

Abstract
In this work, a mixture of a sodium salt of carboxymethylcellulose (CMCNa) and polyethylene glycol diacrylate (PEGDA700) was used for the preparation of a microporous structure by using the combination of two different procedures. First, physical foaming was induced using Pluronic as a blowing agent, followed by a chemical stabilization. This second step was carried out by means of an azobis(2-methylpropionamidine)dihydrochloride as the thermoinitiator (TI). This reaction was activated by heating the sample homogeneously using a microwave generator. Finally, the influence of different CMCNa and PEGDA700 ratios on the final properties of the foams was investigated. The viscosity, water absorption capacity, elastic modulus and porous structure were evaluated for each sample. In addition, preliminary biological characterization was carried out with the aim to prove the biocompatibility of the resulting material. The foam, including 20% of PEGDA700 in the mixture, demonstrated higher viscosity and stability before thermo-polymerization. In addition, increased water absorption capacity, mechanical resistance and a more uniform microporous structure were obtained for this sample. In particular, foam with 3% of CMCNa shows a hierarchical structure with open pores of different sizes. This morphology increased the properties of the foams. The full set of samples demonstrated an excellent biocompatibility profile with a good cell proliferation rate of more than 7 days.
Keywords: foam, cellulose, polyethylene glycol, microwaves
1. Introduction
Foam is a two-phase system obtained by dispersion of a gas bubble phase in a continuous liquid or a solid phase [1]. Cellular solids are quite diffusive in nature, e.g. cork, sponge, coral, wood and pumice stone. Many foods have a cellular structure: bread has cells created by the fermentation of yeast; meringue is a foam of egg whites with sugar [2].
In previous years, different techniques for foaming different precursors, such as polymers, metals and ceramics, were developed with the aim to combine a low-density structure with increased mechanical properties, characteristic of the particular structure of the foams [3]. In particular, the properties of the foam are strictly correlated with the shape of the pores. There are two main groups of structures: closed or open pores. Closed pore-based foams have a cellular structure in which air bubbles are entrapped within a continuous macromolecular phase, whereas open pore foams have an interconnected cellular structure in which air flows through continuous channels. Closed pore foams are generally rigid and exhibit insulating properties, whereas open pore foams are generally flexible and permeable.
In addition to the above-mentioned properties, there are many other secondary characteristics that make this class of material attractive for many technological applications. Among them, properties such as good buoyancy, higher energy dissipation and low cost of production increased the manufacturing of polymer foams for countless applications. In particular, the production of automobile parts, footwear parts and packaging films benefits from all those properties [4].
The growing interest about polymeric foams has enabled understanding of the main principles controlling the foaming processes, starting from nucleation and ending with growth and stabilization of the gas bubbles.
The blowing agents used to generate the gaseous phase can be either chemical or physical blowing agents. In the first case, gas is commonly released from a chemical reaction as a by-product; in the second case, an inert gas is insufflated into the solution [5]. The most common foaming techniques are extrusion, injection moulding, rotational moulding [6] and freeze-drying [7].
Compared with other materials, polymeric foam manufacturing is a growing market. Polyurethane, polyvinyl chloride and polystyrene are the three most commonly used polymers, because they have the capability to form stable foams that are suitable for a number of applications. However, they have a negative environmental impact owing to the intrinsic problem of treating their waste products. For this reason, researchers have focused their attention on the use of biodegradable polymers, such as cellulose, starch, polycaprolactone (PCL), polylactic acid (PLA), aliphatic polyesters and aliphatic–aromatic polyesters [4]. To this aim, several research projects have been carried out using different biodegradable polymers and different foaming techniques. In particular, the combination of the extrusion foaming technique and starch as the natural polymer is widely used in the biodegradable packaging industry. Starch is the second most abundant polysaccharide in nature and it is mainly used in the manufacturing of loose-fill packaging materials owing to its low cost and advantages [8–16].
A relevant example of cellulose foams was obtained by Gao et al. [7], who realized porous bacterial cellulose sponges as a tissue engineering scaffold by using the emulsion freeze-drying technique.
Owing to its good biocompatibility combined with relevant mechanical properties, cellulose and its derivatives were used to develop biomedical application in vivo as tissue engineered scaffolds and in vitro as phantoms able to mimic the acoustic properties of human tissue in diagnostic ultrasound [17–18].
In this study, cellulose was selected as a precursor to create stable foams. Cellulose is the most abundant renewable polysaccharide in nature. It has a remarkable absorption capacity owing to its hydrophilic nature and it is suitable for use in this particular field [19,20]. A sodium salt of carboxymethylcellulose (CMCNa) was mixed with polyethylene glycol diacrylate (PEGDA) because it can be easily thermo-initiated leading to a stable foam. In addition, the PEGDA polymer was selected owing to its higher solubility in water and water solutions, and the capacity to be cross-linked in the presence of free radicals. These properties combined with chemical, mechanical and biological properties of PEG-based systems show the versatility of this material in a wide range of biomedical applications such as drug delivery vehicles, tissue engineered scaffolds and in in vitro applications. It is important to note that the ester linkages introduced upon acrylation of the PEG molecule lead to slow degradation in vivo [21]. However, PEGDA could be easily modified with proper functional groups able to induce a different degradation profile. Mann et al. [22] developed a tissue engineered hydrogel scaffold through photopolymerization of PEG derivatives with proteolytically degradable peptides. Enzymes, such as collagenase and elastane, involved in cell migration degrade these hydrogels.
The aim of this work was to develop an innovative technique leading to the creation of a cellulose-based foam material using the combination of both physical and chemical surfactants as physical blowing agents. Our approach was to study the polymerization initiated by a thermo-initiator activated by microwaves. Different blends of cellulose and PEGDA were tested and the viscosity, absorption capacity and compressibility were evaluated for each sample. Finally, a preliminary biological characterization was carried out in order to prove the suitability of this material for further applications in the field of tissue engineering.
2. Material and methods
2.1. Materials
The two polymers used in this study were the sodium salt of CMCNa (MW 700 000 Da, DS 0.7) and PEGDA (PEGDA700, MW 700 Da). Pluronic F-127 was selected as the blowing agent. All the chemicals were purchased from Sigma-Aldrich and used without further purification.
Azobis(2-methylpropionamidine)dihydrochloride was selected as the thermo-initiator owing to its higher solubility in water; it was purchased from Wako Chemicals USA and used as-received.
2.2. Synthesis of CMCNa–PEGDA foams
The CMCNa water solutions were prepared by using different polymer to water ratios (2 : 3 : 4 wt%). The solutions were gently mixed in a 100 ml batch volume in a glass beaker using a magnetic stirrer. After the CMCNa's complete dissolution, a fixed concentration of thermo-initiator (3% w/w on PEGDA) was added to the CMCNa solution and mixed till completely dissolved (5 min), before the addition of PEGDA. The PEGDA was finally added, in the concentrations shown in table 1, and allowed to mix (1 h). After complete PEGDA dissolution, the Pluronic was added and mixed at 800 r.p.m. for 10 min in order to incorporate and homogeneously distribute air bubbles in a stable form into the mixture. For this study, three independent preparations were performed for each composition. All the measurements were performed in triplicate on these samples.
Table 1.
Composition of CMCNa–PEGDA foams.
| sample ID | CMCNa % (w/V) | PEGDA% (w/w) |
|---|---|---|
| 20P80C2 | 2 | 20 |
| 20P80C3 | 3 | 20 |
| 30P70C2 | 2 | 30 |
| 30P70C3 | 3 | 30 |
| 40P60C2 | 2 | 40 |
| 40P60C3 | 3 | 40 |
2.3. Thermo-polymerization
The microwave reactor (MTS 1200 Mega, Milestone Inc., Shelton, CT 06484) was used to chemically stabilize the foam. The emulsion was poured into cylindrical Teflon moulds and polymerized in the reactor at 900 W for different time points according to the timeline reported in table 2 till complete stabilization. The resulting samples were dried in a vacuum oven at 45°C. The samples were analysed without any further modification.
Table 2.
Curing time of the CMCNa–PEGDA samples. ‘Minus’ symbols, uncross-linked; ‘plus’ symbols, cross-linked.
| sample ID | curing time |
|||
|---|---|---|---|---|
| 45 s | 1 min | 1 min 30 s | 1 min 45 s | |
| 20P80C2 | − | − | + | |
| 20P80C3 | − | − | − | + |
| 30P70C2 | + | |||
| 30P70C3 | − | − | + | |
| 40P60C2 | − | + | ||
| 40P60C3 | − | − | + | |
2.4. Characterization
2.4.1. Measurement of viscosity
The viscosity of solutions 20P80C2, 20P80C3, 30P70C2, 30P70C3, 40P60C2 and 40P60C3 was analysed by means of a rotational rheometer (Ares, Rheometric Scientific) equipped with parallel plates with a diameter of 50 mm. The viscosity was measured on three independent samples resulting from a single preparation for each concentration. The resulting values were calculated as an average of the three measurements. The shear rate was fixed from 0.1 to 100 s−1 with step increments of 0.02 s−1. The Carreau–Yasuda model was used to fit the experimental data resulting from the tested sample. The Carreau–Yasuda model is described in equation (2.1):
| 2.1 |
where 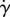 is the shear rate, η∞ the shear rate viscosity at infinite shear rates, η0 the zero shear rate viscosity and λ the relaxation time in seconds. Parameters ‘a’ and ‘n’ can be varied to obtain the power-law region of the shear-thinning behaviour. Relaxation time, λ, when increased while keeping all the other parameters constant, shifts the power-law region to the right. Thus, it dictates the shear-rates at which the power-law transition takes place. Parameter ‘a’ dictates the curvature at the top of the curve. A higher ‘a’ means a sharper transition of viscosity into the power-law regime. The power-law index ‘n’ governs the power-law regime in general. A knowledge of these parameters becomes necessary while implementing a Carreau–Yasuda model into numerical simulations.
is the shear rate, η∞ the shear rate viscosity at infinite shear rates, η0 the zero shear rate viscosity and λ the relaxation time in seconds. Parameters ‘a’ and ‘n’ can be varied to obtain the power-law region of the shear-thinning behaviour. Relaxation time, λ, when increased while keeping all the other parameters constant, shifts the power-law region to the right. Thus, it dictates the shear-rates at which the power-law transition takes place. Parameter ‘a’ dictates the curvature at the top of the curve. A higher ‘a’ means a sharper transition of viscosity into the power-law regime. The power-law index ‘n’ governs the power-law regime in general. A knowledge of these parameters becomes necessary while implementing a Carreau–Yasuda model into numerical simulations.
2.4.2. Measurement of water absorption capacity
The water absorption capacity of the foams was measured by placing four samples for each concentration, previously dried and weighted, into a fixed volume (50 ml) of distilled water for 24 h at room temperature. The samples were removed from the water using a plastic sieve by removing the excess of water, and then weighted. The water absorption was calculated using equation (2.2):
| 2.2 |
where WA is the water absorption capacity value, Wswell is the swollen foam weight and Wdry is the dried foam weight.
2.4.3. Compression test
Four samples for each composition of the foams were tested under the uniaxial compression test till 30% of deformation and a constant deformation rate of 0.002 mm s−1 using the Ares rheometer equipped with parallel plates (uniaxial compression mode test) were achieved. A preload of 20 g was applied in order to start the test at the same value in each sample. Before starting the test, the diameter and the weight of each sample were measured in order to calculate the stress, and to normalize Young's modulus on the average weight basis. Stress and strain were calculated according to equations (2.3) and (2.4)
| 2.3 |
and
| 2.4 |
where σ and ɛ are the stress and strain, respectively, F is the force, Δl is the deformation, A0 and l0 are the original sample's cross-sectional area and the weight, respectively.
The average of Young's modulus was calculated by linear interpolation of the first 10 points (linear range) of the compression curve of each composition. The results were normalized taking into account the weight of the tested sample for each composition. Each composition was tested in triplicate.
2.4.4. Scanning electron microscopy
The morphology of the cross-section for each sample was analysed by environmental scanning electron microscopy (ESEM, XL30 FEI) in low vacuum modality and by applying a tension of 25 kW. The dried samples were cut into slices by using a surgical blade. The samples were placed on the SEM sample holder using double-sided adhesive tape and were observed without any further manipulation.
2.4.5. Biological evaluation
The biocompatibility of the scaffold materials was investigated in vitro using the MG-63 human osteosarcoma cell line, which is widely used to study the biocompatibility of orthopaedic and dental materials (LONZA, Milan, Italy). These cells are at a relatively early stage in the osteoblastic lineage, and therefore they may represent a good model for examining the initial stages in cell osteogenic differentiation induced by biomaterials.
The culture medium was Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum (Gibco-BRL Life Technologies, Italy), 2 mM glutamine and antibiotics (penicillin G sodium 100 U ml−1, streptomycin 100 µg ml−1, Euroclone). Proliferation was evaluated without osteogenic culture medium. The MG-63 cell lines were trypsinized using 0.1% trypsin and 0.1% ethylenediaminetetracetic acid for 5 min, centrifuged at 4000g for 5 min and seeded on material (1 × 103 cells per scaffold). Materials were sterilized by UV irradiation for 1 h. Cells grown on polystyrene plates were used as control.
2.5. Cytotoxic assay
Elution studies were performed by adding 2.5 ml of DMEM solution to each 0.1 g of material according to the ISO10993–5 guidelines. These were placed on a rotating mixer for a period of 3 days; the eluents were removed at different times, and 500 µl was pipetted into sterile 24-well cell culture plates (Corning) previously seeded with 1 × 103cell/scaffold MG63. The negative (non-toxic) control was tissue culture plastic with DMEM solution without material eluent. The plates were further incubated at 37°C, 5% CO2 and 95% air humidity for 24 h. Cell viability was evaluated using the alamarBlue assay.
2.6. alamarBlue assay
Cell viability and proliferation were evaluated by using the alamarBlue assay. This is based on a redox reaction that occurs in the mitochondria of the cells; the coloured product is transported out of the cell and can be measured spectrophotometrically. The cell-material constructs were removed from culture plates at 1, 4 and 7 days, rinsed with PBS (Sigma–Aldrich, Italy), and placed into 24-well culture plates. For each construct, 1 ml of DMEM without Phenol Red (HyClone, UK) containing 10% (v/v) alamarBlue (AbD Serotec Ltd, UK) was added, followed by incubation in 5% CO2 diluted atmosphere for 3 h at 37°C and 5%. Two hundred microlitres of the solution were subsequently removed from the wells and transferred in a 96-well plate. The optical density was immediately measured with a spectrophotometer (multilabel counter, 1420 Victor, Perkin Elmer, Milano, Italy) at wavelengths of 570 and 600 nm. The number of viable cells correlates with the magnitude of dye reduction and is expressed as the percentage of AB reduction (%AB reduction), according to the manufacturer's protocol. Data relate to the per cent reduction between treated cells and a control (media plus alamarBlue but no cells) in the viability assay. Each experiment was performed three times.
The percentage of reduction of alamarBlue is expressed by equation (2.5):
| 2.5 |
where O1 is the molar extinction coefficient (E) of oxidized alamarBlue (Blue) at 570 nm; O2, E of oxidized alamarBlue at 600 nm; R1, E of reduced alamarBlue (Red) at 570 nm; R2, E of reduced alamarBlue at 600 nm; A1, absorbance of test wells at 570 nm; A2, absorbance of test wells at 600 nm; N1, absorbance of negative control well (media plus alamarBlue but no cells) at 570 nm and N2, absorbance of negative control well (media plus alamarBlue but no cells) at 600 nm.
2.7. Statistical analysis
One-way analyses of variance (ANOVA) were performed to detect significant effects among treatments with the post-Bonferroni t-test. Results were considered to be significant at p < 0.05.
3. Results and discussion
3.1. Foam formation
The whole set of samples was prepared by adding the same amount of Pluronic. Different concentrations of cellulose and PEGDA were tested as listed in table 1. During the foaming procedure, solution 20P80C2 increased in volume by about 150% of the initial volume after the addition of Pluronic and was quite stable even after agitation was stopped. The increased volume is due to the incorporation and persistency of stable air bubbles inside the solution. Sample 20P80C3 was stable and showed an increased volume of about 100%. All the foams including the 2% CMCNa were homogeneous; by increasing both the CMCNa and PEGDA concentrations, the resulting solution became quite inhomogeneous and unstable (significant reduced volume after the agitation was stopped). Only sample 20P80C3 was comparable in terms of stability and homogeneity with those of 2% CMCNa. All the above-mentioned effects are probably owing to the differences in the viscosity due to the different polymer concentrations.
3.2. Thermo-polymerization after foam formation
The curing time for each sample is reported in table 2. It is evident that the curing time is different for the tested samples. In particular, there is not a clear trend that can confirm the influence of PEGDA concentration on the time of curing. On the contrary, looking at the effect of CMCNa concentration within each group in which the PEGDA concentration is constant, it seems that it is proportional to an increased curing time. This behaviour is due to decreased chain mobility in relation to a higher CMCNa concentration (i.e. increasing in viscosity).
3.3. Characterization
3.3.1. Measurement of viscosity
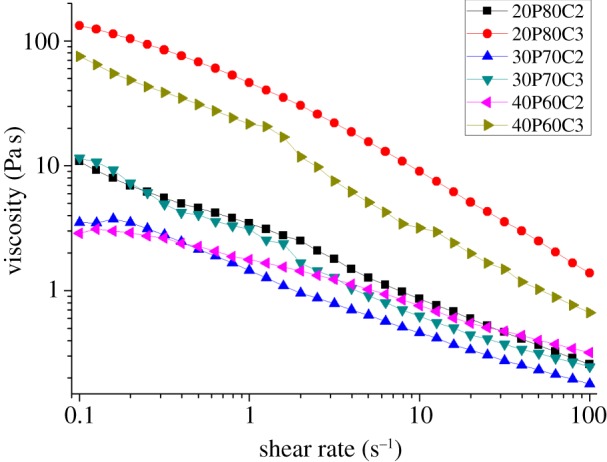
Based on the previous discussion, it is evident that the stability of the foam was highly influenced by viscosity of the solutions. Different ratios of PEGDA–CMCNa were tested in order to check the viscosity of the starting solution. Figure 1 displays the results of the viscosity measurements of PEGDA–CMCNa ratios. The solutions 20P80C2 and 20P80C3 exhibit higher viscosity when compared with other compositions within the same group (2% or 3% CMCNa), resulting in more stable foams when compared with the rest of the samples. The viscosity decreases by increasing PEGDA and increases with increasing CMCNa concentration. The solutions 30P70C2 and 30P70C3 show lower viscosity leading to a reduction of the stability of the foams. The viscosity curves of the solutions with 30% and 40% of PEGDA present regions of nonlinearity owing to the non-homogeneous solution. In these cases, the presence of lumps in the solution was observed. In particular, this effect could be noted at the 3% of CMCNa probably owing to an electrostatic effect between the polymers chains. In fact, the CMCNa is a polyelectrolyte and in water solutions exhibits a dissociate form, with the formation of –CH2COOH– groups and Na+ ions. The negative-charged chains, with the presence of non-polyelectrolyte polymer, for example, the PEGDA, creates a phase separation between the two polymers [23,24]. In general, the data show the characteristic shear thinning behaviour of a polymer over a wide range of shear rates. This behaviour is confirmed by applying the first approximation of the Carreau–Yasuda model. Figure 2 shows overlapping of the 20P80C2 sample with the corresponding numerical fitting obtained using the above-mentioned model. It is interesting to note that there is a good correlation between the experimental data and the model, especially between 1 and 20 s−1.
Figure 1.
Effects of different ratios of PEGDA and 2% and 3% CMCNa solution on viscosity versus shear rate. (Online version in colour.)
Figure 2.
Overlapping of 20P80C2 sample with the corresponding numerical fitting obtained using the Carreau–Yasuda model (λ = 3, a = 1, n = 0.2).
3.3.2. Measurement of water absorption capacity
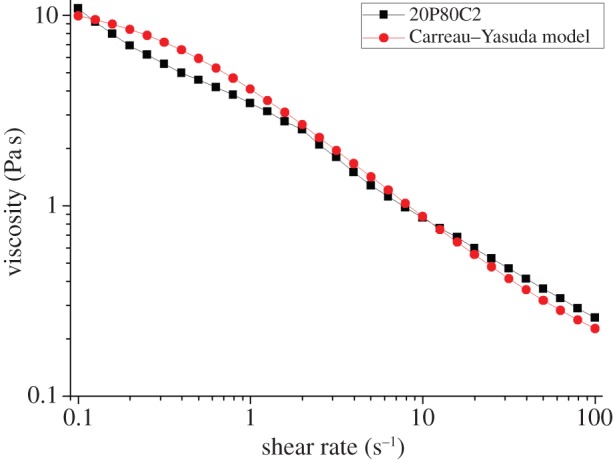
The water absorption capacity values of PEGDA–CMCNa foams at different concentrations are shown in figure 3. The thermo-initiated foams are swelled by immersion in distilled water and allowed to soak in the media for 24 h at room temperature till equilibrium is achieved. The water absorption capacity resulted from a combination of three different effects: elastic response of the cross-linked polymer, polymer solvent interaction and the Donnan effect. This effect consists of an imbalance of charges that is established between the inside and the outside of the gel, following which more water enters the gel generating an osmotic pressure. In addition to those effects, the water uptake is and principally owing to the capillarity effect of the porous structure.
Figure 3.
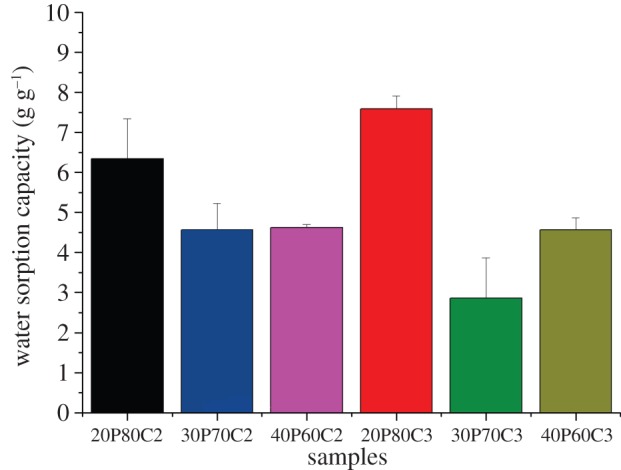
Effects of different ratio of PEGDA and 2% and 3% CMCNa solution on water absorption capacity.
The maximum swelling ratio was approximately 7.5 g g−1 and it was measured for sample 20P80C3. The water absorption capacity decreases with the increase in PEGDA because of an increased degree of cross-linking on one side and due to a reduction of the Donnan effect on the other [17].
It is evident that the samples with a concentration of PEGDA above the limit of 20% show reduced water retention owing to a reduced Donnan effect. In particular, sample 30P70C3, having the lower absorption capacity, has shown lower stability combined with higher inhomogeneity.
In contrast, the higher water absorption capacity reported in figure 3 for the sample 20P80C3 is addressed to the increasing of water absorption effect of the cellulose.
3.3.3. Compression test
The compressibility of a structure is an indicator of the rigidity of the foam. Different blends of PEGDA–CMCNa were tested using an Ares rheometer at room temperature. Figures 4 and 5 display the results of the mechanical test. Foam containing 30% PEGDA has an increased Young's modulus compared with the others (as can be seen in table 3), probably because of the presence of a second, more rigid phase (PEGDA regions). In contrast, the rigidity decreases with the increase in PEGDA because an excess of uncross-linked polymer can function as plasticizer. The increase in CMCNa increases the rigidity owing to a higher polymer density leading to the presence of physical entanglements resulting in a higher degree of cross-linking [18].
Figure 4.
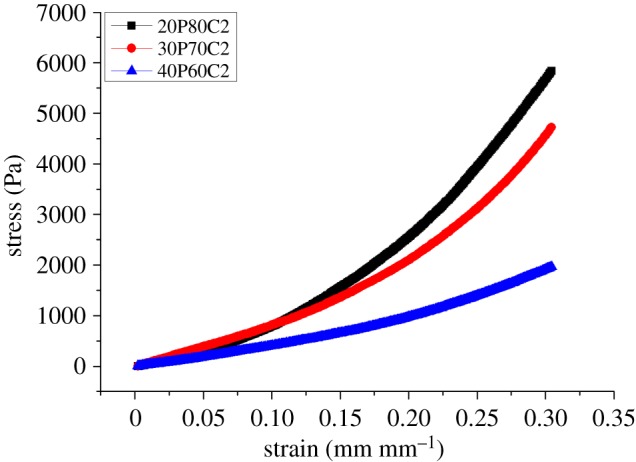
Effects of different ratios of PEGDA and 2% CMCNa solution on compressibility.
Figure 5.
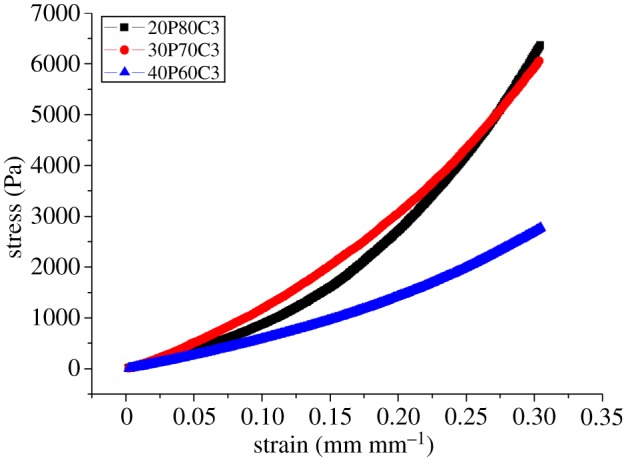
Effects of different ratio of PEGDA and 3% CMCNa solution on compressibility.
Table 3.
Young's modulus of each composition.
| sample ID | E (Pa) | s.d. (Pa) |
|---|---|---|
| 20P80C2 | 5032 | ±95 |
| 20P80C3 | 6747 | ±183 |
| 30P70C2 | 8227 | ±246 |
| 30P70C3 | 7564 | ±24 |
| 40P60C2 | 3614 | ±43 |
| 40P60C3 | 4422 | ±58 |
Figure 6 shows the results of the normalization of Young's modulus based on the weight of each sample. With the exception of the foam containing 30% of PEGDA, owing to the above-mentioned considerations, the graph confirmed that foams containing 20% PEGDA have greater mechanical properties, in terms of compressibility.
Figure 6.
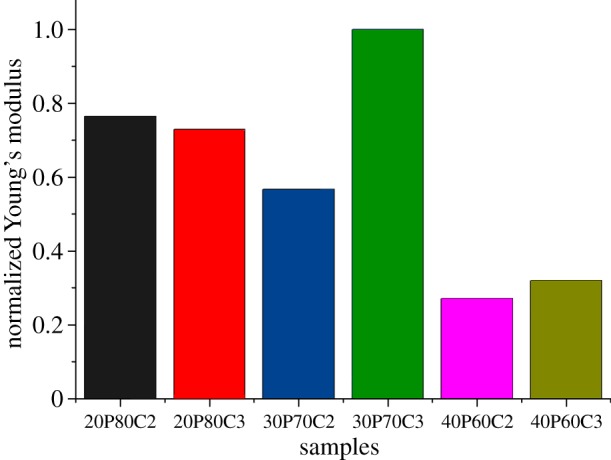
Normalization of Young's modulus taking into account the effective weight of each sample.
3.3.4. Scanning electron microscopy
Figure 7 shows the SEM images of foam 20P80C2. It is evident that this foam shows a hierarchical structure with open pores of different sizes. In particular, the shape of the pore is shown in figure 7d. By increasing the CMCNa concentration (at fixed PEGDA%), it is possible to observe a tighter structure where each pore presents thicker walls according to the increased cellulose concentration (figure 8). In particular, it is possible to observe a secondary structure inside the walls of the pore. In figure 9, this structure could be easily detected. It is remarkable that this secondary structure follows the same template of the primary one. Moreover, in figure 9c,d, it is also possible to observe a third order of structure with the same rules. This is owing to the higher viscosity of the solution that was able to incorporate and retain a larger amount of small air bubbles, creating a more stable solution. This morphology increases the microporosity of the network, thus leading to higher water absorption capacity of this foam. In addition to this, a higher porosity creates an enhanced exposure of the CMCNa chains creating an increased Donnan effect.
Figure 7.

SEM micrographs of sample 20P80C2: cross section at (a) 24×, (b) 103×, (c) 200×, (d) 500×.
Figure 8.

SEM micrographs of the sample 20P80C3: cross-section at (a) 21×, (b) 96×, (c) 200×, (d) 500×.
Figure 9.

SEM micrographs of the sample 20P80C3: cross-section at (a) 800×, (b) 1000×, (c) 2500×, (d) 6500×.
3.3.5. Biological evaluation
The results of the indirect contact assay (alamarBlue) after 24 h exposure of the cells to the supernatants from the leaching studies are shown in figure 10. The scaffold materials did not show a significant release of toxic compounds; in fact, the cells exposed for 24 h to eluents (3 days) reached values similar to the negative control. This behaviour is probably owing to the absence of the biodegradation phenomenon in the short time of cell culture. Moreover, the results of the direct tests are shown in figure 11. They indicated that the scaffold materials have no negative effect on the metabolic activity of the cells. Cell viability and proliferation increase in a statistically significant manner with culture time. Moreover, it is possible to observe that even if 20P80C2 material shows low values of alamarBlue at day 1, it becomes higher at 4 and 7 days, probably owing to the presence of a greater amount of cellulose in the material that improves the cell proliferation [20].
Figure 10.
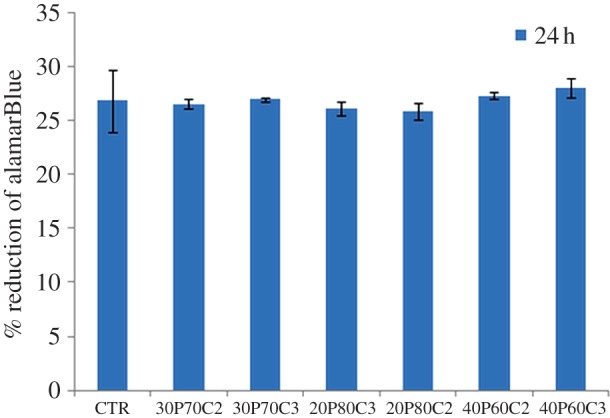
Cytotoxicity assays: alamarBlue after 24 h. (Online version in colour.)
Figure 11.
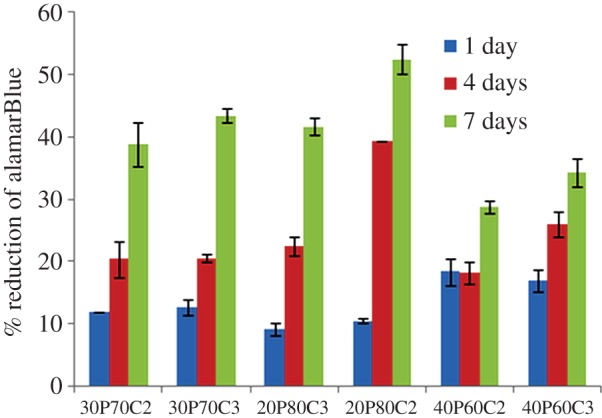
Cell Proliferation: short times of 1, 4 and 7 days.
4. Conclusion
An innovative procedure for the preparation of CMCNa and PEGDA foams was developed, and the effects of different mixtures of PEGDA–CMCNa on physical and mechanical properties were characterized. Microporous PEGDA–CMCNa-based foams have been successfully prepared by using a combination of physical foaming and thermo-activated polymerization with microwaves. Different concentrations of PEGDA and CMCNa influence the viscosity, and the stability of the foam before thermo-polymerization. The more stable foams were those with 20% PEGDA700, resulting in both greater mechanical properties and greater water absorption capacity. In particular, the foams comprising 3% CMCNa show a hierarchical structure composed of three orders. This morphology is probably due to a perfect balance between the viscosity and the electrostatic repulsion that takes place between the polymer chains. This work demonstrated that physical foaming followed by thermo-polymerization induced by microwave is a quick and cheap foaming method to realize structures with open porosity. In addition, biological characterization demonstrates that the materials have no negative effect on the metabolic activity of MG63 cells. Cell viability and proliferation increase with culture time for all the samples.
In future, this method may be optimized to better control the size of the pores (thus increasing the water absorption and the mechanical properties) in order to create and to obtain completely biodegradable foam.
References
- 1.Karim AA, Wai CC. 1999. Characteristics of foam prepared from starfruit (Averrhoa carambola L.) puree by using methyl cellulose. Food Hydrocolloid. 13, 203–210. ( 10.1016/S0268-005X(98)00086-1) [DOI] [Google Scholar]
- 2.Nussinovitch A, Gershon Z, Peleg L. 1988. Characteristics of enzymatically produced agar-starch sponges. Food Hydrocolloid. 12, 105–110. ( 10.1016/S0268-005X(98)00047-2) [DOI] [Google Scholar]
- 3.Klempner D, Sendijarevic V. 2004. Handbook of polymeric foams and foams technology, pp. 5–53. Cincinnati, OH: Hanser. [Google Scholar]
- 4.Khemani KC. 1997. ACS Symposium Series Polymeric Foams: An Overview. In pp. 53–70. Washington, DC: American Chemical Society. [Google Scholar]
- 5.Eaves D. 2004. Handbook of polymeric foams, pp. 180–189. Shawbury UK: Rapra Technology; limited. [Google Scholar]
- 6.Greco A, Maffezzoli A, Manni O. 2005. Development of polymeric foams from recycled polyethylene and recycled gypsum. Polym. Degrad. Stabil. 90, 256–263. ( 10.1016/j.polymdegradstab.2005.01.026) [DOI] [Google Scholar]
- 7.Gao C, Wan Y, Yang C, Dai K, Tang T, Luo H, Wang J. 2011. Preparation and characterization of bacterial cellulose sponge with hierarchical pore structure as tissue engineering scaffold. J. Porous Mater. 18, 139–145. ( 10.1007/s10934-010-9364-6) [DOI] [Google Scholar]
- 8.Fang Q, Hanna MA. 2000. Functional properties of polylactic acid starch-based loose-fill packaging foams. Cereal Chem. 77, 779–783. ( 10.1094/CCHEM.2000.77.6.779) [DOI] [Google Scholar]
- 9.Fang Q, Hanna MA. 2001. Preparation and characterization of biodegradable copolyester-starch based foams. Bioresource Technol. 78, 115–122. ( 10.1016/S0960-8524(01)00013-X) [DOI] [PubMed] [Google Scholar]
- 10.Willett JL, Shogren RL. 2002. Processing and properties of extruded starch/polymer foams. Polymer 43, 5935–5947. ( 10.1016/S0032-3861(02)00497-4) [DOI] [Google Scholar]
- 11.Guan J, Eskridge KM, Hanna MA. 2004. Functional properties of extruded acetylated starch–cellulose foams. J. Polym. Environ. 12, 113–121. ( 10.1023/B:JOOE.0000038542.98493.2c) [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Hanna MA. 2005. Preparation and properties of biodegradable foams from starch acetate and poly(tetramethylene adipate-co-terephthalate). Carbohydrate Polym. 59, 521–529. ( 10.1016/j.carbpol.2004.11.007) [DOI] [Google Scholar]
- 13.Guan J, Hanna MA. 2006. Selected morphological and functional properties of extruded acetylated starch–cellulose foams. Bioresource Technol. 97, 1716–1726. ( 10.1016/j.biortech.2004.09.017) [DOI] [PubMed] [Google Scholar]
- 14.Lee SY, Chen H, Hanna MA. 2008. Preparation and characterization of tapioca starch–poly(lactic acid) nanocomposite foams by melt intercalation based on clay type. Ind. Crops Prod. 28, 95–106. ( 10.1016/j.indcrop.2008.01.009) [DOI] [Google Scholar]
- 15.Salgado PR, Schmidt VC, Ortiz SEM, Mauri AN, Laurindo JB. 2008. Biodegradable foams based on cassava starch, sunflower proteins and cellulose fibers obtained by a baking process. J. Food Eng. 85, 435–443. ( 10.1016/j.jfoodeng.2007.08.005) [DOI] [Google Scholar]
- 16.Stagner J, Narayan R. 2011. Preparation and properties of biodegradable foams. J. Polym. Environ. 19, 598–606. ( 10.1007/s10924-011-0309-1) [DOI] [Google Scholar]
- 17.Sannino A, Madaghiele M, Demitri C, Scalera F, Esposito A, Esposito V, Maffezzoli A. 2010. Development and characterization of cellulose-based hydrogels for use as dietary bulking agents. J. Appl. Polym. Sci. 115, 1438–1444. ( 10.1002/app.30956) [DOI] [Google Scholar]
- 18.Demitri C, Del Sole R, Scalera F, Sannino A, Vasapollo G, Maffezzoli A, Ambrosio L, Nicolais L. 2008. Novel superabsorbent cellulose-based hydrogels crosslinked with citric acid. J. Appl. Polym. Sci. 110, 2453–2460. ( 10.1002/app.28660) [DOI] [Google Scholar]
- 19.Sannino A, Demitri C, Madaghiele M. 2009. Biodegradable cellulose-based hydrogels: design and applications. Materials 2, 253–273. ( 10.3390/ma2020353) [DOI] [Google Scholar]
- 20.Raucci MG, Alvarez-Perez MA, Demitri C, Sannino A, Ambrosio L. 2012. Proliferation and osteoblastic differentiation of hMSCs on cellulose-based hydrogels. J. Appl. Biomater. Funct. Mater. 10, 302–307. ( 10.5301/JABFM.2012.10366) [DOI] [PubMed] [Google Scholar]
- 21.Browning MB, Cosgriff-Hernandez E. 2012. Development of a biostable replacement for PEGDA hydrogels. Biomacromolecules 13, 779–786. ( 10.1021/bm201707z) [DOI] [PubMed] [Google Scholar]
- 22.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. 2001. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: synthetic ECM analogs for tissue engineering. Biomaterials 22, 3045–3051. ( 10.1016/S0142-9612(01)00051-5) [DOI] [PubMed] [Google Scholar]
- 23.Sannino A, Nicolais L. 2005. Concurrent effect of microporosity and chemical structure on the equilibrium sorption properties of cellulose-based hydrogels. Polymer 43, 4676–4685. ( 10.1016/j.polymer.2005.03.072) [DOI] [Google Scholar]
- 24.Sannino A, Netti PA, Madaghiele M, Coccoli V, Luciani A, Maffezzoli A, Nicolais L. 2006. Synthesis and characterization of macroporous poly(ethylene glycol)-based hydrogels for tissue engineering application. J. Biomed. Mater. Res. 79A, 229–236. ( 10.1002/jbm.a.30780) [DOI] [PubMed] [Google Scholar]