamine and palmitate. The Journal of biological chemistry. 1999;274:18503–18514. doi: 10.1074/jbc.274.26.18503. [DOI] [PubMed] [Google Scholar]
- Raetz CR, et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. Journal of lipid research. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]