

Abstract
Rationale
Hypertension is the most prevalent life-threatening disease worldwide and is frequently associated with chronic kidney disease (CKD). However, the molecular basis underlying hypertensive CKD is not fully understood.
Objective
We sought to identify specific factors and signaling pathways that contribute to hypertensive CKD and thereby exacerbate disease progression.
Methods and Results
Using high-throughput quantitative reverse-transcription polymerase chain reaction profiling, we discovered that the expression level of 5′-ectonucleotidase (CD73), a key enzyme that produces extracellular adenosine, was significantly increased in the kidneys of angiotensin II–infused mice, an animal model of hypertensive nephropathy. Genetic and pharmacological studies in mice revealed that elevated CD73-mediated excess renal adenosine preferentially induced A2B adenosine receptor (ADORA2B) production and that enhanced kidney ADORA2B signaling contributes to angiotensin II–induced hypertension. Similarly, in humans, we found that CD73 and ADORA2B levels were significantly elevated in the kidneys of CKD patients compared with normal individuals and were further elevated in hypertensive CKD patients. These findings led us to further discover that elevated renal CD73 contributes to excess adenosine signaling via ADORA2B activation that directly stimulates endothelin-1 production in a hypoxia-inducible factor-α–dependent manner and underlies the pathogenesis of the disease. Finally, we revealed that hypoxia-inducible factor-α is an important factor responsible for angiotensin II–induced CD73 and ADORA2B expression at the transcriptional level.
Conclusions
Overall, our studies reveal that angiotensin II–induced renal CD73 promotes the production of renal adenosine that is a prominent driver of hypertensive CKD by enhanced ADORA2B signaling–mediated endothelin-1 induction in a hypoxia-inducible factor-α–dependent manner. The inhibition of excess adenosine-mediated ADORA2B signaling represents a novel therapeutic target for the disease.
Keywords: adenosine, hypertension, chronic renal disease
Hypertension is a leading cause of morbidity and mortality in the United States and worldwide. The condition affects ≈ 1 in 3 adults in the United States and 25% of the adult population worldwide.1 The estimated total number of adults with hypertension in 2000 was 972 million, and it is predicated to increase by ≈60% to a total of 1.56 billion by 2025.1 In the United States, hypertension accounts for 1 in 7 deaths, >$93.5 billion in medical costs per year, and immeasurable human suffering.1 Because of the increasing morbidity, mortality, and extensive medical costs associated with hypertension, novel therapeutic strategies are desperately needed to reduce hypertension and delay disease progression. Defining the molecular mechanisms underlying the disease is important for developing novel strategies for disease prevention and treatment.
Chronic kidney disease (CKD) is a devastating disease including kidney injury, progression to renal fibrosis, and end-stage renal disease.2-4 It affects 26 million American adults and is the ninth leading cause of mortality in the United States.2 CKD is a major cause of hypertension, and hypertension also can promote progression of CKD to end-stage renal disease.5 Despite intensive research, the molecular basis for pathogenesis of hypertension and subsequent progression from CKD to end-stage renal disease is still not fully understood.3 Among identified factors and signaling pathways involved in hypertensive CKD, the renin–angiotensin system is considered as a key signaling cascade contributing to hypertension, CKD, and its progression to renal fibrosis.6 In both humans and animals with hypertensive nephropathy, circulating angiotensin II (Ang II), the end effector of the renin–angiotensin system, is elevated7 and therapies that inhibit this signaling cascade are effective.8 Ang II can stimulate multiple signaling pathways, but it is unclear which of these pathways drives hypertensive CKD and, when inhibited, which results in disease amelioration. Here, we sought to identify specific factors and signaling pathways that contribute to hypertension and CKD, and thereby exacerbate disease progression in both humans and mice.
Methods
Detailed Methods are available in the Online Data Supplement.
Human Subjects
Kidney biopsy specimens were collected from normal control individuals (n=12), those with CKD alone without hypertension (n=24), and hypertensive patients with CKD (n=32). CKD patients without hypertension and hypertensive CKD patients admitted to the First XiangYa Hospital were identified by nephrologists of the Central South University at Changsha, Hunan, China. Normal individuals were selected on the basis of having normal blood pressure and kidney function before acute kidney rupture resulting from trauma. CKD patients without hypertension were selected on the basis of displaying kidney damage (structural or functional abnormalities of the kidney) with glomerular filtration rate <60 mL/min per 1.73 m2 for >3 months in the absence of hypertension. Individuals with hypertensive CKD were selected on the basis of presenting kidney damage (structural or functional abnormalities of the kidney) with glomerular filtration rate <60 mL/min per 1.73 m2 for >3 months in the presence of hypertension (systolic blood pressure ≥140 mm Hg; diastolic blood pressure ≥90 mm Hg). The research protocol was approved by the Central South University Ethics Committee for the Protection of Human Subjects. Clinical data for normal individuals and patients with mild or severe CKD are listed in the Table.
Table.
Clinic Information of Human Subjects
| Control (n=12) | Mild CKD Without Hypertension (n=24) | Severe CKD With Hypertension (n=32) | |
|---|---|---|---|
| Age, y | 43.6±12.2 | 44.2±11.3 | 46.4±10.5 |
| Sex, M/F | 7/5 | 14/10 | 22/10 |
| Proteinuria/hematuria | ND | 20/16 | 32/24 |
| SBP, mm Hg | 102.3±10.6 | 108.7±7.69 | 159.2±20.1* |
| DBP, mm Hg | 75.4±12.1 | 70.8±7.3 | 96.2±12.7* |
| HB, g/L | 129.76±17.68 | 129.2±17.3 | 125.1±18.63 |
| HCT, % | 38.80±2.51 | 37.38±5.29 | 35.63±5.06 |
| TP, g/L | 61.20±3.05 | 59.03±13.42 | 58.43±10.79 |
| ALB, g/L | 37.67±3.46 | 36.89±8.94 | 35.29±10.89 |
| GLO, g/L | 24.53±2.47 | 23.75±4.09 | 23.14±3.06 |
| TG, mmol/L | 1.92±0.46 | 2.07±0.38 | 1.98±0.26 |
| TC, mmol/L | 5.10±1.70 | 5.74±1.72 | 5.41±1.89 |
| HDL, mmol/L | 2.13±0.41 | 2.07±0.75 | 1.93±0.53 |
| LDL, mmol/L | 3.16±0.47 | 3.57±1.15 | 4.13±1.94 |
| BUN, mmol/L | 5.06±1.10 | 5.72±2.73 | 9.09±1.54* |
| Bcr, μmol/L | 68.31±14.22 | 71.37±22.23 | 109.12±24.89* |
| eGFR, mL/min | 86.31±17.16 | 77.79±15.87 | 52.01±19.61* |
ALB indicates plasma albumin; Bcr, blood creatinine; BUN, blood urea nitrogen; CKD, chronic kidney disease; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; F, female; GLO, plasma globulin; HB, hemoglobin; HCT, hematocrit; HDL, high-density lipoproteins; LDL, low-density lipoproteins; M, male; SBP, systolic blood pressure; TC, total plasma cholesterol; TG, total plasma triglyceride; TP, total plasma protein. Kidneys of controls with normal kidney function and blood pressure were collected after 3 hours of acute kidney rupture.
Compared with control, P<0.05.
Animals
Wild-type (WT) 8- to 10-week-old C57BL/6 mice were purchased from Harlan Laboratories (Indianapolis, IN). Ecto-5’-nucleotidase (CD73)–deficient mice and A2B adenosine receptor (ADORA2B)–deficient mice congenic on a C57BL/6 background were generated and genotyped as described.9 All protocols involving animal studies were reviewed and approved by the Institutional Animal Welfare Committee of the University of Texas Houston Health Science Center. Six to 10 mice for each group were used.
Reverse-Transcription Polymerase Chain Reaction Gene Expression Profiling of Kidneys of Mice With or Without Ang II Infusion
RNA was extracted using total RNA isolation reagent (Invitrogen, Carls-View, CA) from the kidneys after 14-day infusion with saline or Ang II. Genomic DNA contamination was eliminated by DNase treatment with RNeasy Micro Kit (Qiagen GmbH, Hidden, Germany). High-throughput mouse RT2 profiler polymerase chain reaction (PCR) array and RT2 real-timer SyBR Mix were purchased from SuperArray Bioscience Corporation (Frederick, MD). PCR was performed on an ABI Prism 7700 sequence Detector (Applied Biosystems). For data analysis, the ΔCt method was used. For each gene, the fold changes were calculated as difference in kidney gene expression between Ang II–infused and saline-infused mice. P<0.05 is considered significant.
Results
Elevated Renal CD73 Is a Novel Factor Underlying Chronic Increased Kidney Adenosine Production and Contributing to Hypertension and Kidney Injury in Ang II–Infused Mice
In an effort to identify specific renal factors contributing to the pathogenesis of CKD and hypertension, we used a high-throughput quantitative reverse-transcription PCR (RT-PCR) array to compare gene expression profiles in the kidneys of controls and Ang II–infused mice, an animal model of hypertensive CKD10-12 (gene expression changes are listed in Online Table I). Among all of the transcripts screened, CD73 mRNA levels were among the most highly induced in the kidneys of Ang II–infused mice compared with controls (Figure 1A). We further confirmed that CD73 mRNA and protein levels, as well as enzyme activity, were significantly elevated in the kidneys of Ang II–infused mice (Figure 1B and 1C). Intriguingly, CD73 mRNA levels were not significantly increased in the hearts of Ang II–infused mice (Online Figure I). Moreover, mRNA levels of CD38 and CD39, 2 other 5′-ectonucleotidases, were similar between mice with and without Ang II infusion (Online Figure II). In addition, adenosine deaminase (ADA) activity in the kidneys of Ang II–infused mice was reduced ≈ 15% (Online Figure III), which is much lower than CD73 activity induced by Ang II that increased ≈2-fold (Figure 1C). Thus, these studies revealed that CD73 is a key ectonucleotidase induced in the kidneys of Ang II–infused mice.
Figure 1. Detrimental effects of elevated kidney 5′AMP ectonucleotidase (CD73) in hypertension by inducing excess renal adenosine in angiotensin (Ang II)-infused mice.

Ang II was delivered by mini-pump for 14 days. Polyethyleneglycol-modified adenosine deaminase (PEG-ADA) injections were initiated after implantation of the minipump. A, Gene expression profile revealed that CD73, hypoxia-inducible factor-1α (HIF-1α), and prepro-endothelin-1 (prepro-ET-1) mRNA levels were significantly elevated in kidneys of Ang II–infused mice compared with controls infused with saline. The red circles represent transcripts elevated >2-fold at a significant level (P<0.05) in the kidneys of Ang II–infused mice compared with the controls. Green circles represent transcripts reduced >2-fold at a significant level (P<0.05) in kidneys of Ang II–infused mice compared with the controls (n=6 for each group). B, Immuohistochemical analysis of CD73 in the kidneys of CD73−/− mice, wild-type (WT) mice, and WT mice with 14-day Ang II infusion (scale bar, 400 μm). C, CD73 gene expression levels (left), expression of CD73 in the kidneys quantified using Image-Pro Plus image analysis software (middle), and CD73 activity measured by enzyme-based assay in the kidneys of WT and Ang II–infused mice. Data are expressed as mean±SEM (n=6). *P<0.05 vs WT without Ang II infusion. Renal (D) and plasma (E) adenosine levels were determined on day 14. Data are expressed as mean±SEM. *P<0.05 for Ang II–infused mice vs control mice infused with saline; **P<0.05 for Ang II–infused mice with PEG-ADA treatment vs Ang II–infused mice and CD73-deficient mice with Ang II infusion vs Ang II–infused WT mice (n=8–12 per group). F, Systolic blood pressure was measured at daily intervals by the tail-cuff method until mice were euthanized (n=8–12 per group).
Because CD73 is responsible for production of extracellular adenosine,13,14 it is possible that the elevation in kidney CD73 resulting from Ang II infusion promotes the production of elevated renal adenosine. Consistent with this possibility, we found that adenosine levels increased significantly in the mouse kidney (Figure 1D) but not in the plasma (Figure 1E) of Ang II–infused mice, indicating that elevated renal CD73 induced chronic elevation of adenosine locally in the kidney, but not systemically. To determine whether CD73 contributes to elevation of adenosine and subsequent Ang II–induced hypertension and CKD in vivo, we took a genetic approach by infusion of Ang II into WT and CD73-deficient mice. As expected, we found that CD73 deficiency significantly reduced Ang II–mediated adenosine induction in the mouse kidneys (Figure 1D). These findings indicate that elevated CD73 leads to chronic elevation of renal adenosine production in Ang II–infused mice. Functionally, we found that infusion of Ang II into WT mice led to chronic hypertension and kidney injury characterized with proteinuria and decreased urine osmolality (Figure 1F; Online Figure IVA–IVC), features similar to those observed in hypertensive patients with CKD. In contrast, these features were significantly attenuated when Ang II infusion was performed in CD73-deficient mice (Figure 1F; Online Figure IVA–IVC).
To determine whether CD73 deficiency attenuated hypertension and whether renal damage is caused by effects on angiotensin receptor expression, we used quantitative RT-PCR to compare the abundance of transcripts encoding the 3 angiotensin receptors, angiotensin receptor type 1A, angiotensin receptor type 1B, and angiotensin receptor type 2 receptor, in kidneys of WT and CD73-deficient mice. The results (Online Figure VA) show no significant difference in the abundance of these receptor transcripts in the kidneys of WT mice and CD73-deficient mice. Thus, changes in angiotensin receptor expression do not account for the ability of CD73 deficiency to protect against chronic kidney damage and persistent hypertension in Ang II–infused mice.
Elevated Adenosine Contributes to Chronic Hypertension in Ang II–Infused Mice
To assess the direct effect of elevated renal adenosine in hypertension, we took a pharmacological approach of treating Ang II–infused mice with polyethyleneglycol-modified ADA to lower adenosine levels.15,16 Polyethyleneglycol itself showed no effects on blood pressure (Online Figure VI). However, polyethyleneglycol-modified ADA treatment significantly inhibited the increase in renal adenosine levels observed in Ang II–infused mice (Figure 1D). Likewise, Ang II–induced hypertension was significantly attenuated in mice chronically treated with polyethyleneglycol-modified ADA (Figure 1F; Online Figure VIA–VIC). These findings provide direct in vivo evidence that chronically elevated adenosine is a previously unrecognized detrimental mediator to drive Ang II–induced hypertension and kidney disease.
Excessive ADORA2B Adenosine Receptor Activation Underlies Hypertension in Ang II–Infused Mice
In an effort to determine the potential contribution of adenosine receptor signaling to Ang II–induced hypertensive CKD, we measured mRNA levels for the 4 adenosine receptors in WT and CD73-deficient mice with or without Ang II infusion.17,18 Unexpectedly, we found that only Adora2b mRNA levels were significantly elevated in the kidneys of Ang II–infused mice (Figure 2A-2D). Intriguingly, we found that CD73 deficiency significantly reduced Ang II–mediated elevation of Adora2b mRNA levels in the mouse kidneys (Figure 2A-2D), indicating that CD73-dependent elevated renal adenosine preferentially induces Adora2b gene expression in the kidneys of Ang II–infused mice.
Figure 2. Preferentially elevated A2B adenosine receptor (ADORA2B) underlies excessive renal adenosine-mediated hypertension in angiotensin II (Ang II)–infused mice.
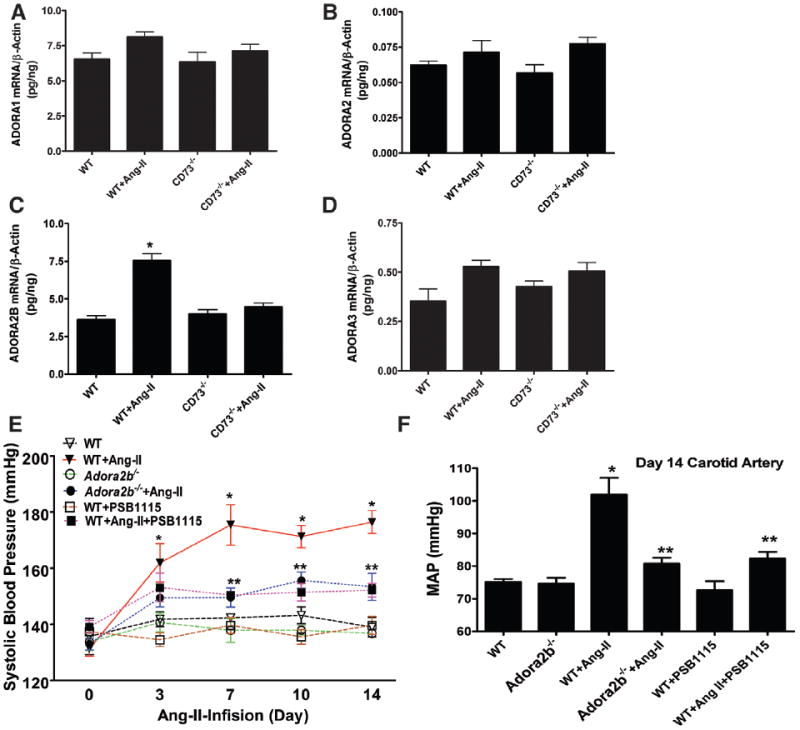
Ang II was delivered to the mice by mini-pump for 14 days. Injection of PSB1115, an ADORA2B-specific antagonist, was initiated after the mini-pump was implanted. A–D, Expression profiles of ADORA1, ADORA2A, ADORA2B, and ADORA3 in the kidneys of wild-type (WT) mice and CD73−/− mice with or without Ang II infusion. Data are expressed as mean±SEM. *P<0.05 for Ang II–infused mice vs control mice infused with saline; **P<0.05 for CD73-deficient mice with Ang II infusion vs Ang II–infused WT mice (n=8–12 per group). E, Systolic blood pressure was measured at daily intervals by the tail-cuff method until mice were euthanized. F, Intracarotid mean arterial blood pressure was measured on day 14 (n=8–10 per group). Data are expressed as mean±SEM. *P<0.05 for Ang II–infused mice vs WT control mice infused with saline; **P<0.05 for Ang II–infused WT mice with PSB1115 treatment vs Ang II–infused WT mice and Adora2b−/− mice with Ang II infusion vs Ang II–infused WT mice (n=8–10 per group).
Next, we used both genetic and pharmacological approaches to determine the role of ADORA2B in chronic hypertension. Genetically, we found that ADORA2B deficiency significantly reduced Ang II–induced hypertension (Figure 2E and 2F). Consistently, we further demonstrated that selectively interfering with ADORA2B activation by an ADORA2B-specific antagonist, PSB1115 significantly attenuated Ang II–induced hypertension (Figure 2E and 2F). Altogether, we showed that elevated renal CD73 is associated with the chronic accumulation of renal adenosine and enhanced ADORA2B signaling, which underlies Ang II–induced hypertension.
CD73 and ADORA2B Expression Levels Are Increased in the Kidneys of Mild CKD Patients Without Hypertension and Are Further Elevated in Severe CKD Patients With Hypertension
To extend our mouse studies to humans, we first examined CD73 and ADORA2B protein levels in kidney biopsy specimens collected from normal controls (n=12), CKD patients without hypertension (n=24), and severe CKD patients with hypertension (n=32; Table shows clinical information of human subjects). Like the expression pattern seen in mice, immunostaining revealed that CD73 and ADORA2B were expressed in both glomeruli and tubules in normal control individuals. CD73 and ADORA2B levels were elevated in both glomeruli and tubules of kidneys isolated from CKD patients with or without hypertension (Figure 3A). Quantitative image analysis demonstrated that increased CD73 and ADORA2B staining in the kidneys of CKD patients was significantly higher than that in the controls, and that CD73 and ADORA2B levels were further elevated in severe CKD patients with hypertension compared with mild CKD patients without hypertension (Figure 3D and 3E). Intriguingly, the elevated CD73 and ADORA2B levels were significantly correlated to disease severity by clinical symptoms (Table), levels of kidney injury quantified by histological score based on hematoxylin and eosin staining (Figure 3A and 3C), and degrees of renal fibrosis by collagen score based on trichrome staining (Figure 3A and 3B). Thus, our human studies demonstrate, for the first time, that elevated CD73 and ADORA2B levels in the kidneys are associated with the severity of the disease.
Figure 3. Elevated renal 5′AMP ectonucleotidase (CD73) and A2B adenosine receptor (ADORA2B) levels seen in chronic kidney disease (CKD) patients with or without hypertension.
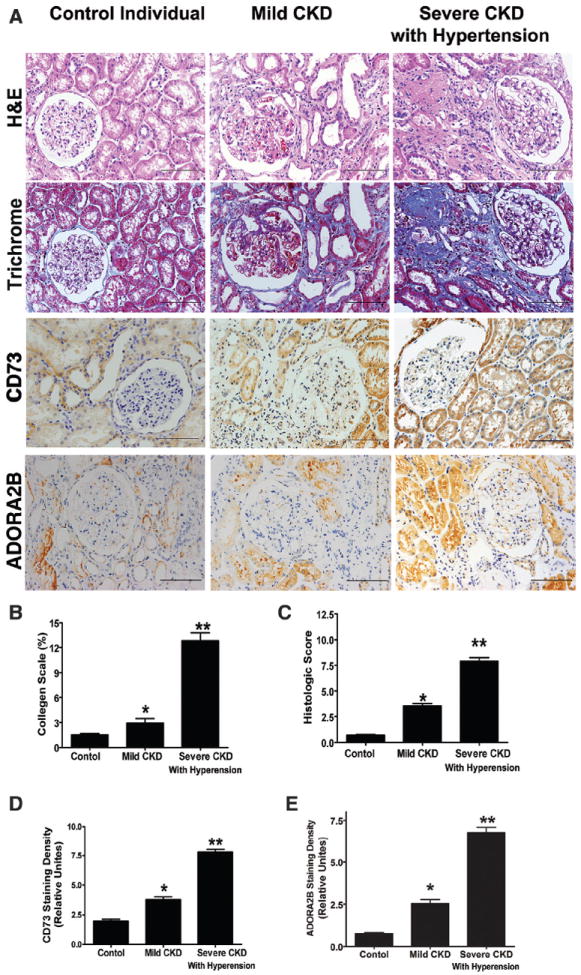
A, Renal histology and immunohistochemical analysis of CD73 and ADORA2B expression in control individuals (n=12), mild CKD patients without hypertension (n=24), and hypertensive patients with CKD (n=32; scale bar, 400 mm). B, Quantitative image analyses showed significantly increased collagen staining in kidneys of hypertensive patients with CKD. C, Histological analysis showed increased kidney injury in hypertensive patients with CKD. Expression of CD73 (D) and ADORA2B (E) in kidneys of control individuals, mild CKD patients without hypertension, and severe CKD patients with hypertension. Signal intensity was quantified using Image-Pro Plus image analysis software. Data are expressed as mean±SEM; *P<0.05 vs control individuals; **P<0.05 vs mild CKD.
ADORA2B Signaling Contributes to Increased Endothelin-1 Production in the Kidneys of Ang II–infused Mice
To identify signaling molecules functioning downstream of ADORA2B that contribute to hypertensive CKD, we re-examined the kidney gene expression profiles of control and Ang II–infused mice, with a particular focus on hypertensive mediators. We found that prepro-endothelin-1 (prepro-ET-1), a precursor of ET-1, a potent vasoconstrictor, was elevated most in the kidneys of Ang II–infused mice (Online Table I; Figure 1A). We confirmed our screening results by showing that prepro-ET-1 mRNA levels were significantly elevated in the kidneys of Ang II–infused mice (Figure 4A). Immunostaining and quantitative image analysis revealed that ET-1 protein levels were significantly elevated in the endothelial cells of glomeruli of Ang II–infused mice (Figure 4B and 4C). Previous studies showed that elevated ET-1 signaling plays an important role in hypertension, kidney dysfunction, and fibrosis in Ang II–infused mice.19-22 However, the role of elevated ADORA2B signaling in Ang II-mediated induction of ET-1 was not determined.
Figure 4. A2B adenosine receptor (ADORA2B)–mediated induction of hypoxia-inducible factor-1α (HIF-1α) regulates endothelin-1 (ET-1) production in the kidneys of angiotensin II (Ang II)–infused mice.

A, Quantitative reverse-transcription polymerase chain reaction (RT-PCR) measurement of prepro-ET-1 mRNA levels in wild-type (WT) mice, CD73−/− mice and Adora2b−/− mice with saline or Ang II infusion, and WT mice with PSB1115 treatment alone or with Ang II infusion. B, Immunohistochemical analysis of ET-1 expression in the kidneys of mice listed. C, Expression of ET-1 in the kidneys of mice listed in (B) was quantified using Image-Pro Plus image analysis software. D, HIF-1α mRNA levels were measured by quantitative RT-PCR in the kidneys of mice listed in A. E, Immunohistochemical analysis of HIF-1α expression in the kidneys of mice listed in (A). F, Expression of HIF-1α in the kidneys of mice listed in (E) was quantified using Image-Pro Plus image analysis software. Data are expressed as mean±SEM. *P<0.05 for Ang II–infused mice vs the control mice infused with saline; **P<0.05 for Adora2b−/−, CD73−/−, and WT mice with PSB11115 treatment infused with Ang II vs Ang II–infused WT mice (n=8–10 per group).
To test this possibility, we again used both genetic and pharmacological approaches. We found that Ang II–induced prepro-ET-1 mRNA production was significantly inhibited in the kidneys of ADORA2B-deficient mice and PSB1115-treated mice (Figure 4A). Similarly, immunostaining and quantitative image analysis further confirmed that ADORA2B deficiency and PSB1115 treatment significantly attenuated Ang II–induced ET-1 protein levels in endothelial cells of glomeruli of these mice (Figure 4B and 4C). Consistent with the findings of an important role for ADORA2B signaling in Ang II–induced production of ET-1 from kidneys, genetic deletion of CD73 also led to a significant reduction in prepro-ET-1 mRNA and protein levels in the kidneys of Ang II–infused mice (Figure 4A-4C). However, the endogenous ET system, including prepro-ET-1, ET receptor type 1A, ET receptor type 1B, and ET-converting enzyme, is intact in CD73-deficient mice (Online Figure VB). Taken together, these results revealed that CD73 activity and ADORA2B signaling contribute to the renal production of ET-1, a likely contributor to hypertension, CKD, and progression of fibrosis.
ADORA2B Signaling Via Hypoxia-Inducible Factor-1α Underlies Ang II–induced ET-1 Production in Mouse Kidneys
To determine what intracellular molecules functioning downstream of ADORA2B underlie Ang II–mediated ET-1 induction, we re-examined the kidney gene expression profiles. We found that hypoxia-inducible factor-1α (HIF-1α) was among the transcripts highly elevated in the kidneys of Ang II–infused mice (Online Table I; Figure 1A). Ang II–mediated induction of renal HIF-1α mRNA was confirmed by RT-PCR analysis (Figure 4D). Immunohistochemical analysis and image quantification studies demonstrated that HIF-1α protein levels were also significantly elevated in the glomeruli of kidneys of Ang II–infused mice (Figure 4E and 4F). Ang II–induced HIF-1α mRNA and protein levels in kidneys were significantly reduced in CD73-deficient mice, ADORA2B-deficient mice, and PSB1115-treated mice (Figure 4E and 4F). These studies provide in vivo evidence that elevated CD73-mediated adenosine induction and excess ADORA2B signaling are required for Ang II–mediated induction of HIF-1α.
Next, to assess the direct renal effect of excess adenosine signaling on HIF-1α and ET-1 induction, we isolated kidneys from WT mice, CD73-deficient mice, and ADORA2B-deficient mice to conduct experiments using kidney explant cultures.4,23 First, we treated kidney explants isolated from WT, CD73-deficient, and ADORA2B-deficient mice in the presence or absence of Ang II. Quantitative RT-PCR analysis showed that Ang II–induced prepro-ET-1 and HIF-1α mRNA levels were significantly reduced in the kidney explants of CD73−/− and Adora2b−/− mice compared with the WT mice (Figure 5A). These findings provide direct evidence for the importance of CD73 and ADORA2B in Ang II–induced HIF-1α and ET-1 production in the mouse kidneys.
Figure 5. A2B adenosine receptor (ADORA2B) functions downstream of angiotensin II (Ang II) responsible for prepro-endothelin-1 (prepro-ET-1) elevation in a hypoxia-inducible factor-1α (HIF-1α)–dependent manner in cultured mouse kidney explants and human endothelial cells.

A, Analysis of prepro-ET-1 and HIF-1α mRNA from cultured kidney explants isolated from wild-type (WT), CD73−/−, and Adora2b −/− mice in response to Ang II treatment. Data are expressed as mean±SEM. *P<0.05 vs without treatment (n=4). **P<0.05 vs WT treated with Ang II. B, 5′-N-ethylcarboxamidoadenosine (NECA) increased the expression of both prepro-ET-1 and HIF-1α mRNA levels in the WT mouse kidney explants, which were completely abolished in the Adora2b−/− mice. *P<0.05 vs without treatment. **P<0.05 vs NECA-treated controls (n=4–6). C, NECA-induced prepro-ET-1 mRNA expression in WT mouse kidney explants was attenuated by the HIF-1α antagonist (chrysin) and enhanced by the HIF-1α stabilizer dimethyloxalylglycine (DOMG). Data are expressed as mean±SEM. *P<0.05 vs without treatment; **P<0.05 vs NECA-treated controls (n=4–6). D, Ang II–mediated induction of HIF-1α and prepro-ET-1 mRNA. Data are expressed as mean±SEM. *P<0.05 vs untreated cells. **P<0.05 vs Ang II–treated cells. E, ADORA2B antagonist (MRS1706) but not A2A adenosine receptor antagonist (SCH442416) attenuated NECA-mediated elevation of both HIF-1α and prepro-ET-1 mRNA levels in human microvascular endothelial cells (HMECs). Data are expressed as mean±SEM. *P<0.05 vs the control without NECA treatment; **P<0.05 vs with NECA treatment alone (n=4–6). F, NECA-mediated induction of prepro-ET-1 mRNA was inhibited by stably knocking down HIF-1α in HMECs (HIF-1α-KD). Insert is a Western blot for HIF-1α and β-actin in control HMECs and HIF-1α-KD. Data are expressed as mean±SEM. *P<0.05 vs untreated control cells (n=4–6). **P<0.05 vs NECA-treated control cells (n=4–6).
Next, to determine whether adenosine signaling via ADORA2B activation directly induces HIF-1α and ET-1 production, we treated kidney explants isolated from both WT and Adora2b−/− mice with 5′-N-ethylcarboxamidoadenosine (NECA), a potent nonmetabolized adenosine analog.17 We found that NECA was capable of inducing both prepro-ET-1 and HIF-1α gene expression in cultured kidney explants from WT mice but not from Adora2b−/− mice (Figure 5B). Finally, to determine whether HIF-1α functioning downstream of ADORA2B is responsible for adenosine-induced ET-1 production, we treated kidney explants from WT mice with either chrysin (HIF-1α inhibitor) or dimethyloxalyl glycine (HIF-1α stabilizer). We found that chrysin significantly reduced NECA-induced prepro-ET-1 gene expression in WT mouse kidney explants (Figure 5C). In contrast, dimethyloxalyl glycine significantly enhanced NECA-induced prepro-ET-1 gene expression in WT mouse kidney explants (Figure 5C). Overall, our studies provide the direct evidence that ADORA2B-mediated HIF-1α induction contributes to Ang II–mediated induction of ET-1 in mouse kidneys.
ADORA2B-Mediated Induction of HIF-1α Underlies Ang II–induced ET-1 in Cultured Human Microvascular Endothelial Cells
HIF-1α and ET-1 were significantly elevated in the endothelial cells of the capillary lumens of kidneys of Ang II–infused mice (Figure 4B and 4E). These results suggest that microvascular endothelial cells are major cell types responsible for excessive adenosine-induced HIF-1α and ET-1 production. It is difficult to decipher the direct role of ADORA2B-mediated HIF-1α elevation in ET-1 induction in intact animals. Therefore, we extended our mouse studies to human microvascular endothelial cells (HMECs). Although these cells are not derived from kidneys, they represent a clonally derived source of HMECs useful to examine the role of adenosine signaling in Ang II–mediated induction of HIF-1α and ET-1. First, we found that Adora2b transcripts are the predominant adenosine receptor transcript expressed in HMECs (Online Figure VIIA). Next, we found that Ang II–mediated induction of both HIF-1α and prepro-ET-1 gene expression in cultured HMECs was significantly attenuated by pretreatment with either α,β-methylene ADP (α,β-methylene ADP [APCP], a CD73 inhibitor) or MRS1706 (an ADORA2B antagonist; Figure 5D). These findings provide the direct evidence that CD73 and ADORA2B play an important role in Ang II–mediated induction of HIF-1α and ET-1 mRNA in HMECs.
Subsequently, we assessed the direct role of adenosine signaling in HMECs. We demonstrated that NECA was capable of inducing both HIF-1α and prepro-ET-1 gene expression in cultured HMECs in a time-dependent manner (Online Figure VIIB and VIIC). In addition, we found that treatment of HMECs with either theophylline (a general AR antagonist) or MRS1706 (an ADORA2B-specific antagonist) significantly inhibited NECA-induced HIF-1α and prepro-ET-1 gene expression in these cells. However, SCH442416 (an A2AR-specific antagonist) had no effects on NECA-induced HIF-1α and prepro-ET-1 gene expression in HMECs (Figure 5E). These studies indicate that ADORA2B is a major receptor underlying excess adenosine-induced HIF-1α and prepro-ET-1 gene expression in endothelial cells.
Finally, to determine whether HIF-1α is required for adenosine-induced prepro-ET-1 gene expression, we generated HIF-1α–deficient HMECs by stably knocking down endogenous HIF-1α expression by means of a small interfering RNA strategy (HIF-1α-KD cells). First, we found that HIF-1α–specific siRNA significantly reduced HIF-1α protein levels in HIF-1α–KD HMEC cells (Figure 5F). More importantly, we found that NECA-induced prepro-ET-1 mRNA was significantly reduced in HIF-1α-KD cells compared with control cells (Figure 5F). Our findings show that ADORA2B activation can directly induce HIF-1α gene expression, and that HIF-1α is essential for adenosine-mediated ET-1 induction in human endothelial cells.
HIF-1α Contributes to Ang II–induced CD73 and Adora2b Gene Expression in Both Cultured Mouse Kidneys and Human Endothelial Cells at Transcriptional Levels
We have revealed that HIF-1α functioning downstream of ADORA2B is directly responsible for Ang II–induced ET-1 production by mouse kidneys and human endothelial cells. HIF-1α is known to be involved in increased CD73 and Adora2b gene expression under hypoxic conditions.24,25 However, whether Ang II is a hypoxia-independent mediator directly inducing HIF-1α levels and thereby responsible for Ang II-induced CD73 and Adora2b expression remains unknown. To test this possibility, we isolated kidneys from WT mice and conducted kidney organ culture as described.4,23 Quantitative RT-PCR analysis indicated that Ang II treatment directly induced both CD73 and Adora2b gene expression (Figure 6A). Moreover, we found that chrysin, a HIF-1α inhibitor, significantly reduced Ang II–induced CD73 and Adora2b gene expression in WT mouse kidney explants (Figure 6A). In contrast, dimethyloxalyl glycine, a HIF-1α stabilizer, significantly enhanced Ang II–induced CD73 and Adora2b gene expression in WT mouse kidney explants (Figure 6A). Consistently, we found that Ang II significantly induced CD73 and Adora2b mRNA in the control HMEC cells (Figure 6B). However, in the HIF-1α knockdown cells (HMECs-HIF-1α KD), Ang II–induced CD73 and Adora2b mRNA levels were significantly reduced (Figure 6B). Overall, our studies demonstrate that Ang II is a previously unrecognized hypoxia-independent mediator directly inducing HIF-1α levels, and elevated HIF-1α is an important transcription factor responsible for Ang II–mediated induction of CD73 and Adora2b mRNA levels in cultured mouse kidneys and HMECs.
Figure 6. Adenosine-mediated elevation of hypoxia-inducible factor-1α (HIF-1α) is responsible for angiotensin II (Ang II)–induced elevation of CD73 and A2B adenosine receptor (Adora2b) gene expression at the transcriptional level.
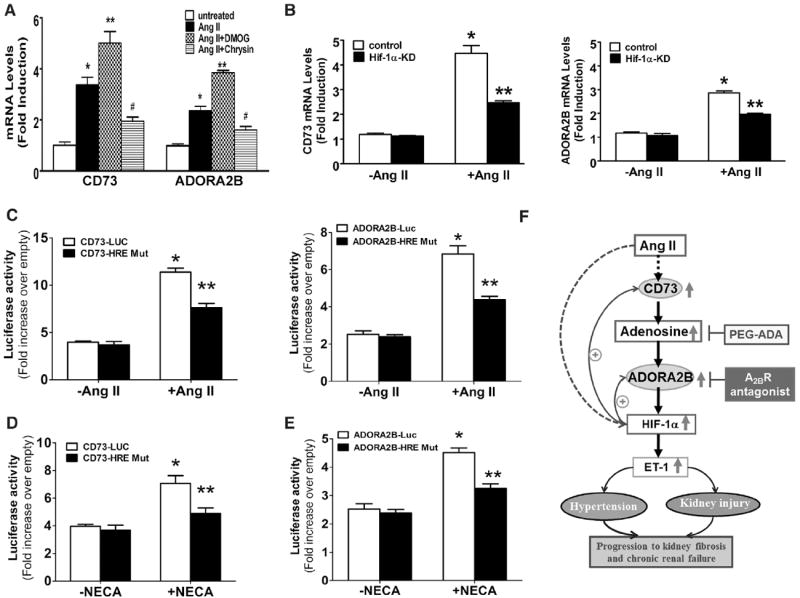
A, Ang II–induced CD73 and Adora2b mRNA expression in wild-type (WT) mouse kidney explants was attenuated by the HIF-1α antagonist (chrysin) and enhanced by HIF-1α stabilizer (DOMG). Data are expressed as mean±SEM. *P<0.05 vs without treatment; **P<0.05 vs treatment with Ang II alone (n=6). B, Ang II–induced elevation of CD73 and Adora2b mRNA were inhibited by stably knocking down HIF-1α in human microvascular endothelial cells (HMECs) (HIF-1α-KD). Data are expressed as mean±SEM. *P<0.05 vs untreated control cells (n=4–6). **P<0.05 vs 5′-N-ethylcarboxamidoadenosine (NECA)-treated control cells (n=6). C–E, CD73 and ADORA2B luciferase reporter assays. Confluent HMECs were transiently transfected with plasmids expressing sequence corresponding to WT CD73 promoter (pGL20.57NT), the ADORA2B promoter (pGL2), mutated CD73 promoter with the mutation of HIF-1α–responsive element (HRE) site (pGL20.57NT), and mutated ADORA2B promoter region with the mutation of HRE site, as well as with the empty pGL2. Twelve hours later, cells were switched to serum-free medium and were treated with or without Ang II (200 nmol/L; C) or NECA (10 mmol/L; D and E) for 24 h. Data are expressed as mean±SEM. *P<0.05 vs without treatment; **P<0.05 vs treatment with Ang II or NECA alone (n=6). F, Working model for excess renal adenosine in hypertension and its progression. Ang II–induced hypertensive nephropathy in mice is associated with a malicious cycle of adenosine signaling in the kidneys characterized by elevated CD73 production and ADORA2B signaling. Excessive ADORA2B signaling stimulates the production of HIF-1α, a positive transcriptional regulator responsible for increased expression of the endothelin-1 (ET-1) gene. HIF-1α, which is stabilized by hypoxia, also promotes increased expression of the genes encoding CD73 and ADORA2B, and in this way serves to perpetuate the malicious cycle of adenosine signaling resulting in excessive ET-1 production. Lowering chronically elevated renal adenosine or interfering ADORA2B activation are likely novel therapeutic possibilities to treat hypertensive renal disease and slow its progression.
Next, we extended mouse studies to cultured HMECs. Specifically, we incubated both control and HIF-1α knockdown cells (HMECs-HIF-1α-KD) with or without Ang II treatment. The results (Figure 6B) show that HIF-1α underlies Ang II–mediated induction of CD73 and Adora2b mRNA levels.
Both CD73 and Adora2b promoters contain a HIF-1α–responsive element (HRE). Thus, we hypothesize that HIF-1α underlies Ang II–induced CD73 and Adora2b gene expression at the transcriptional level. To test this hypothesis, we conducted transfection assays using luciferase reporter genes introduced into in HMECs in the presence or absence of Ang II. We found that Ang II–induced WT CD73 and Adora2b promoter activities compared with untreated controls (Figure 6C). However, deletion of HRE site in either CD73 or Adora2b promoter significantly reduced Ang II–induced CD73 and Adora2b promoter activity (Figure 6C). Thus, these results provide direct evidence that HREs in the promoters of both CD73 and Adora2b are essential for Ang II–induced transcriptional activation of these 2 genes.
HIF-1α Is Responsible for NECA-induced CD73 and Adora2b Gene Expression in Cultured Human Endothelial Cells at Transcriptional Levels
Finally, we determined whether adenosine-induced HIF-1α is responsible for activating CD73 and Adora2b gene expression at transcriptional levels. Similarly, we conducted transfection assays using luciferase reporter genes introduced into HMECs with or without NECA treatment. We found that NECA induced WT CD73 and Adora2b promoter activities compared with untreated controls (Figure 6D-6F). However, deletion of the HRE in either the CD73 or the Adora2b promoter significantly reduced NECA-induced CD73 and Adora2b promoter activity (Figure 6D and 6E). Thus, these results provide direct evidence that the HRE in the CD73 and Adora2b promoters is essential for NECA-induced transcriptional activation of these 2 genes. Taken together, we revealed that adenosine-mediated HIF-1α induction underlies increased CD73 and Adora2b transcriptional activities and functions as a positive feedback to further promote Ang II–mediated elevation of adenosine and to amplify ADORA2B signaling (Figure 6F).
Discussion
In this study, we report that CD73, a critical enzyme in extracellular adenosine production, and ADORA2B, are elevated in kidneys of both mice and humans with hypertension and CKD. Using an Ang II infusion model of hypertensive renal disease in mice, we provide both genetic and pharmacological evidence that elevated renal CD73 contributes to hypertension, proteinuria, decreased tubular concentration ability, and progression of the disease by the excessive production of adenosine in the kidneys. Pharmacological inhibition or genetic disruption of the adenosine receptor ADORA2B significantly attenuates Ang II–induced hypertension. Mechanistically, we determined that HIF-1α is a key downstream mediator underlying excess renal adenosine–induced expression of ET-1, a potent vasoconstrictor. Finally, we revealed that HIF-1α is an important factor responsible for Ang II–induced CD73 and Adora2b expression at the transcriptional level. Without interference, elevated CD73-mediated increased renal adenosine functioning via amplified ADORA2B signaling further enhanced HIF-1α induction, resulting in additional elevation of renal ET-1 production. Thus, AngII–HIF-1α–CD73–ADORA2B functions as a malicious cycle to facilitate ET-1 production in the kidney and leads to hypertension (Figure 6F). Overall, our findings reveal a previously unrecognized detrimental role of chronically elevated renal CD73 levels and ADORA2B signaling in Ang II–induced hypertensive CKD and thereby identify novel and important therapeutic possibilities for the disease.26
Multiple factors are released from locally insulted renal tissue and responding cells. One of the best-known signaling molecules to be induced under hypoxic conditions is adenosine.27 Adenosine protects tissues like the brain,28,29 intestine,30 and heart31 from acute ischemic damage, thereby exhibiting chemoprotective properties.32-35 However, in the setting of repeated or prolonged tissue injury, chronic elevation of adenosine becomes detrimental by promoting or exacerbating tissue injury and dysfunction in the lung,36 penis,24 and kidney,4 and in sickle cell disease.9 Of note, it has been speculated that increased adenosine levels under acute hypoxic conditions may be beneficial by increasing blood flow to ischemic or hypoxic tissues because of the potent vasodilatory effects in most organs. However, in the kidney, adenosine causes afferent arterial constriction and thereby decreases blood flow to the kidney, which is speculated to be a compensatory effect to reduce transport workload in the kidney under acute hypoxic conditions. Thus, persistent elevation of adenosine in the kidney may be detrimental because of prolonged vasoconstriction of afferent arterioles and subsequent severe ischemic renal injury. Notably, previous studies reported that Ang II infusion induces renal adenosine elevation, but not systemically, in rats.37 In addition, a previous report showed that intrarenal adenosine produces hypertension in the dog by activating the sympathetic nervous system.38 More recent studies showed that adenosine leads to increased norepinephrine production from the renal sympathetic nervous system via the A1 adenosine receptor.33 However, specific renal factors causing chronic elevation of adenosine in Ang II–infused mice remained unidentified.
High-throughput quantitative RT-PCR array analysis allowed us to identify elevated renal CD73 accounting for increased kidney adenosine in Ang II–infused mice and revealed the detrimental consequences of enhanced ADORA2B signaling in this mouse model of hypertensive renal disease. Our studies have significantly enhanced our understanding of pathogenesis of hypertensive CKD and are strongly supported by previously studies. For example, previous reports have shown that Ang II infusion results in increased levels of renal interstitial ATP and adenosine.40,41 The adenosine is presumably derived from ATP by the consecutive action of CD39, an ectonucleotidase that converts ATP→ADP→AMP and CD73, which converts AMP to adenosine. Our genetic and pharmacological data showing the importance of CD73 in Ang II–induced accumulation of renal adenosine are in good agreement with previous studies by Franco et al indicating that the Ang II–induced elevation of renal adenosine was significantly inhibited by specific blockade of 5′-nucleotidase.41 It is especially noteworthy that Ang II–induced hypertension was not blocked by drugs that inhibit adenine nucleotide–mediated (ie, ATP, ADP) P2 receptor signaling.40 Consistent with these findings, we present multiple lines of genetic and pharmacological evidence that Ang II–induced hypertension and CKD require adenosine signaling. We show that Ang II–induced hypertensive CKD is reduced in CD73-deficient mice, that Ang II–induced hypertensive nephropathy is reduced by treatment with polyethyleneglycol-modified ADA to lower adenosine, and that Ang II–induced hypertensive renal disease is reduced by pharmacological blockage or genetic deficiency of the adenosine receptor, ADORA2B. Our data are the first to show the importance of adenosine signaling in Ang II–induced hypertensive CKD and, in this way, have identified a previously unrecognized pathway for the pathophysiology of hypertension and CKD.
Considerably more is known about the role of the A1 adenosine receptor and A2A adenosine receptor receptors in renal physiology than is known about ADORA2B receptor signaling.42,43 Previous studies have shown that when rats are maintained on a high-salt diet, adenosine production increases in the medulla associated with an increase in A2 compared with A1 receptor expression.44,45 However, these early studies did not distinguish A2A from A2B receptors. More recently, high levels of ADORA2B expression have been observed in the preglomerular microvessels46 in the cortical thick ascending limb of Henle and in the distal convoluted tubule47 and in inner medullary collecting duct cells.48 The last study shows that adenosine activates A2B receptors and enhances chloride secretion in kidney inner medullary collecting duct cells.48 Several animal studies indicate that CD73-dependent adenosine production plays a protective role in ischemia-induced acute renal injury by antiendothelial leakage and anti-inflammatory effects.27,46,49 However, the role of chronically elevated renal adenosine signaling via adenosine receptors in hypertension and progression of CKD remained largely unknown until our studies reported here. Our genetic and pharmacological studies demonstrate that elevated renal adenosine functioning via ADORA2B signaling is responsible for Ang II–mediated hypertensive nephropathy, likely because of the induction of ET-1, a potent vasoconstrictor known to contribute to hypertensive CKD and its progression.50 Although the ADORA2B has the lowest affinity for adenosine among the 4 adenosine receptors,17,18 in the setting of hypertensive CKD, elevated CD73 produces excessive renal adenosine, resulting in enhanced activation of ADORA2B. Ultimately, excessive ADORA2B signaling becomes detrimental by stimulating HIF-1α production, leading to excessive production of renal ET-1. Although many studies have demonstrated the importance of elevated ET-1 in the pathophysiology of kidney disease,50 the role of adenosine signaling and the importance of CD73 in Ang II–induced hypertensive nephropathy are the important new findings provided in the present article. Thus, our studies raise a novel concept in which, in the short-term or acute setting, elevated adenosine may be beneficial to the kidney by inducing afferent arterial constriction to reduce workload via A1 adenosine receptor signaling51 and stimulating an anti-inflammatory response or maintaining endothelial barrier via A2A adenosine receptor signaling.27 However, in the setting of CKD, chronically elevated renal adenosine coupled with excessive ADORA2B signaling becomes detrimental by inducing renal production of ET-1 to promote hypertension and exacerbate kidney injury and disease progression (Figure 6F).
Ischemia and hypoxia have long been considered to be associated with CKD and are speculated to contribute to progression of the disease, including hypertension and renal fibrosis.52,53 Of note, HIF-1α is well-known to be induced under hypoxia and associated with hypertensive CKD.54 As with adenosine signaling, HIF-1α signaling can be tissue-protective, whereas chronic elevated HIF-1α also can be detrimental.27,55-57 The detrimental effects of prolonged elevation of HIF-1α in CKD has been revealed by studies showing that HIF-1α contributes to renal injury and progression of renal fibrosis in multiple animal models, including Ang II–infused rats.37 Although hypoxia is known to increase HIF-1α, a growing body of evidence indicates that hypoxia-independent mediators, including inflammatory cytokines and growth factors, also stimulate HIF-1α gene activity.58,59 Because baseline oxygen tension in the kidney is already low, Ang II infusion is unlikely to cause substantial additional hypoxia. Here, we show that Ang II treatment directly induces HIF-1α levels in the absence of hypoxia in cultured endothelial cells. Significantly, we further provide in vivo evidence that Ang II infusion leads to elevation of HIF-1α in endothelial cells of glomeruli in mice. Thus, both in vitro and in vivo studies indicate that Ang II is a novel hypoxia-independent mediator directly regulating HIF-1α levels in the kidney. Mechanistically, our studies provide both in vivo animal and in vitro human evidence that elevated CD73-mediated prolonged elevation of renal adenosine coupled with enhanced ADORA2B signaling stimulates the production of HIF-1α, a positive transcriptional regulator responsible for increased expression of the ET-1 gene. Of note, our previous studies revealed that interleukin-6 is an important mediator underlying Ang II–induced ET-1 production in the kidneys and hypertension.23 Thus, it is possible that HIF-1α and interleukin-6 may work together to regulate Ang II–mediated ET-1 induction, an important issue for future studies. Besides ET-1, we further revealed that HIF-1α is important for Ang II–promoted expression of the genes encoding CD73 and ADORA2B at the transcriptional level and serves to perpetuate the malicious cycle of adenosine signaling, resulting in excessive ET-1 production (Figure 6F). Thus, the evidence provided by our human and mouse studies supports a novel working model that chronic elevated renal adenosine contributes to hypertensive CKD via HIF-1α–mediated gene regulation in the following 2 ways: (1) by inducing ET-1 production to promote hypertension and kidney injury; and (2) by inducing CD73 and Adora2b gene expression to further enhance adenosine production and preferentially amplifying HIF-1α signaling in a malicious cycle that promotes the progression of the disease (Figure 6F). Thus, interfering with this detrimental cycle is important for novel therapeutics in the treatment of hypertension and renal disease.
Supplementary Material
Novelty and Significance.
What Is Known?
Hypertension is the most prevalent life-threatening disease worldwide and is frequently associated with chronic kidney disease (CKD).
The renin–angiotensin system is a key signaling cascade contributing to hypertension, CKD, and progression to renal fibrosis.
Elevated levels of angiotensin II (Ang II), the primary effector of the renin–angiotensin system, contribute to hypertension and CKD.
What New Information Does This Article Contribute?
We used an Ang II infusion model of hypertensive renal disease in mice to identify new signaling pathways that contribute to disease pathogenesis.
Our findings reveal important novel opportunities for therapeutic intervention for the treatment of hypertension and renal disease.
We sought to identify new factors and signaling pathways that contribute to hypertensive CKD in an effort to reveal novel therapeutic approaches for treatment. For our studies, we used the well-accepted Ang II infusion model of hypertension in mice. To identify specific factors and signaling pathways contributing to the pathogenesis of CKD and hypertension, we used a high-throughput analytic screening strategy to identify differences in patterns of gene expression in the kidneys of control mice and those infused with Ang II. Unexpectedly, our results revealed that a family of genes associated with a particular signaling pathway (adenosine signaling) was expressed at elevated levels in the Ang II–infused mice. A role for adenosine signaling in Ang II–induced hypertension previously has not been recognized. Our research findings revealed new therapeutic targets that we explored in the Ang II infusion model of hypertensive renal disease in mice. We tested a variety of pharmacological approaches to inhibit the pathophysiological consequences of excessive adenosine signaling and found that each approach resulted in reduced blood pressure and renal damage and reduced progression to renal fibrosis. Thus, our preclinical studies reveal potentially important novel opportunities for therapeutic intervention in the treatment of hypertension and renal disease.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health Grants HL076558, DK077748, DK083559, and HL113574 (to Y. Xia), RC4HD067977, and HD34130 (to Y. Xia and R.E. Kellems), by American Heart Association Grant 10GRNT3760081 (to Y. Xia), by China Scholarship Council Grant 2009637520 (to W. Zhang), HL070952 (to M. R. Blackburn), HL092188 (to H. K. Eltzschig), and DK080236 (to W. Zhang) by China National Natural Science Foundation grant 81228004 (to Y. Xia).
Nonstandard Abbreviations and Acronyms
- ADORA2B
A2B adenosine receptor
- Ang II
angiotensin II
- CD73
5′AMP ectonucleotidase
- CKD
chronic kidney disease
- ET-1
endothelin-1
- HIF-1α
hypoxic-inducible factor 1α
- HMEC
human microvascular endothelial cell
- HRE
HIF-1α–responsive element
- WT
wild-type
Footnotes
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.111.300166/-/DC1.
Disclosures
None.
References
- 1.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 2.Khosla N, Kalaitzidis R, Bakris GL. The kidney, hypertension, and remaining challenges. Med Clin North Am. 2009;93:697–715. doi: 10.1016/j.mcna.2009.02.001. Table of Contents. [DOI] [PubMed] [Google Scholar]
- 3.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 4.Dai Y, Zhang W, Wen J, Zhang Y, Kellems RE, Xia Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J Am Soc Nephrol. 2011;22:890–901. doi: 10.1681/ASN.2010080890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khawaja Z, Wilcox CS. Role of the kidneys in resistant hypertension. Int J Hypertens. 2011;2011:143471. doi: 10.4061/2011/143471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rüster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol. 2011;22:1189–1199. doi: 10.1681/ASN.2010040384. [DOI] [PubMed] [Google Scholar]
- 7.Kalaitzidis RG, Bakris GL. The current state of RAAS blockade in the treatment of hypertension and proteinuria. Curr Cardiol Rep. 2009;11:436–442. doi: 10.1007/s11886-009-0063-3. [DOI] [PubMed] [Google Scholar]
- 8.Sica DA. Pharmacologic Issues in treating hypertension in CKD. Adv Chronic Kidney Dis. 2011;18:42–47. doi: 10.1053/j.ackd.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Dai Y, Wen J, et al. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17:79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou CC, Ahmad S, Mi T, Xia L, Abbasi S, Hewett PW, Sun C, Ahmed A, Kellems RE, Xia Y. Angiotensin II induces soluble fms-like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ Res. 2007;100:88–95. doi: 10.1161/01.RES.0000254703.11154.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hackam DG, Spence JD, Garg AX, Textor SC. Role of renin-angiotensin system blockade in atherosclerotic renal artery stenosis and renovascular hypertension. Hypertension. 2007;50:998–1003. doi: 10.1161/HYPERTENSIONAHA.107.097345. [DOI] [PubMed] [Google Scholar]
- 12.Luft FC, Dechend R, Müller DN. Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med. 2012;44(Suppl 1):S49–S54. doi: 10.3109/07853890.2011.653396. [DOI] [PubMed] [Google Scholar]
- 13.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 15.Blackburn MR, Kellems RE. Adenosine deaminase deficiency: metabolic basis of immune deficiency and pulmonary inflammation. Adv Immunol. 2005;86:1–41. doi: 10.1016/S0065-2776(04)86001-2. [DOI] [PubMed] [Google Scholar]
- 16.Hershfield MS. PEG-ADA replacement therapy for adenosine deaminase deficiency: an update after 8.5 years. Clin Immunol Immunopathol. 1995;76:S228–S232. doi: 10.1016/s0090-1229(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 17.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 18.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 19.Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, Giaid A, Harrison DG. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension. 1997;30:29–34. doi: 10.1161/01.hyp.30.1.29. [DOI] [PubMed] [Google Scholar]
- 20.Kohan DE. Endothelin, hypertension and chronic kidney disease: new insights. Curr Opin Nephrol Hypertens. 2010;19:134–139. doi: 10.1097/MNH.0b013e328335f91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minchenko A, Caro J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Mol Cell Biochem. 2000;208:53–62. doi: 10.1023/a:1007042729486. [DOI] [PubMed] [Google Scholar]
- 22.Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest. 1991;88:1054–1057. doi: 10.1172/JCI115367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59:136–144. doi: 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi T, Abbasi S, Zhang H, Uray K, Chunn JL, Xia LW, Molina JG, Weisbrodt NW, Kellems RE, Blackburn MR, Xia Y. Excess adenosine in murine penile erectile tissues contributes to priapism via A2B adenosine receptor signaling. J Clin Invest. 2008;118:1491–1501. doi: 10.1172/JCI33467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hannedouche T, Krummel T, Parvez-Braun L. Nephroprotection: how to slow the progression of chronic renal insufficiency? Nephrol Ther. 2005;1:135–144. doi: 10.1016/j.nephro.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Adenosine and brain ischemia. Cerebrovasc Brain Metab Rev. 1992;4:346–369. [PubMed] [Google Scholar]
- 29.Schubert P, Rudolphi KA, Fredholm BB, Nakamura Y. Modulation of nerve and glial function by adenosine–role in the development of ischemic damage. Int J Biochem. 1994;26:1227–1236. doi: 10.1016/0020-711x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 30.Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182:4957–4964. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lasley RD, Rhee JW, Van Wylen DG, Mentzer RM., Jr Adenosine A1 receptor mediated protection of the globally ischemic isolated rat heart. J Mol Cell Cardiol. 1990;22:39–47. doi: 10.1016/0022-2828(90)90970-d. [DOI] [PubMed] [Google Scholar]
- 32.Fishman P, Bar-Yehuda S, Farbstein T, Barer F, Ohana G. Adenosine acts as a chemoprotective agent by stimulating G-CSF production: a role for A1 and A3 adenosine receptors. J Cell Physiol. 2000;183:393–398. doi: 10.1002/(SICI)1097-4652(200006)183:3<393::AID-JCP12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 33.Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol. 2005;67:1385–1387. doi: 10.1124/mol.105.011783. [DOI] [PubMed] [Google Scholar]
- 34.Lu Q, Harrington EO, Newton J, Casserly B, Radin G, Warburton R, Zhou Y, Blackburn MR, Rounds S. Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am J Physiol Lung Cell Mol Physiol. 2010;298:L755–L767. doi: 10.1152/ajplung.00330.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. doi: 10.1016/j.it.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franco M, Bautista R, Pérez-Méndez O, González L, Pacheco U, Sánchez-Lozada LG, Santamaría J, Tapia E, Monreal R, Martínez F. Renal interstitial adenosine is increased in angiotensin II-induced hypertensive rats. Am J Physiol Renal Physiol. 2008;294:F84–F92. doi: 10.1152/ajprenal.00123.2007. [DOI] [PubMed] [Google Scholar]
- 38.Katholi RE, Whitlow PL, Hageman GR, Woods WT. Intrarenal adenosine produces hypertension by activating the sympathetic nervous system via the renal nerves in the dog. J Hypertens. 1984;2:349–359. [PubMed] [Google Scholar]
- 39.Jackson EK, Cheng D, Tofovic SP, Mi Z. Endogenous adenosine contributes to renal sympathetic neurotransmission via postjunctional A1 receptor-mediated coincident signaling. Am J Physiol Renal Physiol. 2012;302:F466–F476. doi: 10.1152/ajprenal.00495.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graciano ML, Nishiyama A, Jackson K, Seth DM, Ortiz RM, Prieto-Carrasquero MC, Kobori H, Navar LG. Purinergic receptors contribute to early mesangial cell transformation and renal vessel hypertrophy during angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2008;294:F161–F169. doi: 10.1152/ajprenal.00281.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franco M, Bautista R, Tapia E, Soto V, Santamaría J, Osorio H, Pacheco U, Sánchez-Lozada LG, Kobori H, Navar LG. Contribution of renal purinergic receptors to renal vasoconstriction in angiotensin II-induced hypertensive rats. Am J Physiol Renal Physiol. 2011;300:F1301–F1309. doi: 10.1152/ajprenal.00367.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vallon V, Osswald H. Adenosine receptors and the kidney. Handb Exp Pharmacol. 2009:443–470. doi: 10.1007/978-3-540-89615-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vallon V, Mühlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev. 2006;86:901–940. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 44.Siragy HM, Linden J. Sodium intake markedly alters renal interstitial fluid adenosine. Hypertension. 1996;27:404–407. doi: 10.1161/01.hyp.27.3.404. [DOI] [PubMed] [Google Scholar]
- 45.Zou AP, Wu F, Li PL, Cowley AW., Jr Effect of chronic salt loading on adenosine metabolism and receptor expression in renal cortex and medulla in rats. Hypertension. 1999;33:511–516. doi: 10.1161/01.hyp.33.1.511. [DOI] [PubMed] [Google Scholar]
- 46.Jackson EK, Zhu C, Tofovic SP. Expression of adenosine receptors in the preglomerular microcirculation. Am J Physiol Renal Physiol. 2002;283:F41–F51. doi: 10.1152/ajprenal.00232.2001. [DOI] [PubMed] [Google Scholar]
- 47.Vitzthum H, Weiss B, Bachleitner W, Krämer BK, Kurtz A. Gene expression of adenosine receptors along the nephron. Kidney Int. 2004;65:1180–1190. doi: 10.1111/j.1523-1755.2004.00490.x. [DOI] [PubMed] [Google Scholar]
- 48.Rajagopal M, Pao AC. Adenosine activates a2b receptors and enhances chloride secretion in kidney inner medullary collecting duct cells. Hypertension. 2010;55:1123–1128. doi: 10.1161/HYPERTENSIONAHA.109.143404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoffman A, Grossman E, Abassi ZA, Keiser HR. Renal endothelin and hypertension. Nature. 1994;372:50. doi: 10.1038/372050a0. [DOI] [PubMed] [Google Scholar]
- 51.Franco M, Bell PD, Navar LG. Effect of adenosine A1 analogue on tubuloglomerular feedback mechanism. Am J Physiol. 1989;257:F231–F236. doi: 10.1152/ajprenal.1989.257.2.F231. [DOI] [PubMed] [Google Scholar]
- 52.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74:867–872. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- 53.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nangaku M, Eckardt KU. Hypoxia and the HIF system in kidney disease. J Mol Med (Berl) 2007;85:1325–1330. doi: 10.1007/s00109-007-0278-y. [DOI] [PubMed] [Google Scholar]
- 55.Haase VH. Pathophysiological Consequences of HIF Activation: HIF as a modulator of fibrosis. Ann N Y Acad Sci. 2009;1177:57–65. doi: 10.1111/j.1749-6632.2009.05030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Q, Wang Z, Xia M, Li PL, Van Tassell BW, Abbate A, Dhaduk R, Li N. Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in Sprague-Dawley rats. Hypertension. 2011;58:657–664. doi: 10.1161/HYPERTENSIONAHA.111.177626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou J, Brüne B. Cytokines and hormones in the regulation of hypoxia inducible factor-1alpha (HIF-1alpha) Cardiovasc Hematol Agents Med Chem. 2006;4:189–197. doi: 10.2174/187152506777698344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.