

Abstract
Inflammatory bowel disease (IBD) results from a complex series of interactions between susceptibility genes, the environment, and the immune system. The host microbiome, as well as viruses and fungi, play important roles in the development of IBD either by causing inflammation directly or indirectly through an altered immune system. New technologies have allowed researchers to be able to quantify the various components of the microbiome, which will allow for future developments in the etiology of IBD. Various components of the mucosal immune system are implicated in the pathogenesis of IBD and include intestinal epithelial cells, innate lymphoid cells, cells of the innate (macrophages/monocytes, neutrophils, and dendritic cells) and adaptive (T-cells and B-cells) immune system, and their secreted mediators (cytokines and chemokines). Either a mucosal susceptibility or defect in sampling of gut luminal antigen, possibly through the process of autophagy, leads to activation of innate immune response that may be mediated by enhanced toll-like receptor activity. The antigen presenting cells then mediate the differentiation of naïve T-cells into effector T helper (Th) cells, including Th1, Th2, and Th17, which alter gut homeostasis and lead to IBD. In this review, the effects of these components in the immunopathogenesis of IBD will be discussed.
Keywords: Inflammatory bowel disease, Crohn’s disease, Ulcerative colitis, Microbiome, Autophagy, T helper 17, Innate immune system, Adaptive immune system, Innate lymphoid cells, TL1A
Core tip: Inflammatory bowel disease (IBD) results from the complex interactions between susceptibility genes, the environment, the immune system, and the host’s microbiome. It is thought that either a mucosal susceptibility or a defect in sampling of gut luminal antigen leads to activation of the innate immune system that then recruits cells of the adaptive immune system leading to inflammation. This review will detail the interaction of these components in the immunopathogenesis of IBD.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder that is comprised of both Crohn’s disease (CD) and ulcerative colitis (UC), and is characterized by alternating phases of clinical relapse and remission. CD can affect any part of the gastrointestinal tract and classically presents with fatigue, prolonged diarrhea with or without gross bleeding, abdominal pain, weight loss, and fever. UC characteristically involves the colon and presents with symptoms that usually rectal bleeding, frequent stools, mucus discharge from the rectum, tenesmus, and lower abdominal pain. IBD is thought to be the result of a dysregulated immune system in the context of a genetically susceptible individual. Currently, IBD affects 1.4 million Americans and at a prevalence rate of 396 per hundred thousand individuals worldwide[1]. The incidence of CD in the United States is estimated to be 5 per hundred thousand persons and is characterized by focal and transmural inflammation that can occur anywhere along the length of the gastrointestinal system, that may include B2 stricturing (gut luminal narrowing), B3 penetrating (bowel perforation, fistula, inflammatory mass/abscess), and with possible perianal disease[2,3]. UC affects 8-12 per hundred thousand individuals and is characterized by colonic mucosal inflammation along the entire colon and involving the rectum[3,4]. Also, patients with IBD have an increased risk of developing other chronic inflammatory disorders, such as psoriasis and primary sclerosing cholangitis[5,6].
The exact cause of IBD is still unknown, but is thought to be due to a combination of a patient’s genetics, microbiome, immune response, and the environment that result in an excessive and abnormal immune response against commensal flora in genetically susceptible individuals. Epidemiological data suggest an association between IBD and a number of environmental factors, such as antibiotic use, microbial exposure both early and late in life, and possibly diet[5,7-9]. The genetics of IBD are complex and thought to be polygenic. Genome-wide association studies (GWAS) suggest that dysregulation in innate and adaptive immunity contribute to the development of IBD. Susceptibility variants have been reported in genes associated with autophagy (ATG16L1), the interleukin (IL)-23/Th17 pathway (IL-12B), TGF-beta pathway (SMAD3), T-cell activation (TAGAP), among other immune system genes[10-13].
The identification of these and other loci is only part of a larger picture that aims to understand how polymorphisms in these genes can lead to an increased risk of developing IBD. Here we review the available evidence supporting the role of the microbiome and the innate and adaptive immune responses and their crosstalk in IBD.
THE MICROBIOME
Overview
Th interaction of the host with its abundant microbiota is complex. The luminal surface of the small and large intestine, approximately 300-400 m2, is a unique environment where an enormous population of bacteria exists in close proximity to the immune system of the gut mucosa. This roughly translates to the interactions of 1012 microorganisms per gram of feces with 106 immune cells per gram of enteric tissue[14]. A complex network of interactions exists between gut epithelial cells, immune cells, and foreign bodies that transition along the gut. Functionally, the gut-associated lymphoid tissues generates either an immune response for rejection of pathogens or a clinical immune response of tolerance for dietary and microbial antigens[15]. Data supports the hypothesis that IBD results from a dysregulated immune response to the microbiota. It was found that in CD patients, diversion of feces induces inflammatory remission and mucosal healing in the downstream intestinal segment and infusion of feces reactivates the disease[16]. Furthermore, in UC patients with active disease, treatment with broad-spectrum antibiotics reduced mucosal inflammation[17]. These data support the concept that luminal bacteria provide the stimulus for an inflammatory response leading to mucosal injury. Two main hypotheses have been suggested that might contribute to the loss of tolerance towards the indigenous microbiota in patients with IBD. First, genetic susceptibility leads to a dysregulation of the mucosal immune system that result in excessive immunologic responses to normal flora. Second, an imbalance exists in the composition of the microbiota that elicits a pathologic response from the normal mucosal immune system[18]. In all likelihood, it probably is a combination of both hypotheses.
Advancements in genetic technology, such as 16S ribosomal RNA (rRNA) gene and metagenomic sequencing have allowed researchers to determine the composition of the microbiome[19]. Recently, a number of studies have profiled the “normal” human gut microbiota. Briefly, it is thought that greater than 90% of all phylotypes belong to two divisions, Bacteroidetes and Firmicutes[20]. Other divisions that have consistently been recovered from “normal” individuals include Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia. It is believed that the composition of fecal microbiota remains relatively constant over time, termed resilience, with temporary changes occurring after exposure to food, medicine, and physical environment[21]. In 2010, whole-genome shotgun sequencing revealed 3.3 million nonredundant microbial genes in fecal samples from an adult European cohort[22]. It was found that up to 98% of the genes were bacterial with the rest belonging to archaea, yeasts, viruses, and protists. The three most abundant genera in the fecal samples were Bacteroides, Faecalibacterium, and Bifidobacterium, however the percent composition was found to be highly variable between individuals[23]. Three main enterotypes (independent of gender, age, race, body mass index, or country and continent of residence) were created that are classified based upon the prominent genera represented: enterotype 1-Bacteriodes; enterotype 2-Prevotella; enterotype 3-Ruminococcus[23].
Individuals with IBD have been shown to have changes in the bacterial composition of feces with less bacterial diversity, having fewer numbers of non-redundant bacterial genes, as compared to healthy controls[24,25]. Adding to the complexity, the bacterial profile also differs between individuals with UC and CD. In a twin study, twins with UC were found to have less Bacteroides and more Actinobacteria and Proteobacteria than their healthy twin counterparts. The decrease in Bacteroides was made up for by an increase in the Prevotellaceae family[26]. Also, it has been found that Escherichia coli (E. coli) is to be increased in fecal samples from individuals with UC, with some isolates expressing virulence factors and invading properties[27,28]. Dysbiosis also exists in CD. Many studies have shown a decrease in the abundance of several bacterial species of the phylum Firmicutes in patients with CD[29,30]. Also, the microbiome of individuals with CD predominantly in the ileum was found to differ from those whose disease was found predominantly in the colon. Those with ileal CD had decreased Faecalibacterium and Roseburia and increased amounts of Enterobacteriaceae and Ruminococcus gnavus[31]. Consistent with what is seen in UC patients, E. coli also has been observed in patients with CD[27].
Recently a new pathogenic group called adherent-invasive E. coli (AIEC) has been isolated from the ileum of CD patients[30,32]. Darfeuille-Michaud[33] have accumulated a large body of data showing that AIEC is able to invade epithelial cells and to survive and replicate within macrophages and is associated with ileal mucosal CD pathogenesis. They found that AIEC was present in the inflamed ileum of 22% of chronic CD patients, as compared to only 6% of control patients, as well as 36% of the newly formed terminal ilea of postsurgical CD patients[34]. However, AIEC was found in only a small percentage of affected colons of CD patients and in zero percent of UC patients, suggesting that AIEC strains are associated specifically with ileal mucosa in CD[34]. AIEC facilitate binding and invasion into epithelial cells via type 1 pili and flagella[35]. This interaction is dependent upon epithelial expresseion of CEACAM6, which is a carcinoembryonic antigen upregulated by inflammatory cytokines and possibly by AIEC itself[36]. Furthermore, transgenic mice overexpressing CEACAM6 in epithelial cells are colonized by AIEC and manifest gut inflammation with marked neutrophil infiltration and ulceration[37]. In the lamina propria, AIEC is taken up by macrophages and can survive and proliferate within macrophage vacuoles, which suggest that the bacteria is not readily cleared from the site and may represent a defect in autophagy[38,39].
Also, recently an important role of fungi in gut homeostasis has been found. Besides the massive amount of bacteria known to make up the intestinal microflora, the mammalian gut also contains a rich fungal community that interacts with the immune system via Dectin-1, which is a pattern-recognition receptor expressed by innate immune cells, such as: macrophages, dendritic cells (DCs), and neutrophils[40]. It was found that mice deficient in Dectin-1 exhibited increased susceptibility to dextran sulfate sodium (DSS) colitis that was the result of an altered response to the host’s fungi[41]. Furthermore, polymorphisms in the gene for Dectin-1 have been shown to be strongly linked to a severe form of UC[41]. This data suggests that the interactions between fungi and the innate immune system are important in the development of IBD. Viral infections also impact the gut microflora. In mice, virus-plus-susceptibility gene interactions have been shown to induce colitis that mimics CD. When mice with a specific mutation in a CD susceptibility gene for autophagy (ATG16L1) were infected with murine norovirus they displayed abnormal Paneth cell structure and granule packaging similar to those seen in CD patients homozygous for the risk allele of ATG16L1[42]. These changes were not seen in control mice nor in CD patients homozygous for the nonrisk allele of ATG16L1. Furthermore, when these mice were treated with DSS they displayed worse colitis than control animals[42]. These results demonstrate how a genetic factor and an environmental agent can contribute to the pathogenesis of CD. Also, HIV infection of humans and simian immunodeficiency virus (SIV) infection of rhesus monkeys is known to cause systemic immune activation and associated with damage to the intestinal epithelium and translocation of antigens into the blood[43-45]. Pathogenic SIV infection has been associated with significant expansion of the enteric virome, including adenovirus and parvovirus, that can lead to enteritis without changes in the microbiome[46]. This data suggests that the enteric virome might contribute to AIDS pathogenesis by damaging the intestinal epithelium to allow translocation of microbes and viral antigens into the circulation.
The consequences of these shifts in microbiota are unclear, particularly whether it is cause or effect. Regardless of the inciting cause of IBD, it is apparent that the host-microbiome interaction plays a large part in disease pathogenesis.
Host-microbial interactions
Host-microbe interactions are crucial in the development and modulation of the immune system and protection from pathogenic bacterial invasion. The first line of defense to pathogenic organisms is the innate immune system, which in the gut consists of mucin, the epithelium, and cells of the innate immune system [i.e., neutrophils, DCs, monocytes/macrophages, and innate lymphoid cells (ILCs)]. Interestingly, mice lacking an adaptive immune system, but that have an intact innate immune system, such as recombinase activation gene deficient (RAG-/-) and severe combined immunodeficient (SCID) mice, do not develop spontaneous colitis and co-exist with the microbiota. However, these mice can develop colitis when induced by DSS, anti-CD40 antibody, and Heliobacter hepaticus infection[47-49]. These data suggest that in the absence of an adaptive immune system, the innate immune system is sufficient for the development of IBD. However, the adaptive immune system is still thought to play an important role in the development of IBD, as RAG deficiency can prevent the development of spontaneous colitis that are seen with certain mutant mouse strains[50].
INNATE BARRIERS OF PROTECTION
Mucus
The surface of the intestine is protected by a layer of mucus that is generated by goblet cells in the epithelium (Figure 1). The inner mucus layer is approximately 100 μm thick, firmly adherent, rich in antimicrobials and mucin, and has a low bacterial density. The outer layer of mucus is comprised of mucin, diluted antimicrobials, and some bacteria. A variant in the Muc2 gene, which is the major intestinal secretory mucin, confers susceptibility in humans to IBD and Muc2 deficient mice develop spontaneous colitis[51]. Furthermore, some patients with CD have been found to have goblet cell depletion and an impaired mucus layer, which allows bacteria to adhere directly to epithelial cells, and may contribute to disease progression[52]. It is believed that the ability of commensal bacteria to adhere to the epithelial layer via oligosaccharides helps deter invasion by displacing pathogenic bacteria. FUT2 is a gene that encodes a type alpha (1, 2) fucosyltransferase, which regulates the secretion of the H1 antigen of the ABO antigens into the mucosa. People are either associated as H1 antigen secretors or non-secretors. Twenty percent of the population are non-secretors, which has been associated with a variety of illnesses including recurrent norovirus and encapsulated bacterial infections, duodenal ulcerations, and susceptibility to CD[53-59]. The inability to secrete H1 into the mucosa is thought to affect how commensal and pathogenic flora interact with the epithelial layer and may interfere with the ability of the commensal flora to adhere, which could result in increased susceptibility to infection, invasion, and activation of the immune system.
Figure 1.
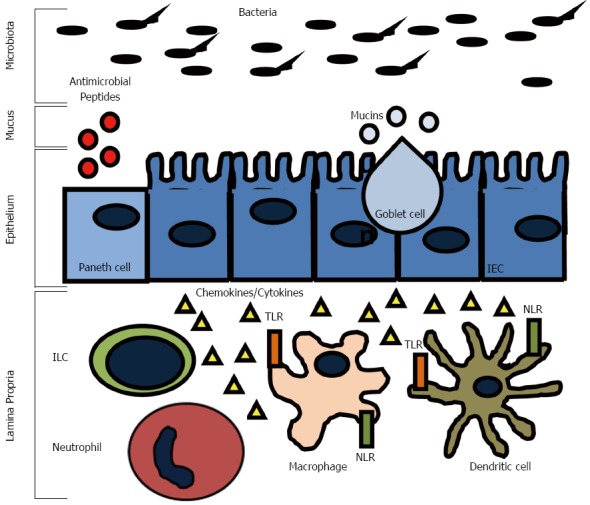
Innate immune system responses in the gut. The intestinal epithelial barrier is equipped with several layers of defense mechanisms to limit luminal antigen translocation. Goblet cells, Paneth cells, and enterocytes secrete mucins and antimicrobial peptides that assemble into a mucus layer. Innate immune system cells, such as macrophages and dendritic cells, can sense invading bacteria through extracellular and intracellular pattern recognition receptors (Toll-like receptors-TRLs and NOD-like receptors-NLRs) and initiate rapid inflammatory reponses mediated by the secretion of cytokines and chemokines. Innate lymphoid cells (ILCs) are also found in the lamina propria where they contribute to cytokine production and inflammatory cell recruitment.
The epithelium
The epithelium of the intestine has many functions, including absorption, secretion, and digestion. There are four main types of epithelial cells: one, absorptive enterocytes; two, mucus producing goblet cells; three, hormone producing enteroendocrine cells; and four, antimicrobial and growth factor producing Paneth cells (Figure 1). The epithelium forms a mucosal barrier with tight junctions between enterocytes that can exclude the entry of most substances. The epithelial layer is renewed every 2-3 d with a balance of proliferation of epithelial cells in the crypts and migration down the villi in the small intestine or onto the surface of the colon and apoptosis and shedding of the enterocytes. Disruption of this process impairs the epithelial integrity and can result in chronic inflammation. Defects in epithelial integrity may contribute to IBD pathogenesis by allowing free passage of organisms across the epithelial layer where they can incite an immune response. For example, mutations in the organic cation transporter (OCTN) gene, which is involved in the transport of cationic proteins, such as amino acids and nutrients, lead to an increased susceptibility to CD[60]. The susceptibility is thought to result from impaired fatty acid oxidation, which can cause colitis in experimental models in the setting of bacterial antigen exposure[61].
The epithelium lies between the immune cells in the lamina propria and the microbiota in the gut lumen and functions to communicate with both. For example, the microbiota signals enterocytes as well as innate cells in the lamina propria via pattern recognition molecules signal receptors, such as toll like receptors (TLRs) and cytosolic NOD-like receptors (NLRs) (Figure 1). These signals have been shown to be necessary for normal homeostasis and resistance to injury[62]. Cytokines, such as interferon (IFN)-γ, interleukin (IL)-17, and IL-22, pathogens, and commensal bacteria have substantial effects on the epithelium by regulating barrier integrity and function[63-66]. Expression of pattern recognition receptors is highly regulated to prevent an inappropriate immune response, but still allow for constant surveillance. Mutations in genes coding for these receptors have been found to be IBD susceptibility genes. Haplotypes of the TLR8 gene can confer protection (H1) or risk (H4) to the development of IBD[67]. Agonists of TLR8 have been shown to cause downstream activation of proinflammatory cytokines such as IFN-γ, IL-12, and tumor necrosis factor (TNF)-α in peripheral blood mononuclear cells[68]. The TLR9 gene is also a CD and UC susceptibility gene[69]. It has been shown that mice infected with Campylobacter jejuni (C. jejuni) or TLR9 agonists have increased susceptibility to mild DSS colitis via a mechanism involving secretion of CXCL8[70].
Defects in the intestinal barrier can lead to persistent immune activation and has been suggested to play a role in IBD[71]. Normally, translocation allows small amounts of luminal antigens to pass transcellularly across the epithelium either through receptor-mediated endocytosis or non-selective endocytosis. A small amount of bacteria is normally allowed to translocate and allows for physiologic sampling of luminal content by the host’s immune system[72]. Animal models that lack components of a healthy epithelial barrier have been shown to develop IBD. Expression of dominant-negative N-cadherin in mouse intestinal epithelium has been shown to lead to CD like symptoms[73]. NOD2 is a protein that acts as an intracellular pattern recognition receptor for muramyl dipeptide (MDP), a component of the bacterial wall peptidoglycans. Mice lacking intracellular pattern recognition receptors, NOD1 and NOD2, were shown to have decreased E-cadherin expression with increased epithelial permeability and decreased antimicrobial production[74]. NOD2 was one of the first CD susceptibility genes, with homozygous mutations found in 15% of patients with CD[75]. Mutations in NOD2, as well as other pattern recognition receptors, might impair the ability of the mucosal immune system to sense organisms thereby leading to defective microbial clearance and persistent antigenic stimulation. This in turn may result in mucosal inflammation and loss of regulatory control over proinflammatory pathways, which could possibly lead to the development of IBD.
AUTOPHAGY
The term autophagy, or “self-eating,” results in the lysosomal degradation of organelles, unfolded proteins, or foreign extracellular material (Figure 2). It is a key process required for maintaining cellular homeostasis after infection, mitochondrial damage, or ER stress. Defects in autophagy have been shown to result in pathological inflammation and GWAS have linked two key genes in autophagy, ATG16L1 and IRGM, to CD[13,76]. An ATG16L1 hypomorphic mouse line that expresses about 1% of the normal level of ATG16L1 was shown to have Paneth cell granule abnormalities that are similar to those found in ileal resections in patients with CD that also carry the ATG16L1 gene variant[77]. While these hypomorphic ATG16L1 mice do not develop spontaneous colitis, they were found to have an increased susceptibility to DSS colitis[42]. However, when rederived virus free, these mice lost the Paneth cell pathology and ability to develop DSS induced colitis, which could be reversed by norovirus infection[42]. A recent study has reported that the ATG16L1 and NOD2 pathways may be interrelated[78]. In 2010, Cooney et al[78] demonstrated that NOD2 stimulation is capable of initiating autophagy in DCs and that for effective autophagy to occur, both intact NOD2 and ATG16L1 functions are required. IRGM belongs to a family of interferon-inducible immunity related GTPases (IRGs) that encodes a protein involved in multiple autophagocytic pathways including intracellular clearance of pathogens[79]. IRGM has been shown to play a role in autophagy during both Salmonella typhimurium and Mycobacterium bovis infections[79,80]. Another study in CD patients has demonstrated that autophagy is also important in the clearance of AIEC and that IRGM and ATG16L1 deficient cells had increased AIEC replication, suggesting that these genes play a significant role in clearance of this organism and intestinal inflammation[39]. These studies implicate that autophagy plays an important role in human inflammatory disorders by direct elimination of intracellular bacteria and activation of pattern recognition receptor signaling which is involved in gut homeostasis and CD pathogenesis.
Figure 2.
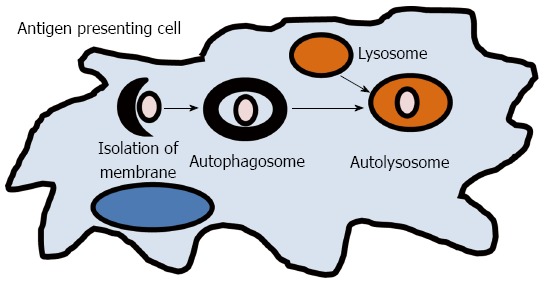
Autophagy. A small volume of cytoplasm is enclosed by the autophagic isolation membrane, which forms the autophagosome. The outer membrane of the autophagosome then fuses with the lysosome where the cytoplasm derived materials are degraded.
INNATE AND ADAPTIVE IMMUNITY
Overview
The immune system has evolved as protection against a wide range of infectious agents. In vertebrates, the immune system is broadly divided into two effector classes, the innate and adaptive immune responses. The innate immune system is the first line of defense and provides an immediate protective response against infections and also helps to initiate the adaptive immune response (Figure 1). The innate immune system is non-specific and does not confer lasting immunity (memory). The innate immune system is comprised of the epithelial barrier, macrophages, monocytes, neutrophils, DCs, and natural killer cells (NK cells), eosinophils, and basophils. These cells act together to initiate inflammation by secreting cytokines, chemokines, and antimicrobial agents. This leads to phagocytosis of infected cells and microorganisms, antigen presentation, and activation of the adaptive immune system.
The adaptive immune response is comprised of lymphocytes (T and B cells) that when activated generate effector responses (cytokines and antibodies). In contrast to the innate immune system, the adaptive immune system is highly specific and confers long lasting immunity (memory). It is generally thought that the adaptive immune system is the main contributor to disease pathogenesis in IBD, either through increased proinflammatory cytokines driven by the T-helper (Th) subsets or by ineffective anti-inflammatory regulatory T-cells (Tregs). Naïve T-cells (Th0) cells after activation are able to differentiate into Th1, Th2, or Th17 cells (Figure 3). In particular, Th1 responses have been thought to drive the pathogenesis of CD, while UC is thought to be driven by Th2 responses. Recent advancements suggest that other cells, such as ILCs and Th17 cells, have emerged as important contributors to IBD pathogenesis.
Figure 3.
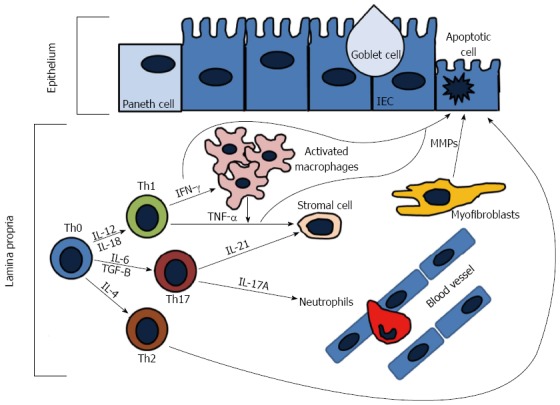
Adaptive immune responses in the gut. During active inflammation, naïve T-cells (Th0) differentiate into T helper cell types (Th1, Th2, Th17) under stimulation of different cytokines. Th1 cells produce interferon (IFN)-γ and tumor necrosis factor (TNF)-α. IFN-γ activated tissue macrophages to produce additional TNF-α, which causes epithelial cell apoptosis and differentiation of stromal cells into myofibroblasts. Activated myofibroblasts produce metalloproteinases (MMPs) that cause tissue degredation. Th2 cells produce interleukin (IL)-13 that can increase intestinal permeability and induce epithelial apoptosis. Th17 cells release IL-17A, which plays a role in recruiting neutrophils to sites of active inflammation, and IL-21 that also induces MMP production that contributes to extracellular matrix degredation.
Role of innate lymphoid cells in IBD
Until recently NK cells were thought to be the only innate cell derived from a lymphoid progenitor. However, recent developments have classified NK cells as a subset of a new family of hematopoietic effector cells called ILCs. ILCs are an emerging and diverse group of immune cells and are part of the new frontier of immunology research. All ILCs derive from an Id2 expressing progenitor and are defined by three main features: One, they are of lymphoid morphology; two, they are cell lineage negative (CD3-, B220-, GR1-, CD11b-, Ter119-); and three, they lack RAG-dependent antigen receptors (Figure 4)[81]. Recently, a unifying ILC classification system has based upon phenotypic and functional characteristics has been proposed (Table 1)[82]. ILCs can be classified into three groups: Group 1 ILCs, which are T-box expressed in T-cells (Tbet) dependent and are comprised of ILC1 and NK cells; group 2 ILCs, which are GATA-binding protein 3 (GATA3) and retinoic acid receptor-related orphan receptor (ROR) dependent, and comprised of ILC2s; and group 3 ILCs, which are RORt dependent and are comprised of ILC3s and lymphoid tissue-inducer (LTi) cells[82].
Figure 4.
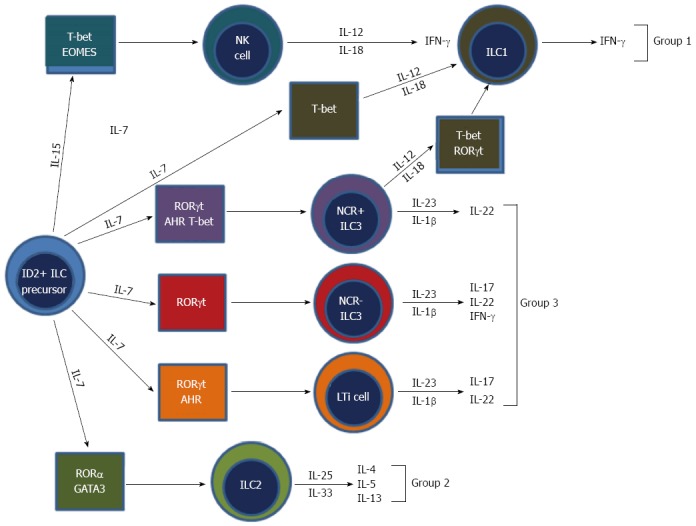
Development and classification of innate lymphoid cells[82]. Innate lymphoid cells (ILCs) all derive from an ID2 positive progenitor cell. Group 1 ILCs make interferon (IFN)-γ. Group 2 ILCs produce IL-5 and interleukin (IL)-13. Group 3 ILCs produce IL-17, IL-22, and IFN-γ. NK require IL-15, whereas all other ILCs require IL-7 for development. Group 2 ILCs depend on transcription factors GATA3 and ROR for development. Group 3 ILCs require RORt for development. Also, subsets of group 3 ILCs require additional transcription factors, such as aryl hydrocarbon receptor (AHR) for development. NK cells, which are group 1 ILCs, require both T-bet and eomesodermin (EOMES). The mechanisms of ILC1 development are not fully elucidated, however are known to require transcription factor T-bet for development.
Table 1.
Innate lymphoid cell subsets
| ILCs | Lineage | Mouse | Human | Cytokines | Function | Disease |
| Group 1 | ILC1s | Lin-Thy1+Sca1+Tbet+ | Lin-CD56+NKp46+NKp30+NKp44+IL-7R- | IFN-γ | Inflammation | IBD? |
| NK cells | NKp46+NK1.1+CD122+NKG2D+CD161+CD16+ | CD122+NKG2D+CD161+KIR+ | IFN-, TNF-α, cytotoxic effectors | Immunity to viruses and intracellular pathogens, tumor surveillance | Inflammatory conditions, IBD | |
| Group 2 | ILC2s | Lin-ICOS+Thy1+Sca1+IL-7R+GATA3+ | Lin-IL-7R+CD45hiCD161+ CRTH2+ | IL-5, IL-9, IL-13 | Immunity to helminthes, wound healing | Allergy, asthma |
| Group 3 | ILC3s | Lin-Thy1+Sca1+RORt+NKp46+IL-7R+CCR6- | Lin-CD56-NKp46+NKp30+NKp44+IL-7R+ | IL-22 | Lymphoid tissue development, intestinal homeostatsis, immunity to extracellular bacteria | IBD |
| LTi cells | Lin-Thy1+Sca1+RORt+NKp46-IL-7R+CCR6+ | Lin-IL-7R+CD45+RORt+ | IL-17A, IL-17F, IL-22 | Homeostasis of epithelia, immunity to extracellular bacteria | IBD |
Lin-: Lineage marker negative (mouse negative for CD3, CD19, B220, CD11b, CD11c, GR1, Ter11; human negative for CD1a, CD3, CD11c, CD34, CD123, TCR, TCR, CD19, CD14, CD16); IBD: Inflammatory bowel disease; ILC: Innate lymphoid cell; IFN: interferon; TNF: Tumor necrosis factor; NK: Natural killer.
The key cytokines secreted by ILCs tend to mirror those secreted by the T-helper cells of the adaptive immune system and therefore ILCs have been thought of as the innate counterparts of T-helper lymphocytes (Table 1). Group 1 ILCs are defined by their ability to produce Th1 cell associated cytokines, in particular IFN-γ. Although under debate, the prototypical cell is the NK cell. Group 2 ILCs are defined by their ability to produce Th2 cytokines, in particular IL-5 and IL-13 and the prototypical cells are the IH2 cells or nuocytes[83-86]. These cells have been shown to play a major role in defense against parasites and in allergy and asthma[87,88]. Group 3 ILCs are defined by their ability to secrete Th17 like cytokines such as IL-17 and IL-22 and the prototypical cells are LTi cells[89]. Group 3 ILCs have been shown to play a major role in autoimmune disease and have been shown to mediate colitis in a mouse model of IBD[47].
Since ILCs have been shown to be important in mucosal immunity it was only logical to examine the role of these cells in IBD. Recent data has implicated ILCs, in particular group 3 ILCs in the development of IBD. While most research into IBD has focused on the role of the adaptive immune system, in particular Th1 and Th17 subsets, as well as ineffective regulatory T-cells, new evidence suggests that IBD can be triggered in RAG-/- mice, which lack all components of the adaptive immune system in an IL-23 dependent manner. Buonocore et al[47] demonstrated that group 3 ILCs, and not NK cells (group 1 ILCs), were increased and produced large amounts of IL-17A and IFN-γ after Hepaticus infection in RAG-/- mice were required for colitogenesis. Accumulating evidence suggests that group 3 ILCs induce colitis via an IL-23R-IL-22 dependent mechanism[47,90,91]. RAG-/- mice can also develop colitis after injection of CD40L. Vonarbourg et al[92] demonstrated that CD40L induced colitis requires the presence of innate lymphocytes because CD40L injection into RAG2-/--IL-2Rg-/- mice, which lack both adaptive immune cells and ILCs, did not develop colitis. Furthermore, group 3 ILC involvement in IBD has been further supported by the observation that RORt-/- mice do not develop CD40L induced colitis[47]. In an elegant set of experiments, it was found that LTi cells were the subset of group 3 ILCs that were colitogenic and required for disease onset, suggesting an important role for these cells in IBD pathogenesis[47]. In individuals with CD, a population of innate lymphocytes that were RORt+ and represent human ILCs were found to be increased in the lamina propria compared to controls and this increase was IL-23 dependent[93,94]. Although a young field, data suggests that group 3 ILCs are an important cell type to study for answering questions about the pathogenesis of IBD as well as possible future therapies.
Th1 and Th2 cells
Th1 cells are induced by IL-12 and characteristically secrete copious amounts of IFN-γ, TNF-α, and IL-12, whereas the signature cytokines secreted from Th2 cells are IL-4, IL-5, and IL-13[95]. CD is thought to be a Th1 mediated disease, while UC is believed to be mediated by Th2 responses[96]. Mucosal T-cells from CD patients have been shown to secrete higher amounts of IFN-γ and IL-2 than from T-cells from UC patients[97,98]. Furthermore, it has been demonstrated that UC patients produce increased amounts of IL-5 and have atypical natural killer T (NKT)-cells that secrete higher amounts of IL-13 as compared to CD patients[99-101]. However, recent data has suggested that the CD-Th1 and UC-Th2 paradigms are not so straight forward. Biopsies from both CD and UC patients have demonstrated high ex vivo levels of IFN-γ and lower levels of IL-13 have been found in UC patients as compared to CD patients[102,103]. Furthermore, data suggests that Th17 cell production of IL-17 and IL-23 play important roles in the pathogenesis of IBD, with DCs isolated from CD patients producing more IL-23 than UC patients[104]. Understanding the complicated interactions underlying the dysregulated adaptive immune response in IBD will ultimately identify novel therapeutic targets.
Th17 cells: Friend or foe
Th17 cells are a subset of helper T-cells that are induced by IL-6 and TGF-B, expanded by IL-23, and characterized by the secretion of copious amounts of IL-17A, IL-17F, IL-21, and IL-22[105-107]. RORt has been identified as the master transcription factor of Th17 differentiation[108,109]. The IL-17 cytokine family includes six members: IL-17A-F[110]. IL-17A and IL-17F are 50% similar in their amino acid structure, while IL-17B, IL-17C, and IL-17D have less homology[111]. IL-17A and IL-17F signal through the same receptor, the IL-17 receptor A (IL-17RA) and act through activation of the NF-κB and MAPK pathways[112,113]. The major proinflammatory effects of IL-17A and IL-17F are the activation of various cellular targets, including the epithelium, endothelium, monocytes/macrophages, fibroblasts, and neutrophils that cause the induction of TNF-α, IL-1B, chemokines (CXCL8, CXCL9, CXCL10), GM-CSF, G-CSF, IL-6, and metaloproteases[114-117]. There are two major subsets of Th17 cells: Th17 cells producing IL-17 and Th1/Th17 cells producing both IFN-γ and IL-17[104,118-122]. IL-17 has been implicated in various immune mediated diseases, including rheumatoid arthritis (RA), asthma, IBD, and experimental autoimmune encephalitis (EAE)[123,124].
Th17 cells and signature cytokines have been extensively studied in IBD. GWAS have identified several genes involved in Th17 differentiation and expansion, including IL-23R, IL-12B, JAK2, STAT3, CCR6 and TNFSF15, as CD susceptibility genes with some overlap in UC[12,58]. As compared to normal, CD and UC patients have increased levels of IL-17A gut mucosal transcripts and the lamina propria contains increased numbers of Th17 and Th1/Th17 cells[102,123,125]. RORt is found to be expressed at higher levels in lamina propria T-cells from CD patients[126].
Th17 pathobiology is complicated by the fact that in different experimental models, Th17 subsets can be distinguished by their function as either “pathogenic” or “nonpathogenic” . Pathogenic Th17 cells are thought to be characterized by their production of IFN-γ and by the expression of specific surface markers, including IL-18R1 and CXCR3[127]. IL-17A deficient mice or those treated with neutralizing antibodies to IL-17A or IL-17RA are resistant to the development of RA and EAE[128,129]. Furthermore, in a trinitrobenzene sulfonic acid (TNBS) mouse model of colitis IL-17RA deficient animals were protected from the development of acute mucosal inflammation[130]. However, in a DSS model of colitis, mucosal inflammation was ameliorated by IL-17F deficiency, but exacerbated by IL-17A deficiency, suggesting an important role for IL-17F and perhaps an alternative role for IL-17A[131-133]. Furthermore, supporting a protective role for IL-17A, it has been shown that IL-17A directly inhibits Th1 cells and suppresses development of inflammation[134]. Additionally, anti-IL-17A monoclonal antibody treatment was shown to exacerbate DSS induced colitis[135]. These studies suggest that IL-17A may protect against the development of mucosal inflammation whereas IL-17F may drive it.
As demonstrated by the data above, the biology of IL-17 deficiency has been complicated, with some studies showing a pathogenic role, while others suggesting a protective role. However, there has been a significant amount of data suggesting that IL-17A has played a pathogenic role in IBD. Therefore, a double-blind, randomized, placebo-controlled study tested whether the anti-IL-17A monoclonal antibody, secukinumab, would be beneficial in CD patients[136]. Surprisingly, the study found that blockade of IL-17A was ineffective and caused a higher rate of adverse advents as compared to placebo, suggesting a protective role of IL-17A[136]. Although the study was halted prematurely, exploratory analysis of CD candidate genetic polymorphisms found that a subset of patients with a minor allele of TL1A actually had an improved clinical score over the course of the treatment[136]. These data suggest that in the right genetic context, secukinumab therapy may be beneficial to some patients, which further supports the concept of treating each individual with IBD based upon their own genetic composition.
TL1A: CONNECTING THE INNATE AND ADAPTIVE IMMUNE SYSTEM
Tumor necrosis factor super family 15 (TNSF15) encodes the protein TL1A, a member of the TNF superfamily, is expressed either membrane bound or secreted by monocytes, macrophages, DCs, fibroblasts, and endothelial cells in response to stimulation by cytokines and microorganisms[137-140]. It binds to death domain receptor 3 (DR3), mainly expressed on T-cells, to initiate a number of immune responses, such as activation of T-cells resulting in the secretion of proinflammatory mediators[141]. TL1A has been implicated in the pathogenesis of many autoimmune diseases, including asthma, rheumatoid arthritis, and IBD[96,140,142-144]. Numerous studies have supported the concept that TL1A is a major regulator of mucosal inflammation at the interface between the innate and adaptive immune system[145-147].
In 2005, in a study of Japanese CD patients, polymorphisms in the TNFSF15 gene was identified as having a strong association with CD[148]. This association has been reproduced in other studies, including European and Jewish CD and UC patients, and has been demonstrated to be the dominant gene in East-Asians with IBD[12,149-153]. Haplotypes within the gene confer either risk or protection, which is dependent upon the ethnicity of the individual. In non-Jewish CD patients, haplotype A is a risk allele, while haplotype B is protective[148-150,153]. However, in Jewish CD patients, haplotype B has a trend towards risk, as these patients had worsened disease as manifested by higher incidents of surgery and increased responses to E. coli outer membrane porin C (OMPC)[154,155]. Furthermore, monocytes isolated from Jewish patients that were haplotype B secreted increased amounts of TL1A than haplotype A carriers after stimulation[156].
Given the information generated from the human GWAS studies, transgenic mice have been created that overexpress TL1A. In 2011, Meylan et al[146] and Taraban et al[147] found that murine colitis driven by TL1A overexpression in T-cells and DCs was found to be dominated by a Th2 response over Th1, with elevation in IL-13 and unchanged levels of IFN-γ. Also, in both models, spontaneous intestinal inflammation developed, with disease severity being greatest in the terminal ileum and correlating to transgene expression level. This observation was abolished with anti-IL-13 treatment. Shih et al[143] reported similar observations in another model of TL1A overexpression. However, they also found that these mice had increased levels of IFN-γ and intestinal fibrosis. The differences in these mouse models may be secondary to differences in the generation of the mice and/or different gut microflora between animal facilities. Regardless, all models demonstrated intestinal inflammation and this supports evidence of TL1A polymorphisms being associated with IBD. Given this information, studies utilizing anti-TL1A antibodies were undertaken. In models of TNBS and DSS colitis anti-TL1A neutralizing antibody treatment was shown to ameliorate weight loss and intestinal inflammation[146,157]. These studies suggest a role for using blocking antibodies to TL1A to ameliorate pathological T-cell responses in IBD.
CONCLUSION
In current immunology there are new Th cell subsets, such as IL-9 producing Th9 cells, IL-22 producing Th22 cells, follicular helper T-cells, and emerging types of Treg cells that are now also all being implicated in the pathogenesis of IBD[158-161]. Furthermore, historically it was thought that terminally differentiated Th cells seldom re-differentiate to other Th subsets, however now the plasticity between Th cells is now extensively under investigation[162].
It has been well documented that the adaptive immune system plays an important role in the development and perpetuation of the inflammatory cascade in IBD. In particular, T-cells have been shown to be key players in driving intestinal inflammation. However, a number of unresolved issues exist that need to be addressed in order to develop successful and appropriate therapeutic strategies. Recent advances have clarified the importance of the innate immune system in IBD pathobiology. Furthermore, besides anti-TNF agents, molecules targeting specific T-cell derived molecules have largely failed. This is likely due to the complexities and redundancies of cytokine networks and highlights how different each individual’s immune system is in the context of their own genetics. The studies of the interactions between the different components of the innate and adaptive immune system, as well as the interactions with the intestinal microbiota, and how these interactions relate in the overwhelming context of an individual’s genetics are areas that will open new horizons in the knowledge of mechanisms of gut inflammation.
Footnotes
Supported by NIH KO8 DK093578; CCFA Career Development Award 3467 (DQS); F Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute
P- Reviewer: Feuerstein JD S- Editor: Cui XM L- Editor: A E- Editor: Ma Ss
References
- 1.Lakatos PL. Recent trends in the epidemiology of inflammatory bowel diseases: up or down? World J Gastroenterol. 2006;12:6102–6108. doi: 10.3748/wjg.v12.i38.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichtenstein GR, Hanauer SB, Sandborn WJ. Management of Crohn’s disease in adults. Am J Gastroenterol. 2009;104:465–83; quiz 464, 484. doi: 10.1038/ajg.2008.168. [DOI] [PubMed] [Google Scholar]
- 3.Satsangi J, Silverberg MS, Vermeire S, Colombel JF. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. doi: 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kornbluth A, Sachar DB. Ulcerative colitis practice guidelines in adults. American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 1997;92:204–211. [PubMed] [Google Scholar]
- 5.Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Huang BL, Chandra S, Shih DQ. Skin manifestations of inflammatory bowel disease. Front Physiol. 2012;3:13. doi: 10.3389/fphys.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bager P, Simonsen J, Nielsen NM, Frisch M. Cesarean section and offspring’s risk of inflammatory bowel disease: a national cohort study. Inflamm Bowel Dis. 2012;18:857–862. doi: 10.1002/ibd.21805. [DOI] [PubMed] [Google Scholar]
- 8.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hviid A, Svanström H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- 10.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 14.Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine. 2012;59:451–459. doi: 10.1016/j.cyto.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol. 2009;70:505–515. doi: 10.1111/j.1365-3083.2009.02319.x. [DOI] [PubMed] [Google Scholar]
- 16.D’Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 17.Casellas F, Borruel N, Papo M, Guarner F, Antolín M, Videla S, Malagelada JR. Antiinflammatory effects of enterically coated amoxicillin-clavulanic acid in active ulcerative colitis. Inflamm Bowel Dis. 1998;4:1–5. doi: 10.1097/00054725-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Fölsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Doré J, Raedler A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141:227–236. doi: 10.1053/j.gastro.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 27.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–1728. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 28.Sokol H, Lepage P, Seksik P, Doré J, Marteau P. Temperature gradient gel electrophoresis of fecal 16S rRNA reveals active Escherichia coli in the microbiota of patients with ulcerative colitis. J Clin Microbiol. 2006;44:3172–3177. doi: 10.1128/JCM.02600-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 30.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15:872–882. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 33.Darfeuille-Michaud A. Adherent-invasive Escherichia coli: a putative new E. coli pathotype associated with Crohn’s disease. Int J Med Microbiol. 2002;292:185–193. doi: 10.1078/1438-4221-00201. [DOI] [PubMed] [Google Scholar]
- 34.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 35.Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol Microbiol. 2003;48:781–794. doi: 10.1046/j.1365-2958.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 36.Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel JF, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest. 2007;117:1566–1574. doi: 10.1172/JCI30504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009;206:2179–2189. doi: 10.1084/jem.20090741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001;69:5529–5537. doi: 10.1128/IAI.69.9.5529-5537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N, Chan AS, Magee AS, Danielson ME, et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature. 2011;472:471–475. doi: 10.1038/nature10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 44.Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. 2012;30:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, Barclay GR, Smedley J, Pung R, Oliveira KM, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 49.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 50.Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol Rev. 2005;206:260–276. doi: 10.1111/j.0105-2896.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 51.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 52.Larsson JM, Karlsson H, Crespo JG, Johansson ME, Eklund L, Sjövall H, Hansson GC. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis. 2011;17:2299–2307. doi: 10.1002/ibd.21625. [DOI] [PubMed] [Google Scholar]
- 53.Blackwell CC, Jonsdottir K, Hanson MF, Weir DM. Non-secretion of ABO blood group antigens predisposing to infection by Haemophilus influenzae. Lancet. 1986;2:687. doi: 10.1016/s0140-6736(86)90193-5. [DOI] [PubMed] [Google Scholar]
- 54.Blackwell CC, Jónsdóttir K, Hanson M, Todd WT, Chaudhuri AK, Mathew B, Brettle RP, Weir DM. Non-secretion of ABO antigens predisposing to infection by Neisseria meningitidis and Streptococcus pneumoniae. Lancet. 1986;2:284–285. doi: 10.1016/s0140-6736(86)92103-3. [DOI] [PubMed] [Google Scholar]
- 55.Carlsson B, Kindberg E, Buesa J, Rydell GE, Lidón MF, Montava R, Abu Mallouh R, Grahn A, Rodríguez-Díaz J, Bellido J, et al. The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 Norovirus infection. PLoS One. 2009;4:e5593. doi: 10.1371/journal.pone.0005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly RJ, Rouquier S, Giorgi D, Lennon GG, Lowe JB. Sequence and expression of a candidate for the human Secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J Biol Chem. 1995;270:4640–4649. doi: 10.1074/jbc.270.9.4640. [DOI] [PubMed] [Google Scholar]
- 57.Marionneau S, Airaud F, Bovin NV, Le Pendu J, Ruvoën-Clouet N. Influence of the combined ABO, FUT2, and FUT3 polymorphism on susceptibility to Norwalk virus attachment. J Infect Dis. 2005;192:1071–1077. doi: 10.1086/432546. [DOI] [PubMed] [Google Scholar]
- 58.McGovern DP, Gardet A, Törkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagacé C, Li C, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorven M, Grahn A, Hedlund KO, Johansson H, Wahlfrid C, Larson G, Svensson L. A homozygous nonsense mutation (428G--& gt; A) in the human secretor (FUT2) gene provides resistance to symptomatic norovirus (GGII) infections. J Virol. 2005;79:15351–15355. doi: 10.1128/JVI.79.24.15351-15355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 61.Roediger WE, Nance S. Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br J Exp Pathol. 1986;67:773–782. [PMC free article] [PubMed] [Google Scholar]
- 62.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 63.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 65.Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–727. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology. 1998;114:657–668. doi: 10.1016/s0016-5085(98)70579-7. [DOI] [PubMed] [Google Scholar]
- 67.Saruta M, Targan SR, Mei L, Ippoliti AF, Taylor KD, Rotter JI. High-frequency haplotypes in the X chromosome locus TLR8 are associated with both CD and UC in females. Inflamm Bowel Dis. 2009;15:321–327. doi: 10.1002/ibd.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ghosh TK, Mickelson DJ, Fink J, Solberg JC, Inglefield JR, Hook D, Gupta SK, Gibson S, Alkan SS. Toll-like receptor (TLR) 2-9 agonists-induced cytokines and chemokines: I. Comparison with T cell receptor-induced responses. Cell Immunol. 2006;243:48–57. doi: 10.1016/j.cellimm.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 69.Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 70.O’Hara JR, Feener TD, Fischer CD, Buret AG. Campylobacter jejuni disrupts protective Toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect Immun. 2012;80:1563–1571. doi: 10.1128/IAI.06066-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 72.Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder TF, Hardt WD, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–1207. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 74.Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, Philpott D, Garcia Rodenas CL, McCoy KD, Verdu EF. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis. 2012;18:1434–1446. doi: 10.1002/ibd.22848. [DOI] [PubMed] [Google Scholar]
- 75.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 76.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 79.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 80.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 82.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 83.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 84.Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- 85.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107:11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eken A, Singh AK, Treuting PM, Oukka M. IL-23R(+) innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2014;7:143–154. doi: 10.1038/mi.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geremia A, Arancibia-Cárcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, Travis SP, Powrie F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208:1127–1133. doi: 10.1084/jem.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Hölscher C, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chinen H, Matsuoka K, Sato T, Kamada N, Okamoto S, Hisamatsu T, Kobayashi T, Hasegawa H, Sugita A, Kinjo F, et al. Lamina propria c-kit+ immune precursors reside in human adult intestine and differentiate into natural killer cells. Gastroenterology. 2007;133:559–573. doi: 10.1053/j.gastro.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 94.Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, Matuzaki Y, Suzuki S, Sugita A, Koganei K, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology. 2010;139:882–892, 892.e1-3. doi: 10.1053/j.gastro.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 95.Romagnani S. Lymphokine production by human T cells in disease states. Annu Rev Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 96.Shih DQ, Targan SR, McGovern D. Recent advances in IBD pathogenesis: genetics and immunobiology. Curr Gastroenterol Rep. 2008;10:568–575. doi: 10.1007/s11894-008-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 98.Noguchi M, Hiwatashi N, Liu Z, Toyota T. Enhanced interferon-gamma production and B7-2 expression in isolated intestinal mononuclear cells from patients with Crohn’s disease. J Gastroenterol. 1995;30 Suppl 8:52–55. [PubMed] [Google Scholar]
- 99.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 100.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Bürgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 102.Rovedatti L, Kudo T, Biancheri P, Sarra M, Knowles CH, Rampton DS, Corazza GR, Monteleone G, Di Sabatino A, Macdonald TT. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–1636. doi: 10.1136/gut.2009.182170. [DOI] [PubMed] [Google Scholar]
- 103.Vainer B, Nielsen OH, Hendel J, Horn T, Kirman I. Colonic expression and synthesis of interleukin 13 and interleukin 15 in inflammatory bowel disease. Cytokine. 2000;12:1531–1536. doi: 10.1006/cyto.2000.0744. [DOI] [PubMed] [Google Scholar]
- 104.Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology. 2009;137:1736–1745. doi: 10.1053/j.gastro.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 105.Dong C, Nurieva RI. Regulation of immune and autoimmune responses by ICOS. J Autoimmun. 2003;21:255–260. doi: 10.1016/s0896-8411(03)00119-7. [DOI] [PubMed] [Google Scholar]
- 106.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 107.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 108.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 109.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 111.Khalturin K, Panzer Z, Cooper MD, Bosch TC. Recognition strategies in the innate immune system of ancestral chordates. Mol Immunol. 2004;41:1077–1087. doi: 10.1016/j.molimm.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 112.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273:27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 113.Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 114.Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337–5344. [PubMed] [Google Scholar]
- 115.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- 117.Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 118.Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Filì L, Ferri S, Frosali F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903–1916. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 122.Sujino T, Kanai T, Ono Y, Mikami Y, Hayashi A, Doi T, Matsuoka K, Hisamatsu T, Takaishi H, Ogata H, et al. Regulatory T cells suppress development of colitis, blocking differentiation of T-helper 17 into alternative T-helper 1 cells. Gastroenterology. 2011;141:1014–1023. doi: 10.1053/j.gastro.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 123.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 125.Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, Fujiyama Y, Andoh A. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol. 2010;160:386–393. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM, Auernhammer CJ, Brand S. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58:1207–1217. doi: 10.1136/gut.2007.130112. [DOI] [PubMed] [Google Scholar]
- 127.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002;46:802–805. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 129.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–388. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 131.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 132.O’Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sarra M, Pallone F, Macdonald TT, Monteleone G. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–1813. doi: 10.1002/ibd.21248. [DOI] [PubMed] [Google Scholar]
- 134.Awasthi A, Kuchroo VK. IL-17A directly inhibits TH1 cells and thereby suppresses development of intestinal inflammation. Nat Immunol. 2009;10:568–570. doi: 10.1038/ni0609-568. [DOI] [PubMed] [Google Scholar]
- 135.Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 136.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cassatella MA, Pereira-da-Silva G, Tinazzi I, Facchetti F, Scapini P, Calzetti F, Tamassia N, Wei P, Nardelli B, Roschke V, et al. Soluble TNF-like cytokine (TL1A) production by immune complexes stimulated monocytes in rheumatoid arthritis. J Immunol. 2007;178:7325–7333. doi: 10.4049/jimmunol.178.11.7325. [DOI] [PubMed] [Google Scholar]
- 138.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 139.Prehn JL, Thomas LS, Landers CJ, Yu QT, Michelsen KS, Targan SR. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J Immunol. 2007;178:4033–4038. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]
- 140.Shih DQ, Kwan LY, Chavez V, Cohavy O, Gonsky R, Chang EY, Chang C, Elson CO, Targan SR. Microbial induction of inflammatory bowel disease associated gene TL1A (TNFSF15) in antigen presenting cells. Eur J Immunol. 2009;39:3239–3250. doi: 10.1002/eji.200839087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Screaton GR, Xu XN, Olsen AL, Cowper AE, Tan R, McMichael AJ, Bell JI. LARD: a new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Natl Acad Sci USA. 1997;94:4615–4619. doi: 10.1073/pnas.94.9.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Barrett R, Zhang X, Koon HW, Vu M, Chang JY, Yeager N, Nguyen MA, Michelsen KS, Berel D, Pothoulakis C, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. 2012;180:636–649. doi: 10.1016/j.ajpath.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shih DQ, Barrett R, Zhang X, Yeager N, Koon HW, Phaosawasdi P, Song Y, Ko B, Wong MH, Michelsen KS, et al. Constitutive TL1A (TNFSF15) expression on lymphoid or myeloid cells leads to mild intestinal inflammation and fibrosis. PLoS One. 2011;6:e16090. doi: 10.1371/journal.pone.0016090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zheng L, Zhang X, Chen J, Ichikawa R, Wallace K, Pothoulakis C, Koon HW, Targan SR, Shih DQ. Sustained tl1a (TNFSF15) expression on both lymphoid and myeloid cells leads to mild spontaneous intestinal inflammation and fibrosis. Eur J Microbiol Immunol (Bp) 2013;3:11–20. doi: 10.1556/EuJMI.3.2013.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011;244:188–196. doi: 10.1111/j.1600-065X.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Meylan F, Song YJ, Fuss I, Villarreal S, Kahle E, Malm IJ, Acharya K, Ramos HL, Lo L, Mentink-Kane MM, et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol. 2011;4:172–185. doi: 10.1038/mi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Taraban VY, Slebioda TJ, Willoughby JE, Buchan SL, James S, Sheth B, Smyth NR, Thomas GJ, Wang EC, Al-Shamkhani A. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011;4:186–196. doi: 10.1038/mi.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, Cardon L, Takazoe M, Tanaka T, Ichimori T, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14:3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 149.Kakuta Y, Kinouchi Y, Negoro K, Takahashi S, Shimosegawa T. Association study of TNFSF15 polymorphisms in Japanese patients with inflammatory bowel disease. Gut. 2006;55:1527–1528. doi: 10.1136/gut.2006.100297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Picornell Y, Mei L, Taylor K, Yang H, Targan SR, Rotter JI. TNFSF15 is an ethnic-specific IBD gene. Inflamm Bowel Dis. 2007;13:1333–1338. doi: 10.1002/ibd.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Thiébaut R, Kotti S, Jung C, Merlin F, Colombel JF, Lemann M, Almer S, Tysk C, O’Morain M, Gassull M, et al. TNFSF15 polymorphisms are associated with susceptibility to inflammatory bowel disease in a new European cohort. Am J Gastroenterol. 2009;104:384–391. doi: 10.1038/ajg.2008.36. [DOI] [PubMed] [Google Scholar]
- 152.Tremelling M, Berzuini C, Massey D, Bredin F, Price C, Dawson C, Bingham SA, Parkes M. Contribution of TNFSF15 gene variants to Crohn’s disease susceptibility confirmed in UK population. Inflamm Bowel Dis. 2008;14:733–737. doi: 10.1002/ibd.20399. [DOI] [PubMed] [Google Scholar]
- 153.Yang SK, Lim J, Chang HS, Lee I, Li Y, Liu J, Song K. Association of TNFSF15 with Crohn’s disease in Koreans. Am J Gastroenterol. 2008;103:1437–1442. doi: 10.1111/j.1572-0241.2007.01752.x. [DOI] [PubMed] [Google Scholar]
- 154.Arnott ID, Landers CJ, Nimmo EJ, Drummond HE, Smith BK, Targan SR, Satsangi J. Sero-reactivity to microbial components in Crohn’s disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am J Gastroenterol. 2004;99:2376–2384. doi: 10.1111/j.1572-0241.2004.40417.x. [DOI] [PubMed] [Google Scholar]
- 155.Mow WS, Vasiliauskas EA, Lin YC, Fleshner PR, Papadakis KA, Taylor KD, Landers CJ, Abreu-Martin MT, Rotter JI, Yang H, et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology. 2004;126:414–424. doi: 10.1053/j.gastro.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 156.Michelsen KS, Thomas LS, Taylor KD, Yu QT, Mei L, Landers CJ, Derkowski C, McGovern DP, Rotter JI, Targan SR. IBD-associated TL1A gene (TNFSF15) haplotypes determine increased expression of TL1A protein. PLoS One. 2009;4:e4719. doi: 10.1371/journal.pone.0004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D, Braun J, Targan SR. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.King C, Sprent J. Emerging cellular networks for regulation of T follicular helper cells. Trends Immunol. 2012;33:59–65. doi: 10.1016/j.it.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 161.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 162.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]