

Abstract
Ischemia and reperfusion–elicited tissue injury contributes to morbidity and mortality in a wide range of pathologies, including myocardial infarction, ischemic stroke, acute kidney injury, trauma, circulatory arrest, sickle cell disease and sleep apnea. Ischemia-reperfusion injury is also a major challenge during organ transplantation and cardiothoracic, vascular and general surgery. An imbalance in metabolic supply and demand within the ischemic organ results in profound tissue hypoxia and microvascular dysfunction. Subsequent reperfusion further enhances the activation of innate and adaptive immune responses and cell death programs. Recent advances in understanding the molecular and immunological consequences of ischemia and reperfusion may lead to innovative therapeutic strategies for treating patients with ischemia and reperfusion–associated tissue inflammation and organ dysfunction.
Ischemia and reperfusion is a pathological condition characterized by an initial restriction of blood supply to an organ followed by the subsequent restoration of perfusion and concomitant reoxygenation. In its classic manifestation, occlusion of the arterial blood supply is caused by an embolus and results in a severe imbalance of metabolic supply and demand, causing tissue hypoxia. Perhaps surprisingly, restoration of blood flow and reoxygenation is frequently associated with an exacerbation of tissue injury and a profound inflammatory response1 (called ‘reperfusion injury’). Ischemia and reperfusion injury contributes to pathology in a wide range of conditions (Table 1). For example, cardiac arrest and other forms of trauma are associated with ischemia of multiple organs and subsequent reperfusion injury when blood flow is restored. Cyclic episodes of airway obstruction during obstructive sleep apnea also lead to hypoxia with subsequent reoxygenation on arousal2. Similarly, individuals with sickle cell disease have periodic episodes of painful vaso-occlusion and subsequent reperfusion with many characteristics that resemble ischemia and reperfusion3. Exposure of a single organ to ischemia and reperfusion (for example, the liver) may subsequently cause inflammatory activation in other organs (for example, the intestine), eventually leading to multiorgan failure4. However, it is important to point out that ischemic syndromes are a heterogeneous group of conditions. Although there are some similarities in the biological responses among these syndromes, there are important differences between a systemic reduction in perfusion (for example, during shock) compared to regional ischemia and reperfusion of a single organ (or differences between warm ischemia—as occurs, for example, during myocardial ischemia and reperfusion—and cold ischemic conditions—such as those that occur during organ transplantation when the organ is cooled with a cold perfusion solution following procurement).
Table 1.
Examples of ischemia and reperfusion injury
| Affected organ | Example of clinical manifestation |
|---|---|
| Single-organ ischemia and reperfusion
| |
| Heart | Acute coronary syndrome |
| Kidney | Acute kidney injury |
| Intestine | Intestinal ischemia and reperfusion; multiorgan failure |
| Brain | Stroke |
| Multiple-organ ischemia and reperfusion
| |
| Trauma and resuscitation | Multiple organ failure; acute kidney injury; intestinal injury |
| Circulatory arrest | Hypoxic brain injury; multiple organ failure; acute kidney injury |
| Sickle cell disease | Acute chest syndrome; pulmonary hypertension, priapism, acute kidney injury |
| Sleep apnea | Hypertension; diabetes |
| Ischemia and reperfusion during major surgery
| |
| Cardiac surgery | Acute heart failure after cardiopulmonary bypass |
| Thoracic surgery | Acute lung injury |
| Peripheral vascular surgery | Compartment syndrome of extremity |
| Major vascular surgery | Acute kidney injury |
| Solid organ transplantation | Acute graft failure; early graft rejection |
Indeed, a wide range of pathological processes contribute to ischemia and reperfusion associated tissue injury (Fig. 1). For example, limited oxygen availability (hypoxia) as occurs during the ischemic period is associated with impaired endothelial cell barrier function5 due to decreases in adenylate cyclase activity and intracellular cAMP levels and a concomitant increase in vascular permeability and leakage6. In addition, ischemia and reperfusion leads to the activation of cell death programs, including apoptosis (nuclear fragmentation, plasma membrane blebbing, cell shrinkage and loss of mitochondrial membrane potential and integrity), autophagy-associated cell death (cytoplasmic vacuolization, loss of organelles and accumulation of vacuoles with membrane whorls) and necrosis (progressive cell and organelle swelling, plasma membrane rupture and leakage of proteases and lysosomes into the extracellular compartment)7. The ischemic period in particular is associated with significant alterations in the transcriptional control of gene expression (transcriptional reprogramming). For example, ischemia is associated with an inhibition of oxygen-sensing prolylhydroxylase (PHD) enzymes because they require oxygen as a cofactor. Hypoxia-associated inhibition of PHD enzymes leads to the post-translational activation of hypoxia and inflammatory signaling cascades, which control the stability of the transcription factors hypoxia-inducible factor (HIF) and nuclear factor-κB (NF-κB), respectively8. Despite successful reopening of the vascular supply system, an ischemic organ may not immediately regain its perfusion (no reflow phenomenon). Moreover, reperfusion injury is characterized by autoimmune responses, including natural antibody recognition of neoantigens and subsequent activation of the complement system (autoimmunity)9. Despite the fact that ischemia and reperfusion typically occurs in a sterile environment, activation of innate and adaptive immune responses occurs and contributes to injury, including activation of pattern-recognition receptors such as TLRs and inflammatory cell trafficking into the diseased organ (innate and adaptive immune activation)10.
Figure 1.
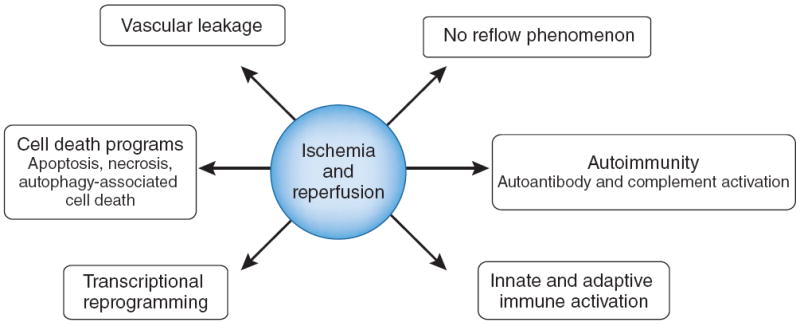
Biological processes implicated in ischemia and reperfusion.
In this review, we highlight recent studies that provide new insight into the molecular and immunological pathways of ischemia and reperfusion, as well as discuss examples of innovative therapeutic approaches based on these mechanistic findings (Table 2).
Table 2.
Examples of promising therapeutic approaches targeting ischemia and reperfusion
| Intervention | Target | Potential downside | Stage | Reference |
|---|---|---|---|---|
| TAK-242 | Inhibition of TLR4 | Immune suppression, worsening of bacterial infections | Phase 2 clinical trial in acute respiratory failure; preclinical studies in ischemia and reperfusion | 25,26 |
| T cell–based approaches | Suppression of γδ T cells; expansion of Treg cells | Unclear | Preclinical | 37,39,40,42 |
| Fibrinogen split product Bβ15–42 | Unclear | Unclear | Phase 2 clinical trial | 15,56,57 |
| Cyclosporine | Inhibition of apoptosis | Immune suppression; worsening of bacterial infection | Phase 2 clinical trial | 66 |
| Chloramphenicol | Activation of autophagy | Bone marrow toxicity (bone marrow suppression or aplastic anemia) | Preclinical (large animal study) | 69 |
| PHD inhibitors | Inhibition of the oxygen sensing PHD enzymes resulting in HIF stabilization | Unclear | Phase 2 clinical trial in renal anemia; preclinical studies in ischemia and reperfusion | 79,92-94 |
| Ischemic preconditioning | Multiple (for example, adenosine signaling, HIF stabilization and attenuation of inflammation) | Unclear | Phase 2 clinical trial | 79-83 |
| Ischemic postconditioning | Multiple | Unclear | Phase 2 clinical trial | 87,88 |
| Remote ischemic conditioning | Multiple | Unclear | Phase 2 clinical trial | 89 |
| Nitric oxide (NO) | Multiple | Elevation of methemoglobin | Phase 2 clinical trial | 106,107 |
| Apyrase | ATP breakdown (attenuation of ATP signaling and promotion of adenosine generation and signaling) | Unclear | Preclinical | 81,120 |
| Nucleotidase | AMP conversion to adenosine; enhanced adenosine generation and signaling | Unclear | Preclinical | 80,122,123 |
| Regadenoson, ATL146e | Specific adenosine receptor agonists targeting Adora2a | Unclear | Phase 1 trial ongoing | 3,131 |
| Bay 60-6583 | Specific adenosine receptor agonist targeting Adora2b | Sickling of red blood cells in individuals with sickle cell disease | Preclinical | 80,129,130 |
| Inhibitors of miR-92a | Promotion of angiogenesis | Unclear | Preclinical | 137 |
| Activators of miR-499 or miR-24 | Inhibition of apoptosis | Unclear | Preclinical | 138,139 |
Ischemia and reperfusion causes sterile inflammation
With a few exceptions, such as bacterial translocation after intestinal injury, ischemia and reperfusion typically occurs in a sterile environment. Nevertheless, the consequences of ischemia and reperfusion share many phenotypic parallels with activation of a host immune response directed toward invading microorganisms10. This sterile immune response involves signaling events through pattern-recognition molecules such as Toll-like receptors (TLRs), recruitment and activation of immune cells of the innate and adaptive immune system and activation of the complement system (Fig. 2). As these responses can have adverse consequences, targeting immune activation is an emerging therapeutic concept in the treatment of ischemia and reperfusion. In contrast, some aspects of the adaptive immune response—particularly the recruitment and expansion of regulatory T cells (Treg cells)—may be beneficial11.
Figure 2.
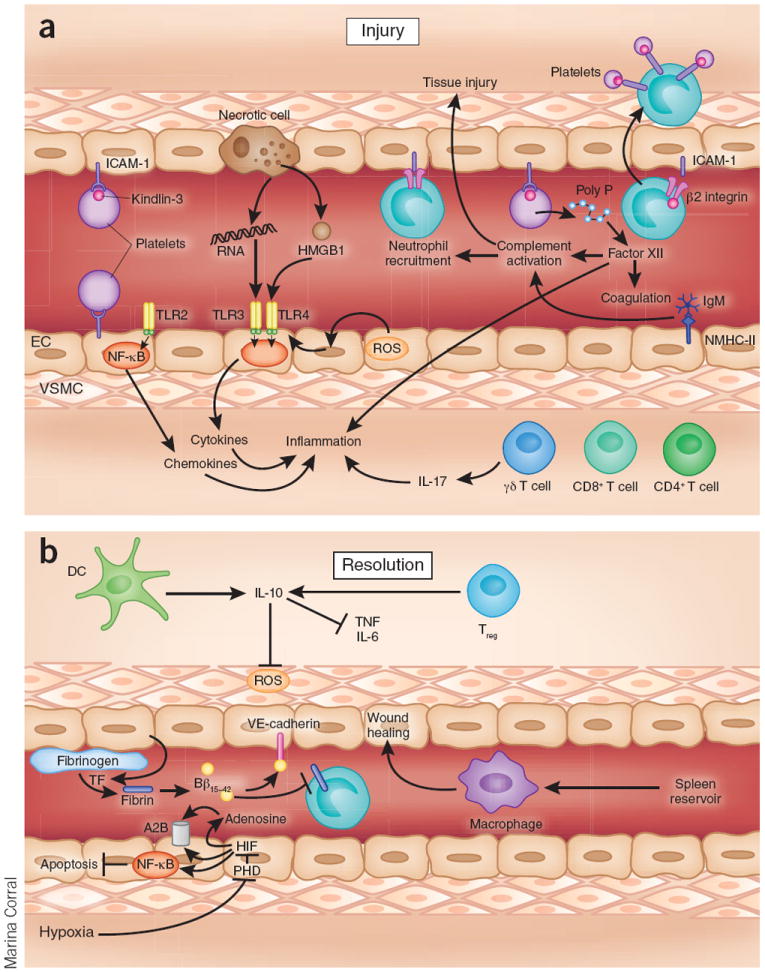
Injury and resolution during ischemia and reperfusion. (a) Ischemia and reperfusion is associated with a pathological activation of the immune system. Tissue hypoxia during the ischemic period results in TLR-dependent stabilization of the transcription factor NF-κB, leading to transcriptional activation of inflammatory gene programs. TLR4 expression can be increased by ROS and can be activated by endogenous ligands such as HMGB1. TLR3 can be activated by RNA released from necrotic cells. After reperfusion, granulocytes such as neutrophils adhere to the vasculature and infiltrate the tissue, and platelets can ‘piggyback’ on neutrophils. Activated platelets can interact with vascular endothelia at the site of injury; this interaction depends on a shift of platelet integrins from a low-affinity state to a high-affinity state (integrin activation or priming), which requires Kindlin-3. Activated platelets release inorganic polyphosphate that directly binds to and activates the plasma protease factor XII, contributing to proinflammatory and procoagulant activation during ischemia and reperfusion. CD4+, CD8+ and γδ T cells contribute to tissue injury; for example, by the release of IL-17 from γδ T cells. (b) Ischemia and reperfusion activates endogenous mechanisms of injury resolution. Tissue hypoxia results in the inhibition of oxygen-sensing PHD enzymes and stabilization of the HIF transcription factor, activating a wide range of transcriptional programs involved in injury resolution, including the production of extracellular adenosine that signals through receptors such as ADORA2B (A2B). In addition, hypoxia-elicited inhibition of PHDs results in NF-κB activation, which contributes to the resolution phase by preventing apoptosis. Regulatory T cells and dendritic cells are important sources of IL-10, which has a crucial role in dampening inflammation and attenuating reactive oxygen production. Splenic reservoir monocytes are recruited from the spleen to the site of tissue energy where they participate in wound healing. Breakdown products from fibrinogen, such as fibrin-derived peptide Bβ15–42, protect the myocardium from injury. NMHC-II, non-muscle myosin heavy chain type II; DC, dendritic cell; EC, endothelial cell; Poly P, polyphosphate; TF, tissue factor; VSMC, vascular smooth muscle cell.
Innate immune responses
The inflammatory response to sterile cell death or injury has many similarities to that observed during microbial infections. In particular, host receptors that mediate the response to microorganisms have been implicated in the activation of sterile inflammation during ischemia and reperfusion10. For example, ligand binding to TLRs leads to the activation of downstream signaling pathways, including NF-κB, mitogen-activated protein kinase (MAPK) and type I interferon pathways, resulting in the induction of proinflammatory cytokines and chemokines10. These receptors can also be activated by endogenous molecules in the absence of microbial compounds, particularly in the context of cell damage or death, as occurs during ischemia and reperfusion10. Such ligands have been termed ‘damage-associated molecular patterns’ (DAMPs). Many of these ligands (for example, high-mobility group box 1 (HMGB1) protein or ATP) are normally sequestered intracellularly; upon tissue damage, they are released into the extracellular compartment where they can activate an immune response12,13. There is also evidence that extracellularly located damage-associated molecular patterns are generated or released in the process of catabolism10. Such catabolic DAMPs can either activate an immune response12 or function as a safety signal to restrain potentially harmful immune responses and promote tissue integrity during ischemia and reperfusion (examples of the latter type of catabolic DAMP are adenosine and the fibrinogen-derived peptide Bβ15–42)14,15.
One of the most widely studied pattern recognition receptors is TLR4, which is known to mediate inflammatory responses to Gram-negative bacteria through its activation by lipopolysaccharide. Mice with targeted gene deletion of Tlr4 are hyporesponsive to lipopolysaccharide, as are humans with missense mutations in TLR4 (ref. 16). TLR4 activation may be enhanced by oxidative stress17, which is generated by ischemia and reperfusion and is known to prime inflammatory cells for increased responsiveness to subsequent stimuli. Alveolar macrophages from rodents subjected to hemorrhagic shock and resuscitation express increased surface levels of TLR4, an effect that was inhibited by adding the antioxidant N-acetylcysteine to the resuscitation fluid17. Moreover, H2O2 treatment of cultured macrophages similarly caused an increase in surface TLR4 expression17. Fluorescent resonance energy transfer between TLR4 and the raft marker GM1, as well as biochemical analysis of raft components, showed that oxidative stress redistributes TLR4 to lipid rafts in the plasma membrane, consistent with the idea that oxidative stress primes the responsiveness of cells of the innate immune system17. Other studies have implicated TLR4 signaling in renal ischemia and reperfusion. Mice with a genetic deletion of Tlr4 are protected from kidney ischemia, and experiments using bone-marrow chimeric mice suggest that kidney-intrinsic Tlr4 signaling has the predominant role in mediating kidney injury18. In addition, a study of patients undergoing kidney transplantation revealed a detrimental role of TLR4 signaling in early graft failure19. Indeed, endogenous TLR4 ligands such as HMGB1 and biglycan were induced during human kidney transplantation, thereby supporting a role for TLR4 in sterile inflammation in the kidney19. Kidneys from individuals with a TLR4 loss-of-function allele (as assessed by diminished affinity of TLR4 for its ligand HMGB1) contained lower levels of proinflammatory cytokines in association with higher rates of immediate graft function after transplantation19. Other TLRs may also have deleterious effects. TLR3, implicated in sensing viral RNA, was proposed to sense RNA released from necrotic cells independent of viral activation, and treatment with a neutralizing antibody to TLR3 was protective in studies of intestinal ischemia and reperfusion in vivo20. TLR2 expression on epithelia is induced by hypoxia21 or inflammation22, and renal TLR2 signaling contributes to acute kidney injury and inflammation during ischemia and reperfusion23. Taken together, these studies suggest that inhibitors of TLR signaling could be effective for the treatment of sterile inflammation induced by ischemia and reperfusion. Accordingly, antagonists for TLR receptors are currently under development24 (Table 2). These antagonists are structural analogs of TLR agonists and likely act by binding the receptor without inducing signal transduction24. For example, experimental studies of TAK-242, a small-molecule inhibitor of TLR4, in large animals have shown efficacy in the treatment of acute kidney injury25. A recent randomized clinical trial of TAK-242 in patients with sepsis and shock or with respiratory failure showed a trend toward improved survival with TAK-242 treatment26, but the findings were not statistically significant. Nevertheless, translational approaches using TLR inhibitors remain promising.
Sterile inflammation during ischemia and reperfusion is also characterized by the accumulation of inflammatory cells. Particularly during the early phase of reperfusion, innate immune cells dominate the cellular composition of the infiltrates. The functional contributions of these cells are not clear: they may contribute to a pathological activation of inflammation and promote collateral tissue injury, or conversely to the resolution of injury. Notably, a recent study showed that monocytes can be recruited from a splenic reservoir to injured tissue after myocardial ischemia and reperfusion to participate in wound healing27. Other studies have found that depletion of conventional dendritic cells increases sterile inflammation and tissue injury in the context of hepatic ischemia and reperfusion injury28. The protection afforded by dendritic cells depends on their production of the anti-inflammatory cytokine interleukin-10 (IL-10), resulting in attenuated levels of tumor necrosis factor-α, IL-6 and reactive oxygen species (ROS). ROS, implicated in the tissue damage that occurs during ischemia and reperfusion11, are toxic molecules that alter cellular proteins, lipids and ribonucleic acids, leading to cell dysfunction or death. NADPH oxidase, an enzyme expressed in virtually all inflammatory cells, contributes to the formation of one such cytotoxic ROS, peroxynitrite. In addition, H2O2 derived from O2− dismutation gives rise to highly toxic hydroxyl radicals through the Haber-Weiss reaction, facilitated by the increased availability of free iron in ischemia11. Peroxynitrite and other reactive species induce oxidative DNA damage and consequent activation of the nuclear enzyme poly (ADP-ribose) polymerase 1 (PARP-1), the most abundant isoform of the PARP enzyme family. Accordingly, PARP inhibitors are in clinical development for the treatment of ischemia and reperfusion injury29.
At sites of sterile inflammation, the accumulation of granulocytes has to be tightly controlled, as too few granulocytes may not allow for adequate tissue repair, whereas too many granulocytes can promote uncontrolled inflammation and tissue injury30. In a clinically relevant mouse model for transplant-mediated lung ischemia and reperfusion, a recent study showed that expression of the Bcl3 protein by the recipient led to inhibition of emergency granulopoiesis and limited acute graft injury30. Inhibition of myeloid progenitor cell differentiation may therefore have promise as a therapeutic strategy for the prevention of tissue injury in the context of sterile inflammation.
Adaptive immune response
Ischemia and reperfusion elicits a robust adaptive immune response that involves, among other cell types, T lymphocytes. The mechanisms by which antigen-specific T cells are activated during sterile inflammation are not well understood, but emerging evidence indicates a contribution of both antigen-specific and antigen-independent mechanisms of activation31,32. Several studies have shown that T cells accumulate during ischemia and reperfusion. For example, T cells are localized to the infarction boundary zone within 24 h of reperfusion of the ischemic brain, accumulate further at 3 and 7 d after reperfusion and are decreased in number after 14 d33. Studies of mouse lines deficient in specific populations of lymphocytes showed that both CD4+ and CD8+ T cells have a detrimental role in ischemia and reperfusion of the brain34, the heart35 and the kidneys36. Further, a recent study suggested a pivotal role for IL-17 produced by γδ T cells in ischemia and reperfusion injury of the brain37. Elevated levels of IL-17 were found both in individuals suffering from stroke38 as well as in mice exposed to brain ischemia and reperfusion37. Notably, subsequent studies identified γδ T cells (as opposed to CD4+ T helper cells) as the main source of IL-17 (ref. 37). In addition, genetic and pharmacologic approaches targeting IL-17 or γδ T cells led to reduced inflammation and robust neuroprotection, indicating that γδ T cells that produce IL-17 are an attractive therapeutic target for ischemic stroke (Table 2).
In contrast, Treg cells appear to have a protective role of in ischemia and reperfusion. For example, a recent study using an experimental stroke model showed that depletion of Treg cells substantially increased delayed brain damage and caused a deterioration in functional outcome39. Based on results including the finding that transfer of wild-type but not IL-10–deficient Treg cells attenuated ischemic brain injury, the authors proposed that Treg cell–dependent production of IL-10 decreases tumor necrosis factor α abundance at early time points and delays interferon γ accumulation39. Although not tested in the setting of ischemia and reperfusion, administration of ex vivo expanded human Treg cells had beneficial effects in a model of transplant atherosclerosis, providing evidence that such an approach is feasible40. Other strategies to enhance Treg cell function following ischemia and reperfusion could involve boosting expression of FOXP3, the key transcription factor for Treg cell differentiation. Previous studies have shown that Foxp3 levels are subject to epigenetic regulation41. Indeed, pharmacological inhibitors of histone/protein deacetylases are effective in treating experimentally induced inflammatory bowel disease and in improving cardiac and islet graft survival in mouse transplantation models through increasing Treg cell numbers and function42.
Innate autoimmunity, complement, platelets and coagulation
During ischemia and reperfusion, innate recognition proteins can be self reactive and initiate inflammation against self tissue in a manner similar to the response triggered by pathogens (known as ‘innate autoimmunity’)9. A series of studies has linked reperfusion injury to the occurrence of so-called ‘natural’ antibodies, leading to activation of the complement system. Natural antibodies are produced in the absence of deliberate immunization and are a major component of the repertoire of B1 cells, which produce IgM and, in some cases, IgG43. For example, a single type of natural antibody prepared from a panel of B1 cell hybridomas (IgMCM-22) restored reperfusion injury in antibody-deficient mice9, suggesting that reperfusion injury can be considered to be an autoimmune type of disorder. Using mouse models of skeletal muscle and intestinal reperfusion injury, a highly conserved region within nonmuscle myosin heavy chain type II A and C was subsequently identified as a self target for natural IgM in the initiation of reperfusion injury44. More recently, additional neoepitopes have been identified, for example, the soluble cytosolic protein annexin IV43. Together, these studies indicate that neoepitopes expressed on ischemic tissues are targets for natural antibody binding during the reperfusion phase with subsequent complement activation, neutrophil recruitment and tissue injury43.
The complement system acts as an immune surveillance system to discriminate among healthy host tissue, cellular debris, apoptotic cells and foreign intruders, varying its response accordingly45. Locally produced and activated, the complement system yields cleavage products that function as intermediaries, amplifying sterile inflammation during ischemia and reperfusion through complement-mediated recognition of damaged cells and anaphylatoxin release, thereby fueling inflammation and the recruitment of immune cells45. Studies in animal models have indicated that inhibition of the complement system might effectively treat ischemia and reperfusion injury; however, results from clinical studies have largely been disappointing46-49. A limitation of the clinical studies could be that one of the inhibitors used, an antibody targeting the complement protein C5, would not affect C3b, which is “upstream” of C5 in the complement cascade and is a key mediator of bacterial opsonization and immune complex solubilization and clearance48. In addition, the complexity of the complement system and incomplete mechanistic insight into the functional consequences of manipulating individual components of the cascade may contribute to difficulties in therapeutic targeting of complement pathways. A recent study of hepatic ischemia and reperfusion injury in mice indicated a dual role of the complement system50: although excessive complement activation is detrimental, a threshold of complement activation is crucial for liver regeneration, and impaired regeneration due to inadequate complement activation can lead to acute liver failure following hepatic resection or liver transplantation.
Excessive platelet aggregation and release of platelet-derived mediators can exacerbate tissue injury following ischemia and reperfusion. Platelet activation can occur through integrin-mediated endothelial interactions51. In addition, platelets can be transported by inflammatory cells across epithelial barriers (by ‘piggybacking’ on polymorphonuclear leukocytes) to sites of injury or inflammation52. A recent study showed a central role for a FERM domain-containing protein (Fermt3, also known as Kindlin-3) in mediating integrin-dependent platelet activation and aggregation51. Fermt3−/− mice were protected in a model of ischemia and infarction after mesenteric arteriole injury with virtually no firm adhesion of platelets to the injured vessel wall51. Other studies have shown that platelets release inorganic polyphosphates, polymers of 60–100 phosphate residues that directly activate plasma protease factor XII and thereby function as proinflammatory and procoagulant mediators in vivo53. Ischemia and reperfusion triggers coagulation by inflammatory mediators and platelet activation in many ways, but several natural anticoagulant mechanisms can inhibit clot formation following ischemia and reperfusion, such as those mediated by antithrombin-heparin, tissue factor inhibitor and protein C54,55. Furthermore, fibrin degradation following ischemia and reperfusion, resulting in the formation of fibrin D fragments, including the peptide Bβ15–42, has been implicated in attenuating inflammation and preserving vascular barrier function during shock56 and in dampening ischemia-reperfusion injury15. Administration of an intravenous bolus of Bβ15–42 attenuated myocardial injury in mice17, and a subsequent randomized clinical trial of patients with acute myocardial infarction with ST-segment elevation showed that treatment with intravenously-administered Bβ15–42 upon reperfusion reduced the size of the necrotic core zone, as assessed using magnetic resonance imaging 5 days after infarction (ref. 57, Table 2).
Cell death during ischemia and reperfusion
Ischemia and reperfusion activates various programs of cell death, which can be categorized as necrosis, apoptosis or autophagy-associated cell death7. Necrosis, characterized by cell and organelle swelling with subsequent rupture of surface membranes and the spilling of their intracellular contents7, is a frequent outcome of ischemia and reperfusion. Necrotic cells are highly immunostimulatory and lead to inflammatory-cell infiltration and cytokine production. In contrast, apoptosis involves an orchestrated caspase signaling cascade that induces a self-contained program of cell death, characterized by the shrinkage of the cell and its nucleus, with plasma membrane integrity persisting until late in the process7. Although this process has traditionally been viewed as less immunostimulatory than necrosis10, recent studies have shown that extracellular release of ATP from apoptotic cells through pannexin hemichannels acts as a ‘find-me’ signal that attracts phagocytes58,59. Inhibition of apoptosis may have promise as a therapeutic strategy for ischemia-reperfusion injury. For example, a study in a mouse model of acute kidney injury identified the matricellullar protein thrombospondin 1 (THBS1, also known as TSP-1), produced by injured proximal tubular cells, as an inducer of apoptosis and found that Thbs1−/− mice are protected from injury60. Other studies have focused on platelet-derived growth factor CC (PDGF-CC), a potent neuro-protective factor that acts by modulating glycogen synthase kinase 3β (GSK-3β) activity61. PDGF-CC gene or protein delivery protected neurons from apoptosis in both the retina and brain in various animal models of neuronal injury, including ischemia-induced stroke. PDGF-CC treatment resulted in increased levels of GSK-3β Ser9 phosphorylation and decreased levels of Tyr216 phosphorylation, consistent with previous findings that Ser9 phosphorylation inhibits and Tyr216 phosphorylation promotes apoptosis61,62.
The transcription factor NF-κB may also modulate apoptosis during ischemia and reperfusion. Limited oxygen availability is associated with activation of NF-κB through a mechanism involving hypoxia-dependent inhibition of oxygen sensors63. Mice with disruption of the gene encoding IKK-β, the catalytic subunit of IKK that is essential for NF-κB activation, offer an opportunity to study the consequences of preventing canonical NF-κB pathway activation. This manipulation, however, results in embryonic lethality owing to massive apoptosis of the developing liver driven by tumor necrosis factor-α (ref. 64). To circumvent this difficulty, studies from the laboratory of Michael Karin examined mice with selective ablation of IKK-β. Study of intestinal ischemia and reperfusion revealed that although IKK-β deficiency in enterocytes is associated with reduced inflammation, severe apoptotic damage occurred in the reperfused mucosa65. NF-κB inhibition can therefore be viewed as a ‘double-edged sword’, in that it is associated with the prevention of systemic inflammation but increased local injury. These results underscore the need for caution in using NF-κB inhibitors for treating intestinal ischemia-reperfusion injury.
The commonly used immunosuppressant cyclosporine is an inhibitor of mitochondrial permeability transition pore opening, an important step in programmed cell death7. In a randomized clinical study of 58 individuals with acute myocardial infarction with ST-segment elevation, treatment with an intravenous bolus of cyclosporine immediately before percutaneous coronary intervention was associated with smaller infarct sizes compared to the saline control (Table 2)66. Although these findings are very encouraging, given the small sample size of this trial, confirmation in a larger clinical trial will be important. In addition, the development of more specific and safer inhibitors of the mitochondrial permeability transition pore could enhance the potential of this approach67.
There is strong evidence supporting the idea that autophagy is an adaptive response to sublethal stress, such as nutrient deprivation, and the deletion of key autophagic genes accelerates rather than inhibits cell death7. The transcription factor HIF, a central mediator of hypoxic responses, also seems to regulate autophagy. The process of mitochondrial autophagy is induced by hypoxia and requires HIF-dependent expression of autophagic genes68, indicating a crucial role for HIF in the metabolic adaptation of hypoxic or ischemic tissues during conditions of limited oxygen. From a therapeutic perspective, a recent study showed that chloramphenicol, traditionally used to treat bacterial infections but more recently recognized as an inducer of autophagy, is protective in a swine model of myocardial ischemia and reperfusion (Table 2)69.
Microvascular dysfunction
Ischemia and reperfusion is associated with a vascular phenotype that includes increased vascular permeability, endothelial cell inflammation, an imbalance between vasodilating and vasoconstricting factors and activation of coagulation and the complement system. Microvascular dysfunction following ischemia and reperfusion in humans can lead to respiratory failure manifesting as hypoxemia and pulmonary edema that is caused not by heart failure but rather by a disruption of the alveolar-capillary barrier function, leading to increased microvascular permeability70. This type of microvascular dysfunction can, for example, occur in patients with graft ischemia and reperfusion during solid organ transplantation71. During the ischemic period, vascular hypoxia can cause increased vascular permeability. Studies using cultured endothelial cells exposed to ambient hypoxia (for example, 2% oxygen over 24 h) showed increased permeability after hypoxia exposure caused by lower cAMP levels5,6. Similarly, mice exposed to ambient hypoxia (8% oxygen over 4–8 h) experienced increases in pulmonary edema, albumin leakage into multiple organs and elevated cytokine levels72-75. Complement system activation, leukocyte-endothelial cell adhesion and platelet-leukocyte aggregation further aggravate microvascular dysfunction after reperfusion76. A study of mouse models of sickle cell disease and transfusion-related lung injury has also implicated neutrophil ‘sandwiches’, in which neutrophil microdomains mediate heterotypic interactions with endothelial cells, red blood cells or platelet, in microcirculation injury77. Mechanistically, E-selectin activation by E-selectin ligand 1 induced polarized, activated αMβ2 integrin clusters at the leading edge of crawling neutrophils, allowing the capture of circulating erythrocytes or platelets. These findings indicate that endothelial selectins can influence neutrophil behavior beyond the canonical rolling step through delayed and organ-damaging activation77.
Attenuated vascular relaxation after reperfusion can result in a ‘no reflow phenomenon’, characterized by increased impedance of microvascular blood flow after the reopening of an infarct-related, occluded blood vessel1, and in a clinical setting is associated with poor outcomes. In a mouse model of ischemic brain injury, ischemia induces sustained contraction of pericytes on microvessels despite successful reopening of the middle cerebral artery78. Suppression of oxidative-nitrative stress relieves pericyte contraction, reduces erythrocyte entrapment and restores microvascular patency with improved tissue survival. Indeed, results from this study showed that the microvessel wall is the major source of oxygen radicals and nitrogen radicals that cause ischemia and reperfusion–induced microvascular dysfunction. Together, these findings indicate that ischemia and reperfusion–induced injury to pericytes may impair microcirculatory reflow and point to the restoration of pericyte function for the treatment of individuals suffering from stroke.
Therapeutic approaches to enhance ischemia tolerance
Therapeutic approaches to render organs more resistant to ischemia could have important clinical uses. Such therapies could be used in a preventive manner during organ transplantation or other types of major surgery associated with ischemia and reperfusion, or after ischemic injury in patients during an intervention aimed at the restoration of blood flow and reperfusion (for example, percutaneous coronary intervention in patients with acute myocardial infarction).
Ischemic conditioning (preconditioning, postconditioning and remote conditioning)
Ischemic preconditioning is an experimental strategy in which exposure to short, nonlethal episodes of ischemia results in attenuated tissue injury during subsequent ischemia and reperfusion. Numerous studies have investigated the underlying mechanisms with the goal of finding pharmacological approaches that would imitate ischemic preconditioning. For example, combinations of genetic and pharmacologic studies have implicated oxygen-dependent signaling pathways79 and purinergic signaling80,81. Other studies have directly applied this experimental strategy to dampen tissue injury from ischemia and reperfusion, for example, by ischemic preconditioning of a transplant graft before liver transplantation or before major liver resections82,83. Although these studies have shown some benefit, they have not been able to reproduce the profound tissue-protective effects of ischemic preconditioning observed in animal studies, perhaps because it is very difficult to systematically identify the most effective preconditioning protocol for a clinical study84-86. Similar to ischemic preconditioning, short episodes of ischemia applied during reperfusion are associated with a reduction in myocardial infarct size, called postconditioning1. A prospective, randomized, controlled, multicenter study investigated whether postconditioning in 30 patients protects the human heart during coronary angioplasty after acute myocardial infarction found beneficial effects87. After reperfusion by insertion of a stent into the occluded coronary artery, postconditioning was initiated within 1 min of reflow by applying four episodes of 1-min inflation and 1-min deflation of the angioplasty balloon. Another clinical study showed that postconditioning was associated with improved cardiac function up to 1 year after an acute myocardial infarction88. Remote ischemic conditioning—induced by repeated brief periods of limb ischemia—was recently found to be effective in myocardial salvage in patients with acute myocardial infarction89. In this study, 333 patients were randomly assigned to remote ischemic conditioning (four cycles of 5-min inflation and 5-min deflation of a blood pressure cuff) or no treatment during transport to the hospital, where they were treated with a percutaneous intervention to achieve reperfusion. Thirty days later, myocardial perfusion imaging revealed increased myocardial salvage with remote conditioning.
Metabolic strategies to increase ischemia tolerance
During ischemia, energy metabolism switches from fatty acid oxidation to more oxidation-efficient glycolysis, allowing tissues to sustain cellular viability during ischemia for a longer amount of time. This metabolic switch is under the direct control of the HIF transcription factor, whose stabilization when oxygen levels fall is responsible for the transcriptional induction of glycolytic enzymes8,90. The stability of HIF is regulated by the oxygen-sensing PHD enzymes, of which there are three isoforms, PHD1–PHD3. Loss of Phd1 lowers oxygen consumption in ischemic skeletal muscle by reprogramming glucose metabolism to a more anaerobic route of ATP production through activation of a peroxisome proliferator–activated receptor-α pathway91. Moreover, treatment with pharmacological PHD inhibitors results in increased ischemia tolerance of the kidneys92 and in cardioprotection similar to that seen with ischemic preconditioning in the heart79. To date, PHD inhibitors seem to be well tolerated in humans93, suggesting that they could be readily tested in larger clinical trials (Table 2).
In addition to these more canonical effects of PHD inhibitors, they could also have other potentially desirable effects, including on vascular normalization of tumors. In mice with heterozygous deletion of the gene encoding the oxygen-sensing PHD2, tumor vessel leakiness and vascular distortion is attenuated, an effect called ‘vascular normalization’—for example, normalization of the architecture of sharply demarcated boundaries and branching points of the tumor vessels. This effect can be mimicked pharmacologically by PHD inhibitors via its stabilizing effects on HIFs8,94.
Among the most well known of HIF target genes is EPO, encoding erythropoietin, the major regulator of red blood cell formation, whose production and secretion are regulated by tissue oxygen levels90. In addition to its role in stimulating red cell production, preclinical studies implicated erythropoietin in tissue protection from ischemia and reperfusion by metabolic adaptation, inhibition of apoptosis or stimulation of angiogenesis95-98. Recently, a prospective, randomized, double-blind, placebo-controlled trial was carried out in patients with acute myocardial infarction with ST-segment elevation to address the efficiency of intravenous treatment with erythropoietin in a clinical setting99. In contrast to many preclinical studies, this trial did not find a protective effect of treatment with intravenous erythropoietin. In fact, erythropoietin treatment of such patients who had successful reperfusion within 4 h of percutaneous coronary intervention did not show reduced infarct size but, rather, higher rates of adverse cardiovascular events99.
Other studies of metabolic adaptation during ischemia and reperfusion have shown that activation of mitochondrial aldehyde dehydrogenase 2 (ALDH2) is associated with robust cardioprotection in rat models100 indicating that pharmacological enhancement of ALDH2 activity applied preventively might increase ischemia tolerance in patients subjected to cardiac ischemia, for example during coronary bypass surgery. Individuals with a genetic defect of ALDH2, as occurs in up to 40% of the Asian population, may particularly benefit from this therapy. Other studies have focused on AMP-activated protein kinase (AMPK), which orchestrates the regulation of energy-generating and energy-consuming pathways and whose activation has been shown to protect the heart against ischemic injury101,102. AMPK activation seems to be an endogenous protective mechanism, as the proinflammatory cytokine macrophage migration inhibitory factor (MIF), whose production is stimulated by ischemia, stimulates AMPK and thereby promotes glucose uptake and cardioprotection101. These results are consistent with findings that human fibroblasts containing a low-activity MIF promoter polymorphism show diminished MIF release and AMPK activation during hypoxia101.
Therapeutic gases
Several therapeutic gases have been used for the treatment of ischemia and reperfusion (Fig. 3), including hydrogen (H2), nitric oxide (NO), hydrogen sulfide (H2S), and carbon monoxide (CO). H2 is a highly diffusible gas and can combine with hydroxyl radicals to produce water, thereby acting as an antioxidant. In a rat model in which oxidative stress damage was induced in the brain by focal ischemia and reperfusion, production of ROS by mitochondria was shown to trigger the mitochondrial permeability transition pore, leading to mitochondrial swelling, rupture and release of cytochrome c, and finally to apoptotic cell death103,104. Inhalation of H2 gas markedly suppressed brain injury by buffering these effects of oxidative stress.
Figure 3.
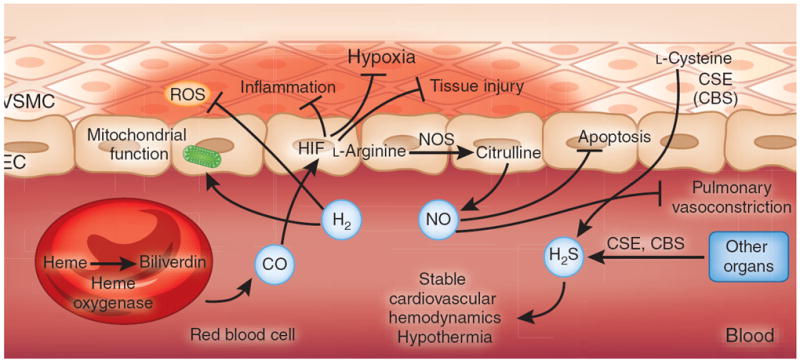
Therapeutic gases for the treatment of ischemia and reperfusion. CO, NO and H2S are considered to be endogenous gas transmitters. The predominant pathway for endogenous CO production involves the conversion of the erythrocyte-derived porphyrin molecule heme to biliverdin by the action of heme oxygenase, liberating CO as a byproduct141. CO has been implicated in attenuating inflammation and tissue injury through the stabilization of HIF. NO is produced predominantly from the endogenous metabolism of l-arginine to citrulline by NO synthase, which is expressed in multiple cell types, including vascular endothelia and neurons (not shown)105. Inhaled NO has been therapeutically used to attenuate hypoxic pulmonary vasoconstriction or to dampen apoptosis during ischemia and reperfusion142. H2S is produced endogenously through the metabolism of l-cysteine by the action of either cystathionine β-synthase (CBS) (expressed predominantly in the brain, nervous system, liver and kidney) or cystathionine γ-lyase (CSE) (expressed predominantly in liver and in vascular and nonvascular smooth muscle)143. Therapeutic use of inhaled H2S has been shown to induce a suspended-animation–like state characterized by hypothermia and stable cardiovascular hemodynamics, and to have protective effects during ischemia and reperfusion. In contrast to endogenous gas transmitters, no biological pathway for the generation of H2 has been described in mammalian cell systems. Therapeutic use of inhaled H2 has been shown to attenuate ischemia and reperfusion–associated accumulation of ROS and to preserve mitochondrial function. EC, endothelial cell; VSMC, vascular smooth muscle cell.
In contrast to H2, NO, H2S and CO are produced endogenously by enzymatically controlled pathways. NO, a soluble gas continuously synthesized in endothelial cells by endothelial NO synthase, regulates basal vascular tone and endothelial function and maintains blood oxygenation through hypoxic pulmonary vasoconstriction. Multiple studies have implicated the endogenous production of NO or its therapeutic application in attenuating ischemia-reperfusion injury105. In a small (n = 10 individuals per group), randomized, placebo-controlled clinical trial using inhaled NO for the treatment of graft ischemia and reperfusion during liver transplantation, NO improved the restoration of liver function and lowered hepatocyte apoptosis106. In contrast, a randomized, placebo-controlled trial for the use of inhaled NO in the acute treatment of sickle cell pain crisis did not find an improvement in the time until crisis resolution107. As the beneficial effects of NO administration may depend on its conversion to nitrite105, a possible explanation for the lack of a beneficial effect in the sickle cell trial is that systemic nitrite was insufficiently generated, perhaps due to the way in which NO was delivered (as a pulse of pure NO in nitrogen at the ‘front’ of the tidal volume).
H2S has also been reported to have therapeutic effects in animal models of ischemia and reperfusion108,109. For example, similar to AMP110, H2S can induce a reversible state of hypothermia and a suspended-animation–like state in rodents111, and treatment with H2S reversibly depresses cardiovascular function without changing blood pressure112. In addition, endogenously produced CO or the administration of CO-releasing molecules have anti-inflammatory and cytoprotective effects that involve HIF stabilization and activation of a HIF-dependent transcriptional response113.
Nucleotide and nucleoside signaling
Nucleotides, particularly in the form of ATP, have been strongly implicated in promoting tissue inflammation during ischemia and reperfusion (Fig. 4). Ischemia and reperfusion results in the release of ATP—whose intracellular concentrations are relatively high (5–8 mM)—into the extracellular compartment. ATP can be spilled by necrotic cells10 or can be released in a controlled fashion from apoptotic cells58,59 or activated inflammatory cells114,115. When it accumulates in the extracellular compartment, ATP acts to recruit phagocytes58,59, activates the Nlrp3 inflammasome during ischemia and reperfusion13 and promotes the chemotaxis of inflammatory cells116. ATP-elicited activation of nucleotide receptors can enhance vascular inflammation; for example, through P2Y6 receptors117 or, following spinal cord injury, through P2X7 receptors118. Pharmacological strategies to block ATP release or ATP receptor signaling may therefore have promise for attenuating sterile inflammation during ischemia and reperfusion. In the extracellular compartment, ATP is enzymatically converted to the nucleoside adenosine119. In animal models of ischemia and reperfusion, pharmacological strategies for spurring ATP breakdown to adenosine—for example, treatment with apyrase, which converts ATP or ADP to AMP, followed by treatment with nucleotidase, which converts AMP to adenosine—are effective in attenuating tissue injury and sterile inflammation (Table 2)82,83,120-125. Beyond alleviating the detrimental effects ATP, ATP conversion to adenosine may be desirable because of the beneficial effects of adenosine itself126. Pharmacological and genetic studies in mouse models of ischemia and reperfusion have shown that signaling through adenosine receptors is protective; for example, through activation of the adenosine A2A receptor (Adora2a) on inflammatory cells3,127,128 or Adora2b on vascular endothelia, epithelia or myocytes72,80,129,130. For example, studies in mouse models of myocardial ischemia and reperfusion80, acute kidney injury130 or intestinal ischemic injury129 have shown promising results for the selective Adora2b agonist BAY 60-6583 in the treatment of ischemia and reperfusion. Moreover, Adora2a activation on invariant natural killer T cells attenuates ischemia and reperfusion in mouse models of sickle cell disease3,131. Due to its potent vasodilatory properties132, the ADORA2A agonist regadenoson (CVT-3146) was approved by the US Food and Drug Administration as a coronary vasodilator for patients requiring pharmacologically-induced stress echocardiography133,134. An ongoing multicenter, dose-finding and safety trial of infused regadenoson has been initiated to study its safety and efficacy in the treatment of ischemia and reperfusion–related tissue injury in patients with sickle cell disease (Table 2)131. Complicating this approach, a recent study showed that adenosine signaling through ADORA2B induces hemoglobin S polymerization, promoting red blood cell sickling, vaso-occlusion, hemolysis and organ damage135,136.
Figure 4.
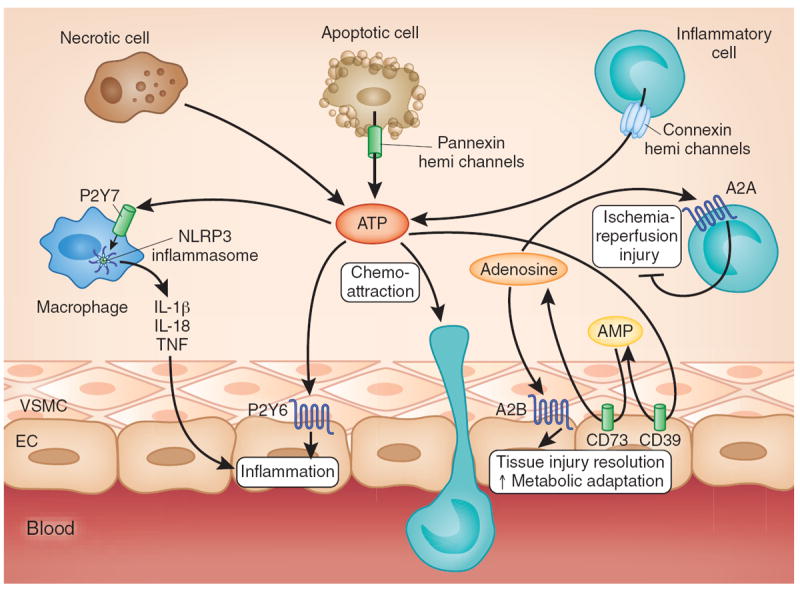
Nucleotide and nucleoside signaling during ischemia and reperfusion. Multiple cell types release ATP during ischemia and reperfusion (for example, spillover from necrotic cells or controlled release through pannexin hemichannels from apoptotic cells or connexin hemichannels from activated inflammatory cells)59,114,119. Subsequent binding of ATP to P2 receptors enhances pathological inflammation and tissue injury, for example, through P2X7-dependent Nlrp3 inflammasome activation13 and P2Y6-dependent enhancement of vascular inflammation117. ATP can be rapidly converted to adenosine through the ecto-apyrase CD39 (conversion of ATP to AMP) and subsequently by the ecto-5′ nucleotidase CD73 (conversion of AMP to adenosine). Adenosine signaling dampens sterile inflammation, enhances metabolic adaptation to limited oxygen availability and promotes the resolution of injury through activation of A2A adenosine receptors expressed on inflammatory cells and activation of A2B adenosine receptors expressed on tissue-resident cells (for example, cardiac myocytes, vascular endothelia or intestinal epithelia). EC, endothelial cell; VSMC, vascular smooth muscle cell.
MicroRNAs (miRNAs) as therapeutic targets
Several studies have suggested a functional role for miRNAs in ischemia and reperfusion (Fig. 5). For example, a recent study showed that the miR-17~92 cluster is highly expressed in human endothelial cells and that miR-92a, a component of this cluster, controls angiogenesis137. Systemic administration of an oligonucleotide antagomir designed to inhibit miR-92a led to enhanced blood vessel growth and functional recovery of damaged tissue in mouse models of limb ischemia or myocardial infarction. MiR-92a seems to target mRNAs corresponding to several proangiogenic proteins, including the α5 integrin. Another study reported that miR-499 administration diminishes apoptosis and the severity of myocardial infarction during ischemia and reperfusion. Inhibition of cardiomyocyte apoptosis by miR-499 was ascribed to direct targeting of a catalytic subunit of the phosphatase calcineurin, attenuating calcineurin-mediated dephosphorylation of dynamin-related protein-1 (Drp1) and thereby decreasing activation of the mitochondrial fission program138. Expression of another miRNA, miR-24, can also be protective in ischemia. miR-24 expression in a mouse model of myocardial ischemia inhibited cardiomyocyte apoptosis, attenuated infarct size and reduced cardiac dysfunction139. In this case, the effect on apoptosis was attributed, in part, through direct repression of the BH3-only domain-containing protein Bim.
Figure 5.
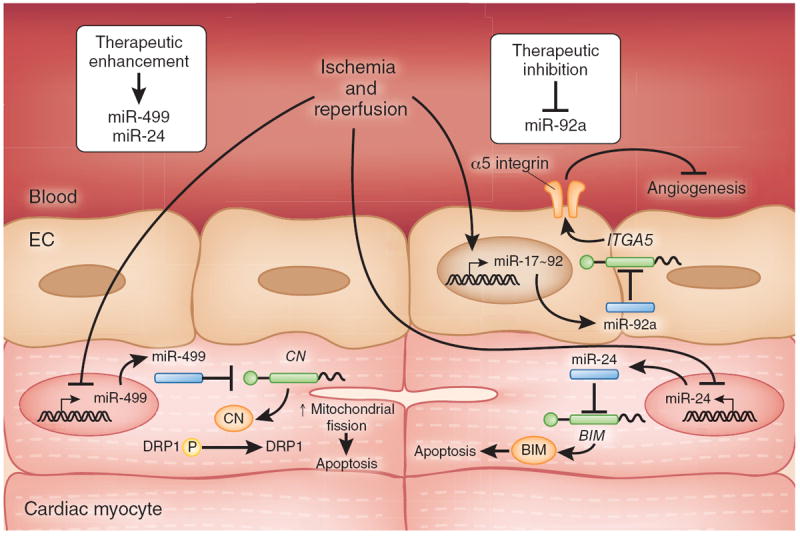
MiRNA pathways implicated in myocardial ischemia and reperfusion. miR-92a (encoded by the miR-17-92a cluster) is highly expressed in vascular endothelia, and blocks ischemic angiogenesis by inhibition of proangiogenic proteins such as the α5 integrin (encoded by ITGA5). In contrast, miR-499 and miR-24 levels are repressed in cardiac tissue following ischemia and reperfusion. MiR-499 suppresses myocyte apoptosis by direct repression of calcineurin subunit synthesis, leading to decreased calcineurin-mediated dephosphorylation of DRP1, thereby interfering with DRP1-mediated activation of the pro-apoptotic mitochondrial fission program. MiR-24 inhibits myocyte apoptosis by direct repression of BIM synthesis. Accordingly, decreasing miR-92a levels (therapeutic inhibition) or increasing miR-499 or miR-24 levels (therapeutic enhancement) might have beneficial effects in the setting of myocardial ischemia and reperfusion. CN, calcineurin; EC, endothelial cell.
Pharmacological approaches to inhibit miRNAs seem likely to become treatment modalities for patients in the near future. For example, liver-expressed miR-122 is essential for hepatitis C virus RNA accumulation in cultured liver cells. Recent studies have shown that administration of a locked nucleic acid complementary to the 5′ end of miR-122 (SPC3649) is effective in silencing miR-122 in nonhuman primates and in the treatment of primates with chronic hepatitis C infection140. Clinical trials in humans are currently being conducted to address the safety and efficacy of SPC3649 in humans (www.clinicaltrials.gov). Similar pharmacological approaches could be developed using locked nucleic acids to target detrimental miRNAs (for example, miR-92a)137 during ischemia and reperfusion (Table 2).
Conclusions
Although rapid reperfusion is needed after ischemia, this reperfusion can paradoxically contribute to tissue injury and destruction. The past decade has seen strong progress in understanding the mechanisms of reperfusion injury and in developing strategies to render tissues more resistant to ischemia or to dampen reperfusion injury. For example, experimental studies of hypoxia-elicited adaptive responses have provided strong evidence for new treatment approaches during ischemia and reperfusion, such as PHD inhibitors or adenosine receptor agonists. Specific therapeutic interventions are now under consideration for clinical safety and efficacy trials, and indeed some agents have already provided promising results in small clinical trials and now require larger follow-up studies to confirm the initial results (Table 2). Unfortunately, other clinical studies have failed to provide evidence for a protective effect of specific therapeutic approaches. It is important to keep in mind that a clinical trial is always based on a specific treatment strategy (for example, the dosage used and the timing of drug delivery), which may lead to the failure of a drug to achieve its desired effect despite its inherent efficacy. Moreover, there remains an urgent need to gain additional mechanistic insight into the molecular events that are triggered by ischemia and reperfusion and that could be exploited therapeutically. For example, highly effective pharmacologic tools to manipulate microRNAs are soon to become available for the treatment of humans. However, our biologic understanding of how microRNAs alter gene expression in ischemic tissues remains rudimentary, and additional mechanistic studies to identify miRNA targets that could be used to treat ischemia and reperfusion will be essential for taking advantage of such pharmacological approaches. Despite the challenges ahead, we are hopeful that new therapies for ischemia and reperfusion will soon be integrated into clinical practice.
Acknowledgments
We thank S.A. Eltzschig for providing artwork during the manuscript preparation. This work is supported by US National Institutes of Health Grants R01-HL0921, R01-DK083385 and R01-HL098294 and a grant from the Crohn’s and Colitis Foundation (H.K.E.) and grant number K08HL102267-01 (T.E.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005;112:2660–2667. doi: 10.1161/CIRCULATIONAHA.105.556746. [DOI] [PubMed] [Google Scholar]
- 3.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010;116:5010–5020. doi: 10.1182/blood-2010-06-290643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Paneth cell–derived IL-17A causes multi-organ dysfunction after hepatic ischemia and reperfusion injury. Hepatology. 2011;53:1662–1675. doi: 10.1002/hep.24253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa S, et al. Hypoxia modulates the barrier and coagulant function of cultured bovine endothelium. Increased monolayer permeability and induction of procoagulant properties. J Clin Invest. 1990;85:1090–1098. doi: 10.1172/JCI114540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogawa S, et al. Hypoxia-induced increased permeability of endothelial monolayers occurs through lowering of cellular cAMP levels. Am J Physiol. 1992;262:C546–C554. doi: 10.1152/ajpcell.1992.262.3.C546. [DOI] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll MC, Holers VM. Innate autoimmunity. Adv Immunol. 2005;86:137–157. doi: 10.1016/S0065-2776(04)86004-8. [DOI] [PubMed] [Google Scholar]
- 10.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 14.Grenz A, Homann D, Eltzschig HK. Extracellular adenosine—a “safety signal” that dampens hypoxia-induced inflammation during ischemia. Antioxid Redox Signal. 2011;15:2221–2234. doi: 10.1089/ars.2010.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petzelbauer P, et al. The fibrin-derived peptide Bβ15–42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 16.Arbour NC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 17.Powers KA, et al. Oxidative stress generated by hemorrhagic shock recruits Toll-like receptor 4 to the plasma membrane in macrophages. J Exp Med. 2006;203:1951–1961. doi: 10.1084/jem.20060943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krüger B, et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA. 2009;106:3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavassani KA, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE. 2007;2:e1364. doi: 10.1371/journal.pone.0001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfs TG, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-γ and TNF-α mediated up-regulation during inflammation. J Immunol. 2002;168:1286–1293. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 23.Leemans JC, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 25.Fenhammar J, et al. Toll-like receptor 4 inhibitor TAK-242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiology. 2011;114:1130–1137. doi: 10.1097/ALN.0b013e31820b8b44. [DOI] [PubMed] [Google Scholar]
- 26.Rice TW, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 27.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bamboat ZM, et al. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010;120:559–569. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kreisel D, et al. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest. 2011;121:265–276. doi: 10.1172/JCI42596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satpute SR, et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. 2009;183:984–992. doi: 10.4049/jimmunol.0801928. [DOI] [PubMed] [Google Scholar]
- 32.Shen X, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. 2009;50:1537–1546. doi: 10.1002/hep.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 34.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-γ in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- 35.Yang Z, et al. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111:2190–2197. doi: 10.1161/01.CIR.0000163586.62253.A5. [DOI] [PubMed] [Google Scholar]
- 36.Day YJ, et al. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-γ. J Immunol. 2006;176:3108–3114. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 37.Shichita T, et al. Pivotal role of cerebral interleukin-17-producing γδ T cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 38.Li GZ, et al. Expression of interleukin-17 in ischemic brain tissue. Scand J Immunol. 2005;62:481–486. doi: 10.1111/j.1365-3083.2005.01683.x. [DOI] [PubMed] [Google Scholar]
- 39.Liesz A, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 40.Nadig SN, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med. 2010;16:809–813. doi: 10.1038/nm.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao R, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 43.Kulik L, et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol. 2009;182:5363–5373. doi: 10.4049/jimmunol.0803980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diepenhorst GM, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg. 2009;249:889–899. doi: 10.1097/SLA.0b013e3181a38f45. [DOI] [PubMed] [Google Scholar]
- 47.Armstrong PW, et al. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. J Am Med Assoc. 2007;297:43–51. doi: 10.1001/jama.297.1.43. [DOI] [PubMed] [Google Scholar]
- 48.Shernan SK, et al. Impact of pexelizumab, an anti–C5 complement antibody, on total mortality and adverse cardiovascular outcomes in cardiac surgical patients undergoing cardiopulmonary bypass. Ann Thorac Surg. 2004;77:942–949. doi: 10.1016/j.athoracsur.2003.08.054. discussion 949–950. [DOI] [PubMed] [Google Scholar]
- 49.Verrier ED, et al. Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: a randomized trial. J Am Med Assoc. 2004;291:2319–2327. doi: 10.1001/jama.291.19.2319. [DOI] [PubMed] [Google Scholar]
- 50.He S, et al. A complement-dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice. J Clin Invest. 2009;119:2304–2316. doi: 10.1172/JCI38289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 52.Weissmüller T, et al. PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J Clin Invest. 2008;118:3682–3692. doi: 10.1172/JCI35874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Müller F, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Lupu F, Esmon CT. Inflammation, innate immunity and blood coagulation. Hamostaseologie. 2010;30:5–6. 8–9. [PubMed] [Google Scholar]
- 55.Esmon CT. Coagulation inhibitors in inflammation. Biochem Soc Trans. 2005;33:401–405. doi: 10.1042/BST0330401. [DOI] [PubMed] [Google Scholar]
- 56.Groger M, et al. Peptide Bβ15–42 preserves endothelial barrier function in shock. PLoS One. 2009;4:e5391. doi: 10.1371/journal.pone.0005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atar D, et al. Effect of intravenous FX06 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction results of the F.I.R.E. (efficacy of FX06 in the prevention of myocardial reperfusion injury) trial. J Am Coll Cardiol. 2009;53:720–729. doi: 10.1016/j.jacc.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 58.Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chekeni FB, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thakar CV, et al. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J Clin Invest. 2005;115:3451–3459. doi: 10.1172/JCI25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang Z, et al. Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3β phosphorylation. J Exp Med. 2010;207:867–880. doi: 10.1084/jem.20091704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- 63.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc Natl Acad Sci USA. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 65.Chen LW, et al. The two faces of IKK and NF-κB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 66.Piot C, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 67.Hausenloy DJ, Yellon DM. Time to take myocardial reperfusion injury seriously. N Engl J Med. 2008;359:518–520. doi: 10.1056/NEJMe0803746. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Sala-Mercado JA, et al. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–S184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klausner JM, et al. Reperfusion pulmonary edema. J Am Med Assoc. 1989;261:1030–1035. [PubMed] [Google Scholar]
- 71.de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med. 2003;167:490–511. doi: 10.1164/rccm.200207-670SO. [DOI] [PubMed] [Google Scholar]
- 72.Eckle T, et al. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008;111:5571–5580. doi: 10.1182/blood-2007-11-126763. [DOI] [PubMed] [Google Scholar]
- 74.Thompson LF, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosenberger P, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 76.Eltzschig HK, Collard CD. Vascular ischaemia and reperfusion injury. Br Med Bull. 2004;70:71–86. doi: 10.1093/bmb/ldh025. [DOI] [PubMed] [Google Scholar]
- 77.Hidalgo A, et al. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15:384–391. doi: 10.1038/nm.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 79.Eckle T, Kohler D, Lehmann R, El Kasmi KC, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 80.Eckle T, et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 81.Köhler D, et al. CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation. 2007;116:1784–1794. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 82.Petrowsky H, et al. A prospective, randomized, controlled trial comparing intermittent portal triad clamping versus ischemic preconditioning with continuous clamping for major liver resection. Ann Surg. 2006;244:921–928. doi: 10.1097/01.sla.0000246834.07130.5d. discussion 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Azoulay D, et al. Effects of 10 minutes of ischemic preconditioning of the cadaveric liver on the graft’s preservation and function: the ying and the yang. Ann Surg. 2005;242:133–139. doi: 10.1097/01.sla.0000167848.96692.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eckle T, et al. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–H2540. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]
- 85.Grenz A, et al. Use of a hanging-weight system for isolated renal artery occlusion during ischemic preconditioning in mice. Am J Physiol Renal Physiol. 2007;292:F475–F485. doi: 10.1152/ajprenal.00275.2006. [DOI] [PubMed] [Google Scholar]
- 86.Hart ML, et al. Use of a hanging-weight system for liver ischemic preconditioning in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1431–G1440. doi: 10.1152/ajpgi.00083.2008. [DOI] [PubMed] [Google Scholar]
- 87.Staat P, et al. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 88.Thibault H, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]
- 89.Bøtker HE, et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet. 2010;375:727–734. doi: 10.1016/S0140-6736(09)62001-8. [DOI] [PubMed] [Google Scholar]
- 90.Semenza GL. Life with oxygen. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 91.Aragonés J, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 92.Hill P, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:39–46. doi: 10.1681/ASN.2006090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bernhardt WM, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. 2010;21:2151–2156. doi: 10.1681/ASN.2010010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mazzone M, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brines ML, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA. 2000;97:10526–10531. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai Z, et al. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 97.Parsa CJ, et al. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999–1007. doi: 10.1172/JCI18200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fantacci M, et al. Carbamylated erythropoietin ameliorates the metabolic stress induced in vivo by severe chronic hypoxia. Proc Natl Acad Sci USA. 2006;103:17531–17536. doi: 10.1073/pnas.0608814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Najjar SS, et al. Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. J Am Med Assoc. 2011;305:1863–1872. doi: 10.1001/jama.2011.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen CH, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miller EJ, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 102.Qi D, et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119:3807–3816. doi: 10.1172/JCI39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ohsawa I, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 104.Wood KC, Gladwin MT. The hydrogen highway to reperfusion therapy. Nat Med. 2007;13:673–674. doi: 10.1038/nm0607-673. [DOI] [PubMed] [Google Scholar]
- 105.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 106.Lang JD, Jr, et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J Clin Invest. 2007;117:2583–2591. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gladwin MT, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. J Am Med Assoc. 2011;305:893–902. doi: 10.1001/jama.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 109.Elrod JW, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daniels IS, et al. A role of erythrocytes in adenosine monophosphate initiation of hypometabolism in mammals. J Biol Chem. 2010;285:20716–20723. doi: 10.1074/jbc.M109.090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 112.Volpato GP, et al. Inhaled hydrogen sulfide: a rapidly reversible inhibitor of cardiac and metabolic function in the mouse. Anesthesiology. 2008;108:659–668. doi: 10.1097/ALN.0b013e318167af0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chin BY, et al. Hypoxia-inducible factor 1α stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci USA. 2007;104:5109–5114. doi: 10.1073/pnas.0609611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Eltzschig HK, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 115.Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 117.Riegel AK, et al. Selective induction of endothelial P2Y6 nucleotide receptor promotes vascular inflammation. Blood. 2011;117:2548–2555. doi: 10.1182/blood-2010-10-313957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peng W, et al. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci USA. 2009;106:12489–12493. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eltzschig HK, et al. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113:224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eltzschig HK, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 122.Grenz A, et al. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol. 2007;18:833–845. doi: 10.1681/ASN.2006101141. [DOI] [PubMed] [Google Scholar]
- 123.Hart ML, et al. Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186:4367–4374. doi: 10.4049/jimmunol.0903617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Hart ML, et al. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008;22:2784–2797. doi: 10.1096/fj.07-103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK. SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol. 2010;184:4017–4024. doi: 10.4049/jimmunol.0901851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2011 Sep 19; doi: 10.1146/annurevphysiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 128.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85:1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hart ML, Jacobi B, Schittenhelm J, Henn M, Eltzschig HK. Cutting edge: A2B adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol. 2009;182:3965–3968. doi: 10.4049/jimmunol.0802193. [DOI] [PubMed] [Google Scholar]
- 130.Grenz A, et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Field JJ, Nathan DG, Linden J. Targeting iNKT cells for the treatment of sickle cell disease. Clin Immunol. 2011;140:177–183. doi: 10.1016/j.clim.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gao Z, et al. Novel short-acting A2A adenosine receptor agonists for coronary vasodilation: inverse relationship between affinity and duration of action of A2A agonists. J Pharmacol Exp Ther. 2001;298:209–218. [PubMed] [Google Scholar]
- 133.Hendel RC, et al. Initial clinical experience with regadenoson, a novel selective A2A agonist for pharmacologic stress single-photon emission computed tomography myocardial perfusion imaging. J Am Coll Cardiol. 2005;46:2069–2075. doi: 10.1016/j.jacc.2005.05.097. [DOI] [PubMed] [Google Scholar]
- 134.Thompson CA. FDA approves pharmacologic stress agent. Am J Health Syst Pharm. 2008;65:890. doi: 10.2146/news080038. [DOI] [PubMed] [Google Scholar]
- 135.Gladwin MT. Adenosine receptor crossroads in sickle cell disease. Nat Med. 2011;17:38–40. doi: 10.1038/nm0111-38. [DOI] [PubMed] [Google Scholar]
- 136.Zhang Y, et al. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17:79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bonauer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 138.Wang JX, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2011;17:71–78. doi: 10.1038/nm.2282. [DOI] [PubMed] [Google Scholar]
- 139.Qian L, et al. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;208:549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–743. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 142.Gladwin MT, Schechter AN. Nitric oxide therapy in sickle cell disease. Semin Hematol. 2001;38:333–342. doi: 10.1016/s0037-1963(01)90027-7. [DOI] [PubMed] [Google Scholar]
- 143.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]