

Abstract
Phase diagrams offer a wealth of thermodynamic information on aqueous mixtures of bilayer-forming lipids and micelle-forming detergents, providing a straightforward means of monitoring and adjusting the supramolecular state of such systems. However, equilibrium phase diagrams are of very limited use for the reconstitution of membrane proteins because of the occurrence of irreversible, unproductive processes such as aggregation and precipitation that compete with productive reconstitution. Here, we exemplify this by dissecting the effects of the K+ channel KcsA on the process of bilayer self-assembly in a mixture of Escherichia coli polar lipid extract and the nonionic detergent octyl-β-d-glucopyranoside. Even at starting concentrations in the low micromolar range, KcsA has a tremendous impact on the supramolecular organization of the system, shifting the critical lipid/detergent ratios at the onset and completion of vesicle formation by more than 2-fold. Thus, equilibrium phase diagrams obtained for protein-free lipid/detergent mixtures would be misleading when used to guide the reconstitution process. To address this issue, we demonstrate that, even under such nonequilibrium conditions, high-sensitivity isothermal titration calorimetry can be exploited to monitor the progress of membrane-protein reconstitution in real time, in a noninvasive manner, and at high resolution to yield functional proteoliposomes with a narrow size distribution for further downstream applications.
Integral membrane proteins (IMPs) play key roles in many physiological processes and account for more than half of all drug targets.1 Most approaches to IMPs depend on detergents, which serve as disintegrators of cellular membranes and as dispersing agents during purification and biochemical, biophysical, or structural analyses.2−4 However, vectorial IMP functions such as signaling and transport can be studied only in the context of a membrane that separates two aqueous compartments.5 The most popular model environments for investigating such functions are lipid bilayers, which mimic the most essential features of biological membranes.6 Even in cases where vectorial properties are irrelevant, protein incorporation into bilayer membranes is desirable because these provide a more native-like environment than detergent micelles.7 Thus, purified IMPs need to be reconstituted from a detergent-solubilized state into a well-defined and controllable bilayer system,8,9 which may be achieved by addition of lipid followed by dilution10 or detergent removal through dialysis,5 gel filtration chromatography,11 hydrophobic adsorption,12 or complexation.13 Irrespective of the method used, reconstitution is often an intractable, lengthy trial-and-error procedure.14 On the premise that the development of more efficient and rational reconstitution strategies could benefit from a better understanding and control of the self-assembly process resulting in proteoliposomes, the complex interplay among IMPs, lipids, and detergents has been analyzed in considerable detail.2−17 Since proteoliposomes for functional assays are usually produced at rather low protein densities, the behavior of protein/lipid/detergent mixtures might be expected to be dominated by the properties of the lipid and the detergent,8 whose interactions therefore have been the subject of particular scrutiny.18
The supramolecular assemblies in mixtures of bilayer-forming lipids and micelle-forming detergents can be approximated as pseudophases,19 whose prevalence as a function of composition is captured in a phase diagram (see below). In the simplest case, two ranges featuring either micelles or bilayer structures are separated from one another by a coexistence range.20 At a given detergent concentration, two thresholds referred to as the solubilization (SOL) and saturation (SAT) boundaries indicate the lipid concentrations at which bilayer formation sets in and is completed, respectively. Phase diagrams provide fundamental insights into lipid/detergent interactions21 and have practical applications by enabling tight control of the supramolecular state of, for instance, lipidic drug-delivery systems.22 Their relevance to IMP reconstitution draws from the observation that the reconstitution success depends on the phase ranges explored during proteoliposome formation.8,9,15−17 For example, while several nonionic detergents with sugar-based headgroups can catalyze IMP insertion without requiring vesicle dissolution, Triton and cholate depend on partial or complete solubilization, respectively, before efficient reconstitution can be initiated.9 Thus, knowledge of the pertinent lipid/detergent phase diagram should be expected to be a promising starting point for a judicious approach to IMP reconstitution.23
In practice, however, the wealth of physicochemical data collected on numerous lipid/detergent combinations18 is seldom exploited for the optimization of solubilization and reconstitution protocols for IMPs24 because such efforts are frustrated by the finding that proteins themselves may change the supramolecular behavior of the system.10,25,26 In part, this might be due to a direct influence on the partition equilibrium between micellar and bilayer structures, which is particularly relevant for IMP solubilization from protein-rich membranes after overexpression26 or for reconstitution at high protein densities required for 2D crystallization.10,25 However, even much lower protein concentrations typical of functional reconstitution may render phase diagrams of simple lipid/detergent systems futile. This is because IMP reconstitution is, in the vast majority of cases, a kinetically rather than a thermodynamically controlled process, as productive reconstitution is almost never complete but competes with unproductive reactions, namely, aggregation and precipitation.27−29 These irreversible processes render IMP reconstitution hard to control and, more specifically, result not only in a loss of protein but also in a reduction of the actual concentrations of lipid and detergent, thus rendering useless any phase diagram based on nominal concentrations. These problems are further exacerbated by the scarcity of noninvasive methods capable of monitoring the progress of a reconstitution experiment in real time.
For simple lipid/detergent systems, isothermal titration calorimetry (ITC) is an excellent technique for following vesicle solubilization and reconstitution,20,21 offering unique accuracy, resolution, and sensitivity as well as robustness against light scattering and other experimental issues. Here, we demonstrate the dramatic extent to which phase diagrams of lipid/detergent mixtures may be misleading during IMP reconstitution and establish ITC as a powerful, label-free online method for following proteoliposome assembly even under nonequilibrium conditions. This is exemplified for the reconstitution of the channel protein KcsA30 from the nonionic detergent octyl-β-d-glucopyranoside (OG) into vesicles made from Escherichia coli polar lipid extract. KcsA is a homotetrameric K+ channel that was key to elucidating the mechanism of selective K+ transport31 and is also involved in ultrafast water transport.29,32 We chose KcsA because (i) it is a robust IMP that can be produced in amounts sufficient for extensive in vitro experiments,31,33 (ii) its channel activity and thus successful reconstitution can easily be confirmed,34 and (iii) it has proven useful as a model to test reconstitution protocols.35−37 In spite of these advantageous properties, reconstitution yields are typically low, and protein aggregation is common during reconstitution,28,29 rendering KcsA a more representative example than the few IMPs that seem to incorporate into proteoliposomes almost quantitatively.38E. coli polar lipid extract and OG lend themselves for the reconstitution of bacterial IMPs, and the thermodynamics of solubilization and reconstitution of this system in the absence of protein has been characterized in detail.39
Experimental Section
Protein Production and Purification
A pQE-60 plasmid containing the KcsA gene was kindly provided by Prof. Roderick MacKinnon (Rockefeller University). The KcsA sequence was cloned into a pET-30 Ek/LIC expression vector (Merck-Novagen, Darmstadt, Germany), yielding a DNA construct encoding a fusion protein comprising a hexahistidine tag, a 44-residue spacer, and KcsA residues 1–160 (ref (40)). For fluorescence correlation spectroscopy (FCS), serine at position 6 was mutated to cysteine using a QuikChange mutagenesis kit (Stratagene, La Jolla, USA). Published protocols31,33 were adapted for KcsA production and purification. Overexpression was done in E. coli BL21(DE3)pLysS cells, which were grown at 37 °C for 6 h and at 30 °C for another 18 h. Cell pellets were resuspended in buffer (10 mM KH2PO4/K2HPO4, 150 mM KF, 10 mM imidazole, pH 8.0), disrupted in a high-pressure homogenizer (Avestin, Mannheim, Germany), and incubated with 40 mM decyl-β-d-maltopyranoside (Glycon Biochemicals, Luckenwalde, Germany) on a shaker at 4 °C for 2 h. The cell suspension was centrifuged at 50 000g and 4 °C for 30 min, and the supernatant was subjected to immobilized metal ion affinity chromatography (Bio-Rad, Munich, Germany). Detergent exchange into 80 mM OG (Glycon Biochemicals) and adjustment to pH 7.1 were performed on a P-6 desalting column (Bio-Rad). For fluorescent labeling of the S6C mutant, we applied minor modifications to a published protocol41 as described.29 Final KcsA and OG concentrations were determined by, respectively, spectrophotometry and thin-layer chromatography.42
Vesicle Preparation and Characterization
The molar mass of E. coli polar lipid extract (Avanti Polar Lipids, Alabaster, USA) was taken as 700 g/mol.43 Lipid powder was dissolved in buffer (10 mM KH2PO4/K2HPO4, 150 mM KF, pH 7.1) to a final concentration of 50 mM by vortexing for 15 min. For FCS, E. coli polar lipid extract was supplemented with 0.01% (w/w) N-(lissamine rhodamine B sulfonyl)phosphatidylethanolamine (Rh-PE). The lipid suspension was sonicated for 40 min in a Sonopuls HD 2070 homogenizer (Bandelin electronic, Berlin, Germany). Dynamic light scattering (DLS) was done on a Zetasizer NanoSZ (Malvern Instruments, Worcestershire, UK) equipped with a 50 mW laser emitting at 532 nm and a detector mounted at an angle of 173°. Measurements were performed on protein- and detergent-free lipid vesicles after sonication, on protein/lipid/detergent mixtures immediately after reconstitution, and on proteoliposomes after subsequent removal of aggregates and residual OG by dilution of the mixture recovered from the ITC cell, ultracentrifugation at 160 000g and 8 °C for 1 h, resuspension of the pellet in buffer, and extrusion through two, 100 nm polycarbonate filters (Avestin, Ottawa, Canada). Only monoexponential autocorrelation functions characteristic of narrow size distributions were subjected to cumulants analysis44 to obtain the Z-average size and the second-order polydispersity index and were used to derive intensity-weighted size distribution functions.
Isothermal Titration Calorimetry
ITC was carried out on VP-ITC and iTC200 calorimeters (GE Healthcare) after gentle degassing. Reconstitution was accomplished by titrating 1.5 μM KcsA tetramer solubilized in 30–35 mM OG with 20–50 mM sonicated E. coli polar lipid extract. Injection volumes and stirring speeds were, respectively, 1.5–3 μL and 307 rpm on the VP-ITC and 0.4 μL and 1000 rpm on the iTC200. Automated baseline adjustment and peak integration were done with the public-domain software NITPIC45 to yield normalized reaction heats as a function of lipid concentration. Solubilization and saturation boundaries were derived from the inflection points in these isotherms with the aid of nonlinear least-squares fitting.46
Fluorescence Correlation Spectroscopy
FCS was performed on an LSM 510 META ConfoCor 3 (Carl Zeiss, Jena, Germany) equipped with avalanche photodiodes. A 20 μL sample containing Atto 488-labeled KcsA reconstituted by stepwise addition of rhodamine-labeled vesicles and further processed by dilution, ultracentrifugation, and extrusion as described above was transferred onto a coverslip, and fluorescence signals were acquired at 25 °C for 20 s; 488 nm and 633 nm lasers were used to independently monitor the diffusion characteristics of fluorescently labeled protein and lipid, respectively. Each temporal signal was autocorrelated independently, and autocorrelation functions from quadruplicate experiments were averaged and analyzed using standard models for free 3D-diffusion.47 633 nm traces were fitted with a single component; to account for small amounts of unbound Atto 488 dye, 488 nm autocorrelation functions were analyzed using two components, the diffusion time of one of which was fixed at 0.03 ms. The numbers of proteoliposomes or micelles containing Atto 488-labeled KcsA in the confocal volume were derived from the amplitudes of the autocorrelation functions and were used to calculate the number of KcsA tetramers per proteoliposome, as detailed elsewhere.29
Activity Assay
Free-standing planar lipid membranes were formed according to an established protocol.48 Lipid was dissolved in n-decane to a final concentration of 20 mg/mL and was spread across a circular aperture with a diameter of 100–120 μm in a polytetrafluoroethylene septum separating two compartments filled with buffer (10 mM HEPES, 100 or 150 mM KCl, pH 4.0). The transmembrane current was measured with Ag/AgCl electrodes and an Axon GeneClamp 500 amplifier (Molecular Devices, Sunnyvale, USA) under voltage-clamp conditions. The recording filter was a 4-pole Bessel with a 3-dB corner frequency of 500 Hz. Gaussian filters of 117 Hz were applied to reduce noise. The amplified signal was digitized by a PCI 6025E computer board (National Instruments, Munich, Germany) and analyzed with WinEDR (Strathclyde Electrophysiology Software, Strathclyde, UK). The protein/lipid/detergent mixture recovered from the ITC cell was diluted to 10 mM OG, incubated at 8 °C for 1 h, and ultracentrifuged at 100 000g and 8 °C for 1.5 h. The pellet was washed with buffer, recentrifuged for 1 h, and resuspended in buffer to reach a lipid concentration of 10 mg/mL; 1–2 μL of proteoliposome suspension was injected into the cis compartment, followed by membrane rupture and formation of a new membrane. To increase incorporation efficacy, 250 mM urea was added to the cis side. The current was monitored at a holding voltage of 100–150 mV.
Results
To shed light on the influence of an IMP on the self-assembly of a lipid/detergent mixture, we used ITC to monitor the reconstitution of KcsA from an OG-solubilized state into proteoliposomes and compared it with the transformation of protein-free micelles into bilayer vesicles.
Reconstitution of Protein-Free Vesicles from a Micellar Solution
Figure 1a depicts an ITC experiment in which vesicles composed of 50 mM E. coli polar lipid extract were titrated into 35 mM OG, above the detergent’s critical micellar concentration of 29.6 mM.39 The first injections gave rise to endothermic, smoothly decaying heat signals, which reflected the dissolution of injected vesicles and the incorporation of lipid into mixed micelles. The SOL boundary was reached at a lipid concentration of 3.1 mM, where the micelles became saturated with lipid. Further titration led to the appearance of detergent-saturated bilayer vesicles coexisting with lipid-saturated micelles. Throughout this coexistence range, the reaction heats remained uniform because each injection entailed transfer of a constant amount of lipid and detergent from micelles into vesicles. The SAT boundary at 7.2 mM lipid was marked by a sharp rise in the heat of reaction, indicating the disappearance of micellar structures. Beyond this point, the calorimetric signal smoothly decreased as the lipid concentration rose, thus reducing the difference in composition between newly injected pure lipid vesicles and mixed vesicles in the cell. We have previously performed such reconstitution experiments on E. coli polar lipid extract/OG mixtures over a range of lipid and detergent concentrations.39 Solubilization experiments corresponding to reverse titrations of micelles into vesicles could confirm that this system is under thermodynamic control, allowing for an equilibrium phase diagram to be constructed (cf. Figure 3 below).39
Figure 1.
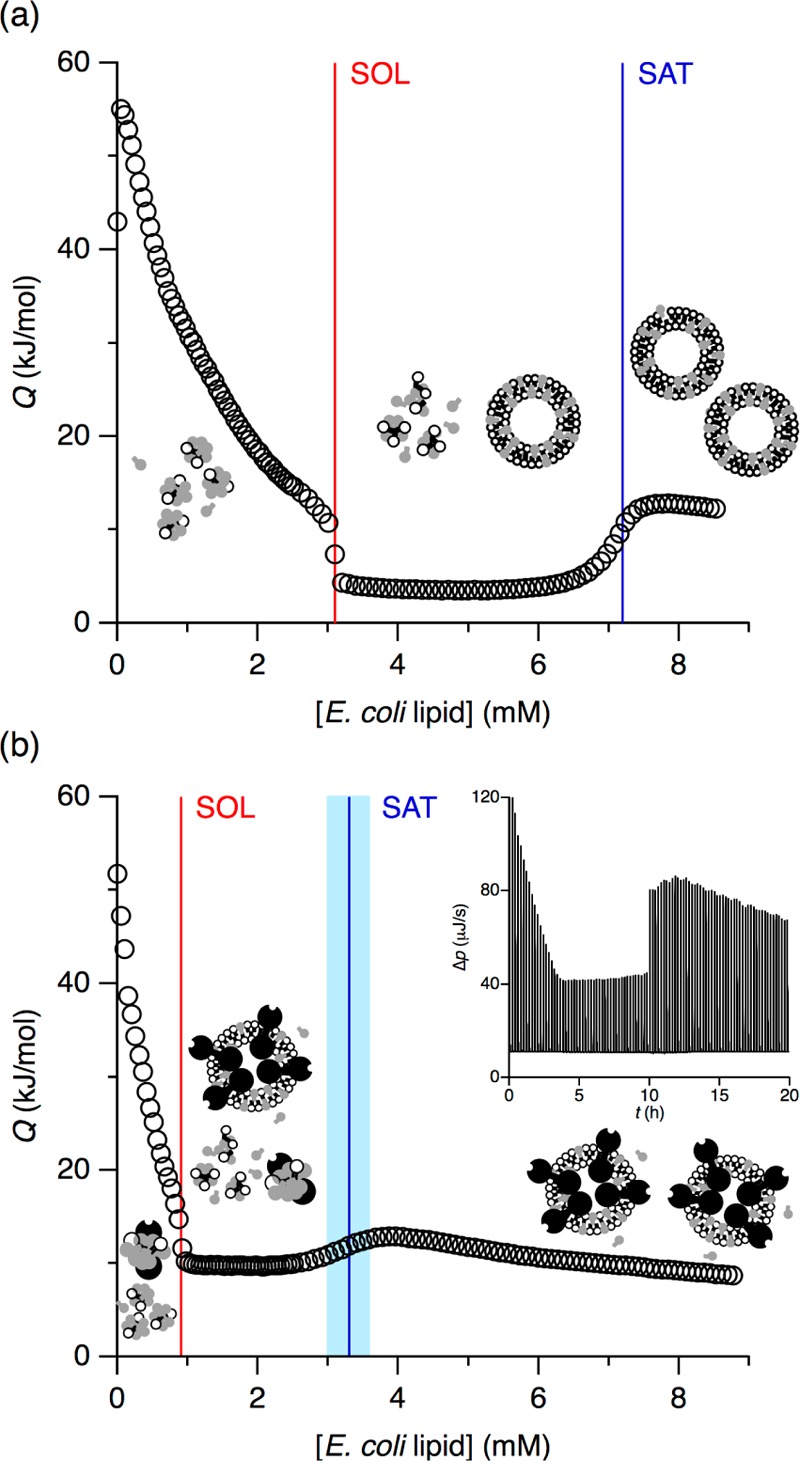
ITC reconstitution isotherms in (a) the absence or (b) the presence of 1.5 μM KcsA tetramer at 8 °C. Both isotherms depict the heats of reaction, Q (circles), obtained upon titration of 35 mM OG with 50 mM E. coli polar lipid extract. Also shown are the SOL and SAT boundaries (red and blue lines, respectively) and the uncertainty in the SAT boundary in the presence of KcsA (light blue band). Inset: Thermogram displaying differential heating power, Δp, versus time, t. The discontinuity at t ≈ 10 h is due to an increase in injection volume.
Figure 3.
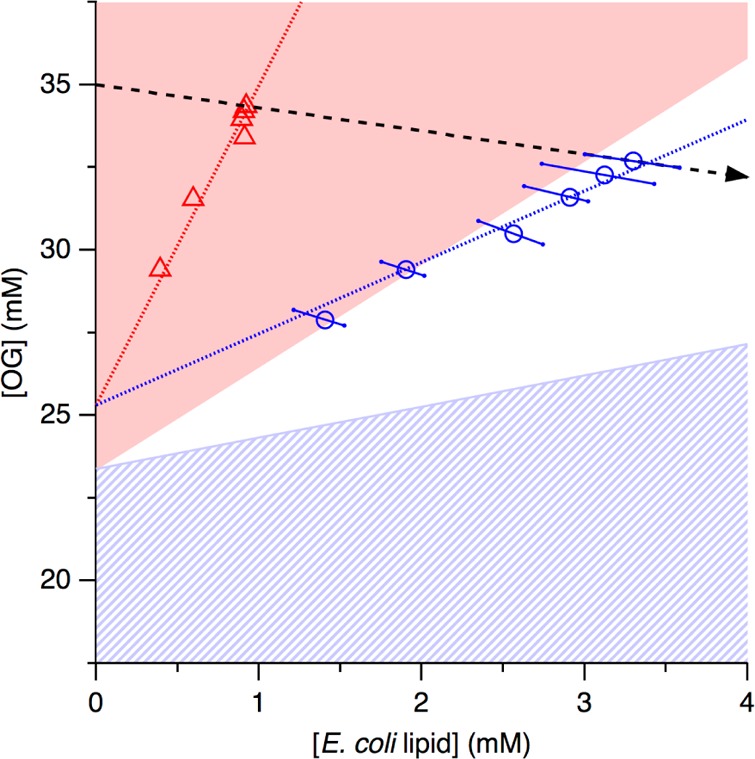
Critical lipid/detergent concentration pairs in the presence of KcsA (open symbols and dotted lines) and phase diagram of E. coli polar lipid extract and OG (colored areas) at 8 °C. Experimental data from Figures 1 and 2 (red triangles and blue circles) and linear regressions (red and blue dotted lines) denoting, respectively, the SOL and SAT boundaries in the presence of 1.5 μM KcsA tetramer. Also shown are the micellar, coexistence, and vesicular ranges for protein-free E. coli polar lipid extract/OG mixtures39 (red, white, and blue hatched areas, respectively), the uncertainties in the SAT boundary in the presence of protein (blue error bars), and the trajectory of the titration depicted in Figure 1b (dashed arrow).
Membrane-Protein Reconstitution by Titration Calorimetry
The isotherm in Figure 1b was obtained by repeating the above reconstitution titration in the presence of 1.5 μM KcsA tetramer in the calorimeter cell under otherwise identical conditions. Even though the overall shape was reminiscent of the protein-free system, the presence of KcsA at a low concentration affected the detailed form of the trace and resulted in substantial quantitative differences. Most notably, both inflection points were shifted to much lower lipid concentrations, with the SOL and SAT boundaries appearing at 0.9 and 3.3 mM lipid, respectively. While the transition from the micellar range to the coexistence range at the SOL boundary was as sharp as it was in the absence of protein, the transition from the coexistence range to the bilayer range broadened in the presence of KcsA, thus creating some leeway in the determination of the SAT boundary. To quantitatively account for this uncertainty, we considered the lipid concentration range over which the first derivative of the isotherm did not deviate significantly from its maximum value and propagated its lower and upper bounds through all subsequent analyses (see below).
To follow the transition from micellar to vesicular protein/lipid/detergent assemblies systematically, we performed ITC reconstitution titrations in the presence of KcsA at various lipid and detergent concentrations, as summarized in Figure 2. While the major hallmarks discussed above were preserved across all thermograms and isotherms, the initial detergent and lipid concentrations used to, respectively, solubilize and reconstitute KcsA were found to have a pronounced influence on the location of the SOL and SAT boundaries.
Figure 2.
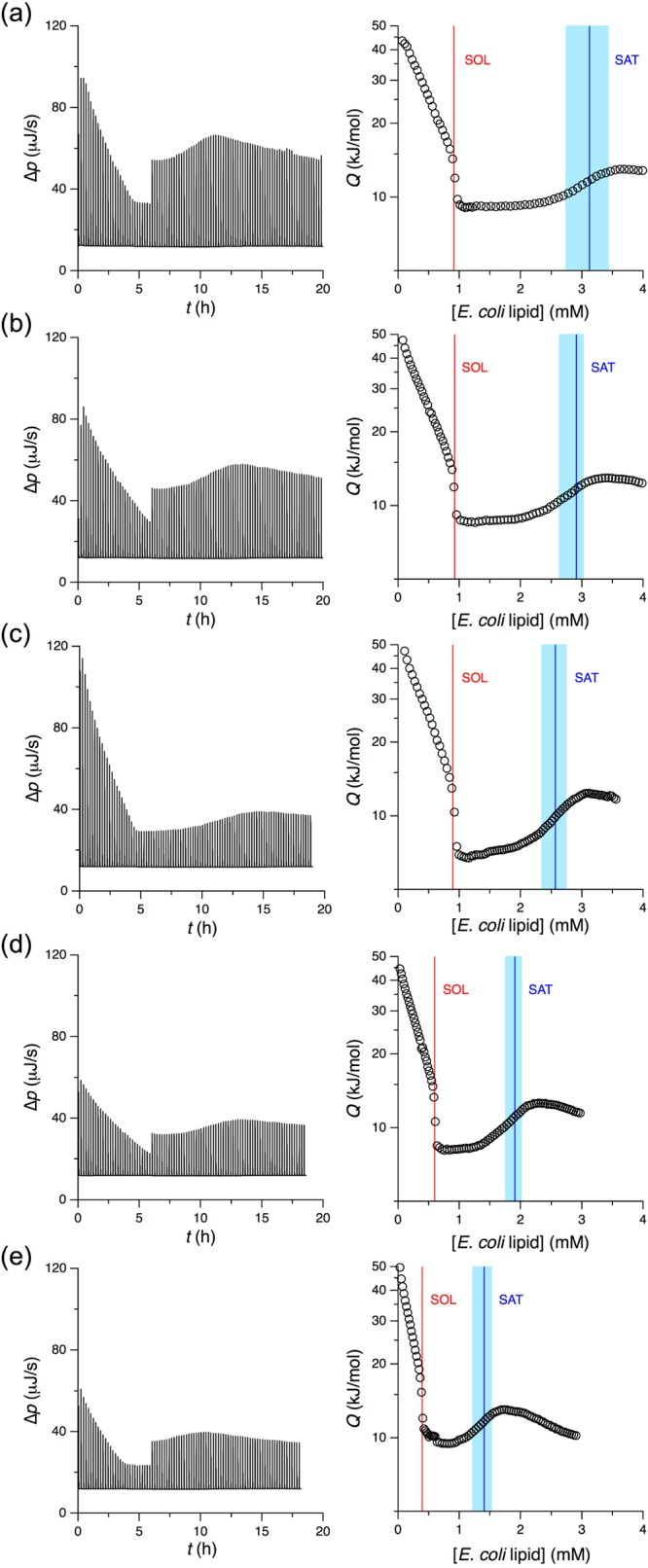
Thermograms (left) and isotherms (right) of reconstitution titrations performed at various lipid and detergent concentrations in the presence of 1.5 μM KcsA at 8 °C. (a) 40 mM lipid into 35 mM OG. (b) 30 mM lipid into 35 mM OG. (c) 20 mM lipid into 35 mM OG. (d) 20 mM lipid into 32.5 mM OG. (e) 20 mM lipid into 30 mM OG. Discontinuities in thermogram amplitudes are due to changes in injection volume. See Figure 1 for details.
Influence of KcsA on Bilayer Formation
Figure 3 depicts the critical lipid/detergent concentration pairs measured in the presence of 1.5 μM KcsA tetramer (Figures 1 and 2) as coordinates in a phase diagram for protein-free E. coli lipid/OG mixtures.39 This phase diagram is characterized by two straight lines separating the coexistence range from the purely micellar and vesicular ranges. The SOL boundary corresponds to a critical OG/E. coli lipid ratio of RDm,SOL = 3.1, while the SAT boundary is given by RDb,SAT = 0.95. These values indicate that, in the absence of protein, micelles contain at least a 3-fold molar excess of OG over lipid, whereas bilayer vesicles can accommodate up to one OG molecule per lipid molecule. The lipid/detergent concentration pairs now obtained in the presence of KcsA do not fall onto these equilibrium phase boundaries, as linear regression would yield critical OG/E. coli lipid ratios of 9.7 and 2.2 for the onset and completion of reconstitution, respectively. However, it is important to realize that these values do not, in general, correspond to the actual compositions of the protein/lipid/detergent assemblies because, in the presence of protein, the axes in Figure 3 do not reflect the actual concentrations of lipid and detergent in solution or suspension but rather indicate their nominal concentrations.
Particle Size Distributions before and after Reconstitution
To confirm that the calorimetric signatures observed upon titration of OG-solubilized KcsA with E. coli polar lipid (Figures 1 and 2) were indeed indicative of the formation of vesicles, we determined hydrodynamic particle diameters both before and after ITC-monitored reconstitution with the aid of DLS. As exemplified in Figure 4a, sonicated E. coli polar lipid vesicles in the absence of protein and detergent yielded monoexponential autocorrelation functions characteristic of narrow size distributions, whereas crude reconstitution mixtures recovered from the calorimeter cell revealed complex autocorrelation functions reflecting the presence of larger aggregates. However, removal of aggregates and residual OG by dilution, ultracentrifugation, and extrusion afforded proteoliposomes with well-defined size distributions, as evidenced in Figure 4b. Cumulants analysis44 returned a Z-average size of (140 ± 10) nm and a second-order polydispersity index of 0.16 ± 0.03 for proteoliposomes as compared with values of, respectively, (70 ± 5) nm and 0.17 ± 0.02 for sonicated E. coli polar lipid vesicles before reconstitution. Thus, the final, detergent-free proteoliposomes were about twice as large as the pure lipid vesicles used as starting material but had a similarly narrow, unimodal size distribution.
Figure 4.
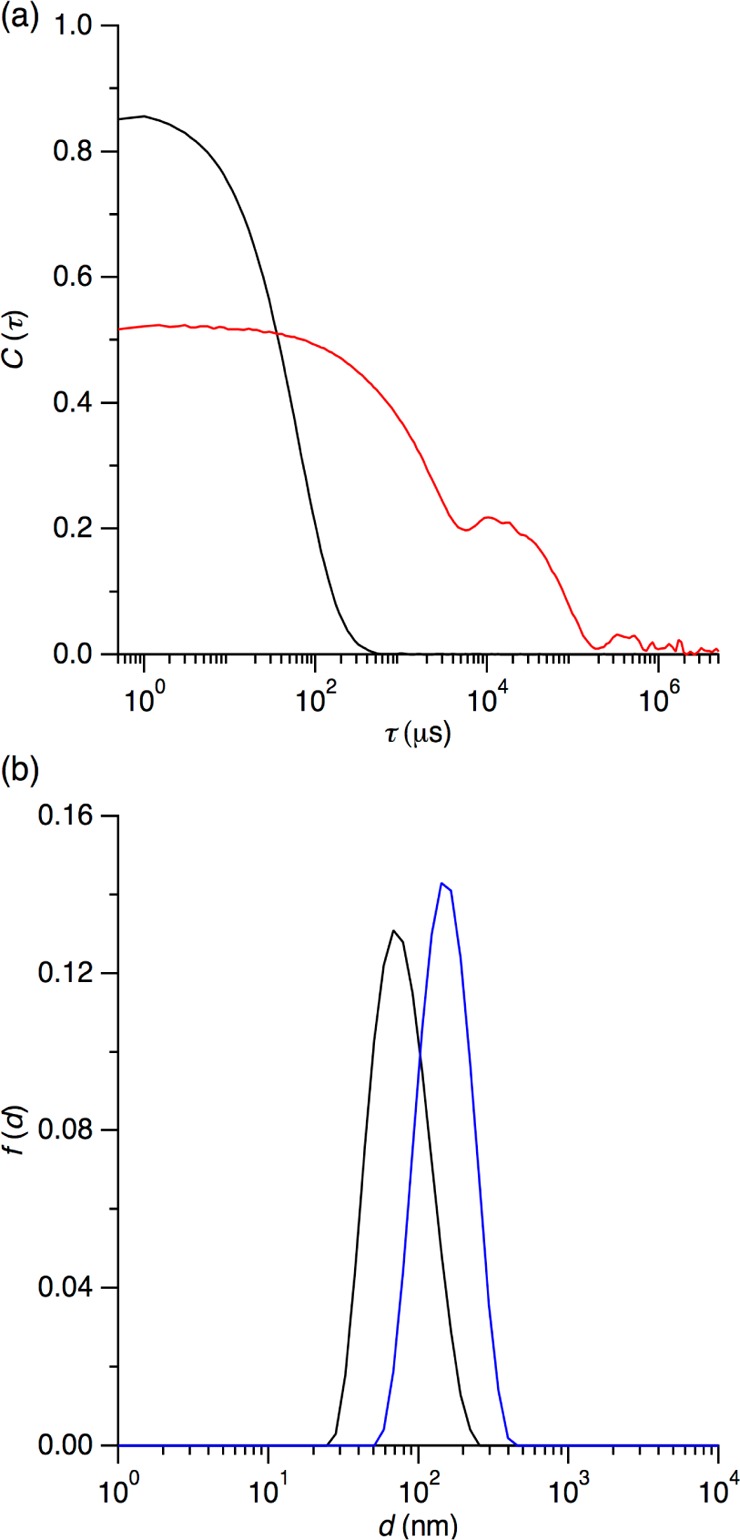
Size characterization of reconstituted proteoliposomes and protein-free lipid vesicles by DLS. (a) Normalized autocorrelation function, C(τ), versus delay time, τ, as determined for sonicated E. coli polar lipid vesicles before reconstitution (black) and the reconstitution mixture after calorimetrically monitored lipid addition to OG-solubilized KcsA (red). (b) Intensity-weighted distribution functions, f(d), of the hydrodynamic diameter, d, of sonicated E. coli polar lipid vesicles before reconstitution (black) and proteoliposomes after reconstitution and removal of aggregates and OG by dilution, centrifugation, and extrusion (blue).
Protein Incorporation and Reconstitution Yield
To quantify the reconstitution yield and verify that both the protein and the lipid were coreconstituted, we tracked the fates of both components using FCS.29,47 For this purpose, we produced a cysteine mutant (S6C) of KcsA labeled with the fluorophore Atto 488 and prepared E. coli polar lipid vesicles supplemented with 0.01% (w/w) Rh-PE as a lipidic fluorescent probe. Titrations of OG-solubilized KcsA(S6C) with labeled vesicles were stopped after the SAT boundary, and aggregates as well as OG were removed by dilution, ultracentrifugation, and extrusion. The autocorrelation functions in Figure 5a were measured independently for the two fluorophores by exciting Atto 488-labeled KcsA(S6C) at 488 nm and Rh-PE at 633 nm, revealing that both existed as single populations with identical diffusion times of 3.05 ms. Resolubilization of proteoliposomes gave rise to a single population having a diffusion time of 0.17 ms characteristic of micelles. Comparison of autocorrelation amplitudes before and after resolubilization29 revealed that the average number of KcsA tetramers per proteoliposome was 2.8 ± 0.5. Generally, this ratio assumed values of 1–4,29 depending on the initial detergent concentration and the amount of lipid added.
Figure 5.
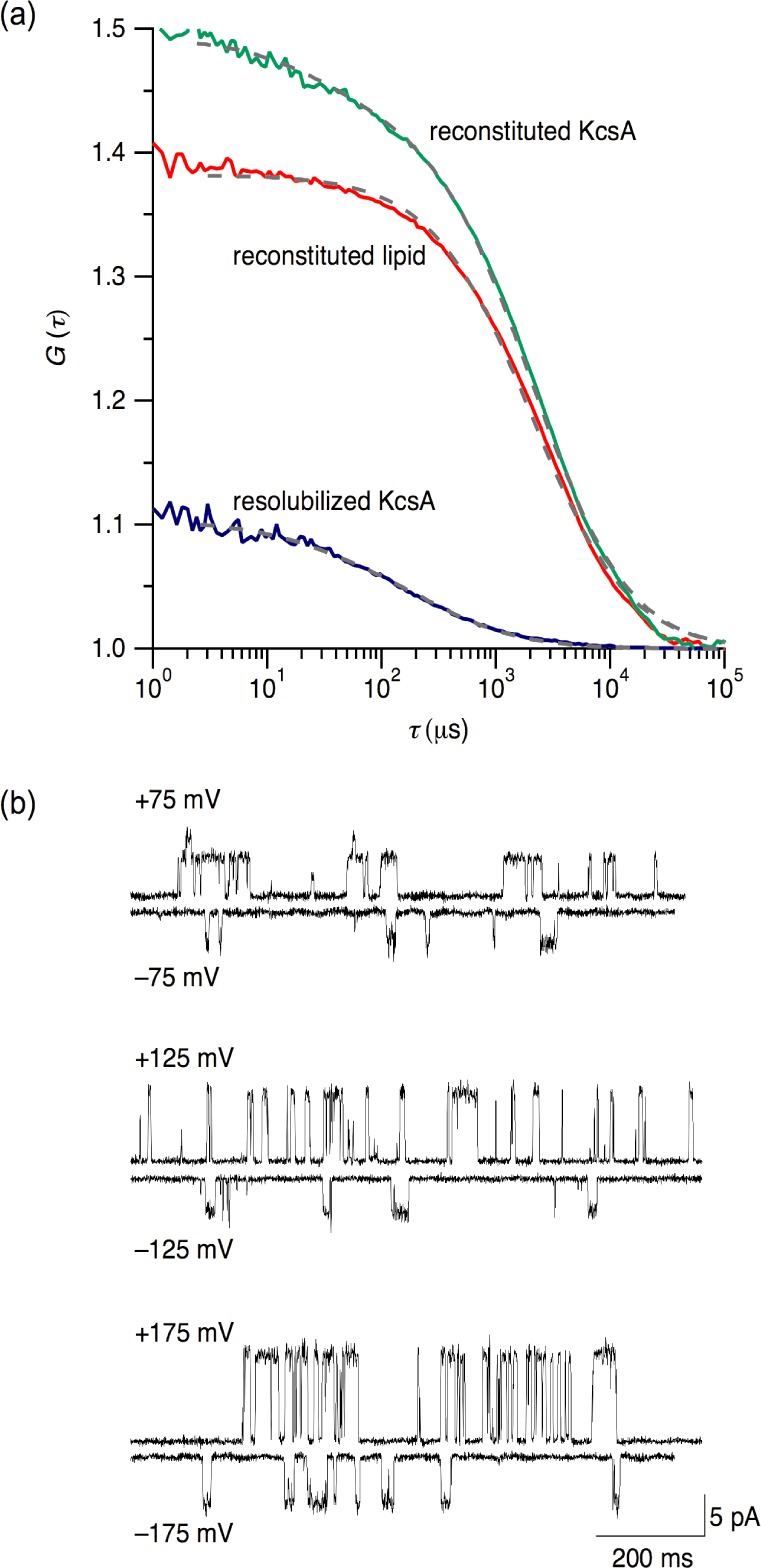
Confirmation of proteoliposome formation and channel activity after reconstitution. (a) FCS autocorrelation functions, G(τ), versus delay time, τ, before and after resolubilization of proteoliposomes with 68 mM OG. Experimental data (colors) and fits (dashed). (b) Single-channel currents recorded after transfer of KcsA from proteoliposomes into planar membranes composed of E. coli polar lipid extract. pH 4.0 on both sides, voltage as indicated.
Protein Activity after Reconstitution
Single-channel experiments corroborated that KcsA not only incorporated into proteoliposomes but also regained its native activity. To this end, we removed aggregates and residual OG by dilution and ultracentrifugation, transferred KcsA into planar lipid membranes,49 and measured the transmembrane flux of K+ ions. As shown in Figure 5b, inward and outward transport was observed with voltage dependencies characteristic of KcsA.34 Other hallmarks of KcsA were also confirmed, including a prolonged lifetime of the open state and the ensuing simultaneous opening of several channels upon increasing the content of anionic lipid (Figure S1a, Supporting Information),50 unilateral or complete inactivation upon shifting to pH 7.0 on, respectively, one or both sides of the membrane (Figure S1b,c, Supporting Information),34 and a reduction in the open-channel amplitude and the opening frequency by tetraethylammonium (Figure S1d, Supporting Information).34
Discussion
Efforts have been made to rationalize and optimize the reconstitution of IMPs by elucidating the influence of lipids and detergents8−17 and experimental parameters such as the rate of detergent extraction8 or the permeability of the bilayer.21,51 By contrast, only little attention is usually paid to the role of the IMP itself. Besides the large amounts of protein required for such systematic investigation, the prime reason for this most likely lies in the assumption that the low protein/lipid ratios commonly employed for functional reconstitution justify treatment of an IMP as an “infinitely diluted solute having almost no influence on the detergent and lipid interactions”.8 The present findings demonstrate that this assumption does not generally hold.
Experimental Challenges during Membrane-Protein Reconstitution
Our experimental conditions29 afford reconstitution yields of 10–20%. For instance, with an average of 3 KcsA channels per proteoliposome (Figure 5a) having a diameter of 140 nm (Figure 4b) and assuming a surface area requirement of 0.7 nm2 per lipid molecule, we arrive at a molar ratio of lipid to KcsA tetramer of ∼60 000. For a final lipid concentration of 9 mM (Figure 1b), this translates to a channel concentration of 0.15 μM, that is, 10% of the initial concentration. Correcting for dilution effects results in a yield of ∼12%. Although such low yields still enable protein densities largely sufficient for functional assays (Figure 5b), they entail two adverse consequences for proteoliposome formation: on the one hand, unproductive reactions competing with reconstitution make phase diagrams for lipid/detergent mixtures (Figure 3) futile in predicting the reconstitution progress; on the other hand, aggregation (Figure 4a) is a serious obstacle to the application of turbidimetry and light scattering methods for following the reconstitution process.
Ideally, a plot of critical lipid/detergent concentration pairs such as the one in Figure 3 would have to be established for the protein/lipid/detergent system of interest under the specific conditions used for IMP reconstitution. In the case of KcsA, the most likely explanation for the shifts observed for the SOL and the SAT boundaries is that aggregation reduces not only the protein yield but also the “active” OG concentration, that is, the detergent fraction that can be transferred from micelles into bilayers during the reconstitution process. Applying systematic reconstitution experiments to a more extensive set of proteins differing in hydrophobicity, size, structure, and topology might afford deeper insights into the underlying molecular determinants. In most cases, however, such an approach would be hampered by the prohibitively high sample requirements of usually precious target protein, emphasizing the need for more practical, online protocols that can be used without previous knowledge of the pertinent SOL and SAT boundaries.
Monitoring Membrane-Protein Reconstitution in Real Time by Titration Calorimetry
In the absence of general rules, a single calorimetrically monitored reconstitution titration such as the one in Figure 1b can be exploited to follow a reconstitution trial in real time and in a label-free and noninvasive manner to yield functional proteoliposomes. If the experiment aims at maximum yield, titration with lipid vesicles has to be continued until the SAT boundary is crossed. Stopping the titration earlier would mean sacrificing protein because of the persistence of micelles, whereas addition of more lipid would dilute the proteoliposome sample with protein-free vesicles. If, by contrast, the focus is on increasing protein density, proteoliposomes should be harvested upon halting the titration after the SOL boundary. The approach does not depend on prior knowledge of concentrations or phase diagrams and, unlike other sophisticated ways of monitoring IMP reconstitution,25 does not require diagnostic sample removal during the titration.
ITC has proven exceptionally useful for quantifying composition-dependent phenomena in aqueous surfactant solutions,52 including membrane solubilization and reconstitution,20,21 partitioning and translocation,51,53 permeabilization,54 and curvature strain.55 The present results show that high-sensitivity ITC can be applied also to more complex systems comprising protein, lipid, and detergent under conditions where irreversible processes impede the application of rigorous formalisms established for equilibrated systems of known concentrations.20,21,51−55 Calorimetry offers a number of advantages over other methods traditionally employed to monitor IMP reconstitution. Light scattering and turbidimetry are insensitive to the disappearance of micelles and thus fail in detecting the SAT boundary. Moreover, because these methods often cannot distinguish vesicles from rodlike micelles or aggregates, they may also lead to erroneous conclusions with respect to the location of the SOL boundary. ITC suffers from neither of these problems because it is sensitive to changes in supramolecular organization (e.g., the appearance of bilayers or the disappearance of micelles) rather than to properties indicative of the existence of certain types of assemblies (e.g., hydrodynamic size). Even if additional information may be required to interpret or confirm calorimetric data, ITC quickly and reliably yields high-resolution information on the concentrations at which major changes in the supramolecular organization of the reconstitution mixture occur and which therefore warrant further investigation.
Conclusions
The strong influence of KcsA on the assembly of bilayer membranes suggests that lipid/detergent phase diagrams are poor guides for the functional reconstitution of membrane proteins. To our knowledge, no other quantitative data on protein-containing systems are currently available, but it is safe to assume that different proteins and experimental conditions will influence reconstitution to various extents. In the absence of such data, we exemplify how reconstitution experiments can be monitored in real time with the aid of high-sensitivity calorimetry, which is independent of spectroscopic or radiolabels and compatible with turbid or heterogeneous samples. Since the reaction heat stems mostly from lipid/detergent interactions, the method also should not depend on the type and concentration of protein. In the future, this approach shall be adapted to related applications such as membrane solubilization26 and 2D crystallization,10,25 where higher protein densities suggest such effects to be even more prominent.
Acknowledgments
We thank Prof. Roderick MacKinnon (Rockefeller University) for the KcsA plasmid, Dr. Annette Diehl (FMP Berlin) for help with protein production, and Prof. Heiko Heerklotz (University of Toronto) for fruitful discussions. This work was supported by Grants KE 1478/1-2 from the Deutsche Forschungsgemeinschaft (DFG), 961-386261/969 from the Stiftung Rheinland-Pfalz für Innovation, and P19716 from the Austrian Science Fund (FWF).
Supporting Information Available
Additional electrophysiological experiments and quantitative comparison with simple lipid/detergent systems. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Overington J. P.; Al Lazikani B.; Hopkins A. L. Nat. Rev. Drug Discovery 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- le Maire M.; Champeil P.; Møller J. V. Biochim. Biophys. Acta 2000, 1508, 86–111. [DOI] [PubMed] [Google Scholar]
- Privé G. G. Methods 2007, 41, 388–397. [DOI] [PubMed] [Google Scholar]
- Duquesne K.; Sturgis J. N. Methods Mol. Biol. 2010, 601, 205–217. [DOI] [PubMed] [Google Scholar]
- Saparov S. M.; Kozono D.; Rothe U.; Agre P.; Pohl P. J. Biol. Chem. 2001, 276, 31515–31520. [DOI] [PubMed] [Google Scholar]
- Bordag N.; Keller S. Chem. Phys. Lipids 2010, 163, 1–26. [DOI] [PubMed] [Google Scholar]
- Fiedler S.; Broecker J.; Keller S. Cell. Mol. Life Sci. 2010, 67, 1779–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollivon M.; Lesieur S.; Grabielle-Madelmont C.; Paternostre M. Biochim. Biophys. Acta 2000, 1508, 34–50. [DOI] [PubMed] [Google Scholar]
- Rigaud J. L.; Lévy D. Methods Enzymol. 2003, 372, 65–86. [DOI] [PubMed] [Google Scholar]
- Dolder M.; Engel A.; Zulauf M. FEBS Lett. 1996, 382, 203–208. [DOI] [PubMed] [Google Scholar]
- Allen T. M.; Romans A. Y.; Kercret H.; Segrest J. P. Biochim. Biophys. Acta 1980, 601, 328–342. [DOI] [PubMed] [Google Scholar]
- Rigaud J. L.; Mosser G.; Lacapere J. J.; Olofsson A.; Levy D.; Ranck J. L. J. Struct. Biol. 1997, 118, 226–235. [DOI] [PubMed] [Google Scholar]
- DeGrip W. J.; VanOostrum J.; Bovee-Geurts P. H. M. Biochem. J. 1998, 330, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloda A.; Lua L.; Hall R.; Adams D. J.; Martinac B. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1540–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternostre M. T.; Roux M.; Rigaud J. L. Biochemistry 1988, 27, 2668–2677. [DOI] [PubMed] [Google Scholar]
- Rigaud J. L.; Paternostre M. T.; Bluzat A. Biochemistry 1988, 27, 2677–2688. [DOI] [PubMed] [Google Scholar]
- Knol J.; Sjollema K.; Poolman B. Biochemistry 1998, 37, 16410–16415. [DOI] [PubMed] [Google Scholar]
- Koynova R.; Caffrey M. Chem. Phys. Lipids 2002, 115, 107–219. [DOI] [PubMed] [Google Scholar]
- Lichtenberg D. Biochim. Biophys. Acta 1985, 821, 470–478. [DOI] [PubMed] [Google Scholar]
- Heerklotz H.; Tsamaloukas A. D.; Keller S. Nat. Protoc. 2009, 4, 686–697. [DOI] [PubMed] [Google Scholar]
- Keller S.; Heerklotz H.; Jahnke N.; Blume A. Biophys. J. 2006, 90, 4509–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S.; Sauer I.; Strauss H.; Gast K.; Dathe M.; Bienert M. Angew. Chem., Int. Ed. 2005, 44, 5252–5255. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Mahmood N. A. B. N.; Schuurman-Wolters G. K.; Poolman B. Nat. Protoc. 2008, 3, 256–266. [DOI] [PubMed] [Google Scholar]
- Zehnpfennig B.; Urbatsch I. L.; Galla H. J. Biochemistry 2009, 48, 4423–4430. [DOI] [PubMed] [Google Scholar]
- Signorell G. A.; Kaufmann T. C.; Kukulski W.; Engel A.; Rémigy H. W. J. Struct. Biol. 2007, 157, 321–328. [DOI] [PubMed] [Google Scholar]
- Maslennikov I.; Krupa M.; Dickson C.; Esquivies L.; Blain K.; Kefala G.; Choe S.; Kwiatkowski W. J. Struct. Funct. Genomics 2009, 10, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heegaard C. W.; le Maire M.; Gulik-Krzywicki T.; Møller J. V. J. Biol. Chem. 1990, 265, 12020–12028. [PubMed] [Google Scholar]
- Saparov S. M.; Tsunoda S. P.; Pohl P. Biol. Cell 2005, 97, 545–550. [DOI] [PubMed] [Google Scholar]
- Hoomann T.; Jahnke N.; Horner A.; Keller S.; Pohl P. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10842–10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrempf H.; Schmidt O.; Kümmerlen R.; Hinnah S.; Müller D.; Betzler M.; Steinkamp T.; Wagner R. EMBO J. 1995, 14, 5170–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle D. A.; Morais C. J.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. Science 1998, 280, 69–77. [DOI] [PubMed] [Google Scholar]
- Saparov S. M.; Pohl P. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 4805–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes D. M.; Perozo E. Biochemistry 1997, 36, 10343–10352. [DOI] [PubMed] [Google Scholar]
- Heginbotham L.; LeMasurier M.; Kolmakova-Partensky L.; Miller C. J. Gen. Physiol. 1999, 114, 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokogawa M.; Takeuchi K.; Shimada I. J. Am. Chem. Soc. 2005, 127, 12021–12027. [DOI] [PubMed] [Google Scholar]
- Leptihn S.; Thompson J. R.; Ellory J. C.; Tucker S. J.; Wallace M. I. J. Am. Chem. Soc. 2011, 133, 9370–9375. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M.; Iwamoto M.; Kato A.; Yoshikawa K.; Oiki S. J. Am. Chem. Soc. 2011, 133, 11774–11779. [DOI] [PubMed] [Google Scholar]
- Urban S.; Wolfe M. S. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 1883–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylova O. O.; Jahnke N.; Keller S. Biophys. Chem. 2010, 150, 105–111. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Nature 2001, 414, 43–48. [DOI] [PubMed] [Google Scholar]
- Blunck R.; Cordero-Morales J. F.; Cuello L. G.; Perozo E.; Bezanilla F. J. Gen. Physiol. 2006, 128, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriks L. R.; Mayor J. A.; Kaplan R. S. Anal. Biochem. 2003, 323, 234–241. [DOI] [PubMed] [Google Scholar]
- Ingram L. O. Appl. Environ. Microbiol. 1977, 33, 1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel D. E. J. Chem. Phys. 1972, 57, 4814–4820. [Google Scholar]
- Keller S.; Vargas C.; Zhao H.; Piszczek G.; Brautigam C. A.; Schuck P. Anal. Chem. 2012, 84, 5066–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmer G.; Keller S. Nat. Protoc. 2010, 5, 267–281. [DOI] [PubMed] [Google Scholar]
- Magde D.; Elson E. L.; Webb W. W. Biopolymers 1974, 13, 29–61. [DOI] [PubMed] [Google Scholar]
- Mueller P.; Rudin D. O.; Tien H. T.; Wescott W. C. J. Phys. Chem. 1963, 67, 534–535. [Google Scholar]
- Labarca P.; Latorre R. Methods Enzymol. 1992, 207, 447–463. [DOI] [PubMed] [Google Scholar]
- Marius P.; Zagnoni M.; Sandison M. E.; East J. M.; Morgan H.; Lee A. G. Biophys. J. 2008, 94, 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S.; Heerklotz H.; Blume A. J. Am. Chem. Soc. 2006, 128, 1279–1286. [DOI] [PubMed] [Google Scholar]
- Olofsson G.; Loh W. J. Braz. Chem. Soc. 2009, 20, 577–593. [Google Scholar]
- Tsamaloukas A. D.; Keller S.; Heerklotz H. Nat. Protoc. 2007, 2, 695–704. [DOI] [PubMed] [Google Scholar]
- Heerklotz H. Biophys. J. 2001, 81, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerklotz H.; Binder H.; Schmiedel H. J. Phys. Chem. B 1998, 102, 5363–5368. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.