

Abstract
Background
The enzyme hexokinase‐2 (HK2) phosphorylates glucose, which is the initiating step in virtually all glucose utilization pathways. Cardiac hypertrophy is associated with a switch towards increased glucose metabolism and decreased fatty acid metabolism. Recent evidence suggests that the increased glucose utilization is compensatory to the down‐regulated fatty acid metabolism during hypertrophy and is, in fact, beneficial. Therefore, we hypothesized that increasing glucose utilization by HK2 overexpression would decrease cardiac hypertrophy.
Methods and Results
Mice with cardiac‐specific HK2 overexpression displayed decreased hypertrophy in response to isoproterenol. Neonatal rat ventricular myocytes (NRVMs) infected with an HK2 adenovirus similarly displayed decreased hypertrophy in response to phenylephrine. Hypertrophy increased reactive oxygen species (ROS) levels, which were attenuated by HK2 overexpression, thereby decreasing NRVM hypertrophy and death. HK2 appears to modulate ROS via the pentose phosphate pathway, as inhibition of glucose‐6‐phosphate dehydrogenase with dehydroepiandrosterone decreased the ability of HK2 to diminish ROS and hypertrophy.
Conclusions
These results suggest that HK2 attenuates cardiac hypertrophy by decreasing ROS accumulation via increased pentose phosphate pathway flux.
Keywords: glucose, hexokinase, hypertrophy, oxidative stress
Introduction
Under normal situations, the heart relies heavily on fatty acid oxidation for its energy metabolism.1 However, during cardiac hypertrophy there is a switch to increased glucose utilization and decreased fatty acid metabolism.2–4 Glucose metabolism is initiated by cellular glucose uptake mediated by glucose transporters (GLUT), and glucose phosphorylation by a hexokinase (HK) to form glucose‐6‐phosphate.5 There are 4 mammalian HK isozymes that exist, with both HK1 and HK2 expressed in the heart.6 As HK2 expression is dynamically regulated by factors such as insulin and hypoxia,7–8 HK2 receives most of the attention as the predominant muscle HK. In addition to the kinase function of HK1 or HK2, both enzymes contain a hydrophobic region of 21 amino acids in their N‐termini, which allows for association with the voltage‐dependent anion channel (VDAC) on the outer mitochondrial membrane.9–10 This mitochondrial localization protects cells against death;11–15 this protection has mainly been attributed to a reduction in reactive oxygen species (ROS).13–16
It is still debated whether the increase in glucose utilization during hypertrophy is an integral pathologic process. However, genetic models mostly argue that altered fatty acid metabolism is the driver of hypertrophy, and that increased glucose utilization is compensatory. For example, mice with cardiac‐specific overexpression of peroxisome proliferator‐activated receptor α (PPARα) demonstrate increased fatty acid metabolism, decreased glucose metabolism, and hypertrophied hearts compared with wildtype littermates.17 Deletion of PPAR‐gamma coactivator 1α (PGC1α) increases hypertrophy and accelerates heart failure after pressure overload.18 Cardiac‐specific overexpression of the GLUT1 transporter attenuated cardiac hypertrophy and improved survival after pressure overload.19 Antithetically, cardiac‐specific deletion of the GLUT4 transporter spontaneously results in hypertrophied hearts.20 Regarding HK2, transverse aortic constriction‐induced hypertrophy is associated with increased HK2 protein, an effect abolished in PGC1β‐deficient animals,21 whereas hearts from mice heterozygous for Hk2 deletion have exacerbated cardiac hypertrophy after pressure overload.22 It was suggested that decreased HK2 expression decreases mitochondrial HK2, and increases ROS production due to mitochondrial permeability transition.22 Thus, these studies suggest that increasing glucose metabolism is likely a compensatory mechanism during hypertrophy.
Little is known about how increased glucose metabolism decreases cardiac hypertrophy. While glycolysis is considered the preferred route for glucose, other glucose utilization pathways consist of glycogen formation, the pentose phosphate pathway, and the hexosamine biosynthetic pathway.23 Importantly, phosphorylation of glucose by HK is the initiating step in all of these pathways. While glycogen does not appear to be altered during hypertrophy,24 flux through the pentose phosphate pathway and the hexosamine biosynthetic pathway increase during hypertrophy.25–27 We therefore hypothesized that mice with cardiac‐specific overexpression of HK2 would demonstrate decreased hypertrophy in response to chronic isoproterenol infusion. Indeed, HK2 overexpression did result in attenuated cardiac and cardiomyocyte hypertrophy in response to isoproterenol. HK2 overexpression also protected against isoproterenol‐induced cardiomyocyte death. Overexpression of HK2 also decreased hypertrophy in cultured neonatal rat ventricular myocytes (NRVMs) treated with phenylephrine. Hypertrophy was associated with an increase in ROS accumulation, which was attenuated by HK2. HK2 overexpression increased glucose‐6‐phosphate dehydrogenase (G6PDH) activity within the pentose phosphate pathway, and inhibition of G6PDH blocked the ability of HK2 to attenuate ROS and hypertrophy. Taken together, these results support that HK2 overexpression is anti‐hypertrophic due to increased glucose shuttling to the pentose phosphate pathway and reduction of ROS accumulation.
Methods
Animals were handled as approved by the University of Missouri Animal Care and Usage Committee in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health.
HK2 Transgenic Mice and Isoproterenol Infusion
The cDNA for mouse HK2 was inserted into the α‐myosin heavy chain (αMHC) promoter cassette and injected into fertilized FVB/N oocytes. Mice were maintained in a pure FVB/N background. Transgenic (TG) mice were identified by PCR. Both male and female mice were used and nontransgenic (NTG) littermates were used as controls. Chronic isoproterenol infusion (60 mg/kg per day for 14 days) was administered by implantation of mini‐osmotic pump (Alzet) into 2‐ to 3‐month‐old mice under isofluorane anesthesia (1.2% to 1.8%, 0.6 L flow of O2). Vehicle‐treated mice were implanted with pumps to perfuse 0.9% saline.
Echocardiography
Echocardiograms were performed under isofluorane anesthesia (1.2% to 1.8%, 0.6 L flow of O2) using a GE Vivid 7 ultrasound system (GE Healthcare) with a 12‐mHz transducer. The echocardiographer was blinded to the treatment group. M‐mode echocardiography was performed using the parasternal short‐axis view of the left ventricle. Images were captured digitally and 6 consecutive cardiac cycles were measured and averaged for each animal.
Histological Assessments
Mice underwent deep inhalation anesthesia (2% to 3% isofluorane) and hearts were perfused through the apex with 4% paraformaldehyde in PBS containing 25 mmol/L KCl and 5% dextrose to stop hearts in end diastole. Hearts were then frozen in OTC and sectioned by microtome. Sections were stained with fluorescently labeled wheat germ agglutinin (WGA) or Gomori's Trichrome. For the WGA labeling, 20 cells per field for a total of 10 fields were planimetered using NIH ImageJ. Sections were also stained for terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling (TUNEL) utilizing a cell death detection kit (Roche).
qRT‐PCR Gene Expression Analysis
RNA was extracted from mouse ventricles with TRIzol (Invitrogen) for first‐strand DNA synthesis (Superscript III First‐Strand Synthesis System; Invitrogen). Quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) was performed using a Bio‐Rad cycler with Sybr green intercalating dye (Takara SYBR Premix Ex Taq). Primer sequences were obtained from Roche's Universal ProbeLibrary.
Mitochondrial Isolation and Measurement of Mitochondrial Respiration
For subcellular fractionation experiments, mouse hearts were subfractionated by differential centrifugation as previously described.28 Mitochondrial respiration was assessed by a Clark type electrode (Qubit) using Vernier LoggerPro software. Isolated mitochondria (125 μg) were suspended in buffer consisting of 150 mmol/L KCl, 5 mmol/L KH2PO4, 10 mmol/L Tris pH 7.4, 2.5 mmol/L MgCl2, and 5 mmol/L glutamate/malate or 5 mmol/L succinate to obtain State2 respiration. Then 200 μmol/L ADP was added to initiate State3 respiration.
Western Blotting Analysis
Mouse tissue or cell culture lysates were solubilized in lysis buffer containing 150 mmol/L NaCl, 10 mmol/L Tris (pH 7.4), 1 mmol/L EDTA, and 1% Triton‐X100. Samples were sonicated and then centrifuged at 17 000 g for 10 minutes to remove insoluble debris. Protein concentration was measured by the Bradford method. Proteins were resolved on 10% SDS/PAGE gels, transferred onto PVDF membranes, and blocked with 10% milk in TBS‐T. The membranes were then immunoblotted using the following commercially available antibodies: anti‐HK2, anti‐HK1, anti‐O‐GlcNAcylation, and anti‐VDAC (Abcam); anti‐LDH (Santa Cruz); and anti‐GAPDH (Millipore). Membranes were then incubated with the appropriate alkaline phosphatase‐linked secondary antibody (Santa Cruz Biotechnology) and visualized by enhanced chemifluorescence (Amersham Biosciences).
HK2 Activity Assay
HK enzymatic activity was assessed as described previously.29 Total HK, HK1, and HK2 activity were measured simultaneously, by means of HK2 denaturation at 45°C as formerly described.30 Briefly, total cytosolic and mitochondrial lysates were divided into 2 aliquots, 1 kept on ice and 1 put into a 45°C water bath for 1 hour to denature HK2. At the end of the denaturing step, hexokinase activity was determined in both aliquots by measuring glucose‐6‐phosphate formation coupled to NADPH formation spectrophotometrically at 340 nm. The assay buffer consisted of 100 mmol/L Tris‐HCl pH 8.0, 10 mmol/L glucose, 0.4 mmol/L NADP+, 10 mmol/L MgCl2, 5 mmol/L ATP, and 0.15 U of G6PDH. Activity was normalized to protein concentration as measured by the Bradford method.
Mitochondrial Swelling
Mitochondria were isolated as described above and resuspended in swelling buffer (150 mmol/L KCl, 5 mmol/L KH2PO4, 10 mmol/L Tris pH 7.4) to a final concentration of 0.25 mg/mL. De‐energized mitochondria received no substrate, and 5 mmol/L glutamate/malate was added to the buffer to energize mitochondria. Mitochondrial swelling, an index of permeability transition, was induced by the addition of 250 μmol/L CaCl2 or 100 μmol/L CaCl2 for de‐energized and energized mitochondria, respectively, and absorbance was measured spectrophotometrically at 520 nm.31
Isolation and Culture of Rat Ventricular Myocytes
Neonatal rat ventricular myocytes (NRVMs) were isolated as previously described.32 Briefly, 1‐ to 3‐day‐old Sprague‐Dawley rat pup heart ventricles were digested with trypsin and then collagenase according to the manufacturer's instructions (Worthington). Isolated cells were preplated on uncoated polystyrene dishes to remove cardiac fibroblasts, and then plated onto gelatin‐coated dishes. Cells were maintained in M199 medium supplemented with 10% BGS for 24 hours, then serum‐free M199 for the duration of experimentation. Hypertrophy was induced by treatment with 25 μmol/L phenylephrine (PE) or 2 μmol/L Angiotensin‐II for a period of 48 hours. Glucose‐6‐phosphate dehydrogenase (G6PDH) activity was inhibited by treatment with 25 μmol/L dehydroepiandrosterone (DHEA) for 48 hours, concurrent with the hypertrophic drug treatment.
Creation and Infection of HK2 Adenovirus
Replication‐deficient adenoviruses for β‐galactosidase and mouse HK2 were generated using the AdEasy adenoviral system (Stratagene). NRVMs were infected with adenovirus at a multiplicity of infection of 100 plaque‐forming units, and cultured for 24 hours before experimentation.
siRNA Transfection
NRVMs were transfected with 100 nmol/L of either a non‐targeting control siRNA or an HK2‐targeting siRNA pool (Dharmacon siGENOME Cat: M‐051128‐01) using Lipofectamine RNAiMax. Cells were transfected 48 hours before experimentation.
Measurement of NRVM Cell Area
For cell surface area determination, NRVMs were immunostained for tropomyosin (Sigma) and all cardiomyocytes in each random field were planimetered using NIH ImageJ to obtain 1 average per field. Data from ≥4 independent isolates with ≥4 random fields per group were summarized.
Measurement of ROS Accumulation
ROS was measured in NRVMs by staining with 1 μmol/L 2′,7′‐dichlorofluorescein (DCF) (Invitrogen) in Hank's buffered saline solution for 30 minutes. Cells were then washed with HBSS and imaged on a fluorescence microscope (Olympus IX51). Average DCF fluorescence per cell was determined with NIH ImageJ.
Measurement of NRVM Cell Death
NRVMs were treated with 50 μmol/L H2O2 for 1 hour, and then costained with Sytox Green (Invitrogen) to label dead cell nuclei and bis‐benzimide to label all cell nuclei. Cells were then imaged on a fluorescence microscope and the percentage of dead cells was determined with NIH ImageJ.
Glutathione Assay
Reduced glutathione (GSH) levels were determined in cardiac and cultured myocyte lysates using a commercially available luciferase‐based assay from Promega. Levels were then corrected for the protein concentration of each sample.
G6PDH Activity Assay
The enzymatic activity of G6PDH was measured as performed previously.33 Briefly, 100 μL of whole cell lysates were added to a cuvette with 900 μL of buffer (50 mmol/L Tris, 1 mmol/L MgCl2, and 100 μmol/L NADP+, pH‐8.1). Enzyme activity was measured by increase in absorbance at 340 nm, due to the conversion of NADP+ to NADPH, which is formed by both G6PDH and 6‐phosphogluconate dehydrogenase (6PGDH) in the pentose phosphate pathway. Thus, 2 reactions were measured for each sample, 1 containing substrates for both G6PDH and 6PGDH (total dehydrogenase activity) and 1 reaction with substrate only for 6PGDH. G6PDH activity was then calculated by subtracting the 6PGDH activity from the total dehydrogenase activity. Substrate concentrations were 200 μmol/L glucose‐6‐phosphate and 200 μmol/L 6‐phosphogluconate. Activity was normalized to protein concentration as measured by the Bradford method.
Statistical Analysis
All data are expressed as mean±SEM. Statistical significance was determined between 2 groups by an unpaired Student's t test or between multiple groups with 1‐way ANOVA followed by Scheffe's post‐hoc test. A P value <0.05 was considered significant.
Results
Characterization of αMHC‐HK2 Mice
Cardiac‐specific HK2 overexpression was performed by cloning the mouse HK2 cDNA into the αMHC promoter cassette. TG mouse hearts displayed ≈3‐fold increased HK2 expression compared to NTG littermates (Figure 1A and 1B). Total HK activity and HK2‐specific activity were accordingly increased in HK2 TG mice (Figure 1C). Importantly, neither HK1 expression nor HK1 activity was altered by HK2 overexpression (Figure 1A and 1C). TG hearts were normal in appearance and displayed HW/BW ratios similar to NTG littermates (Figure 1D and 1E). Moreover, fractional shortening, ejection fraction, and chamber size were not different in the TG hearts when compared to the NTG controls (Table).
Figure 1.
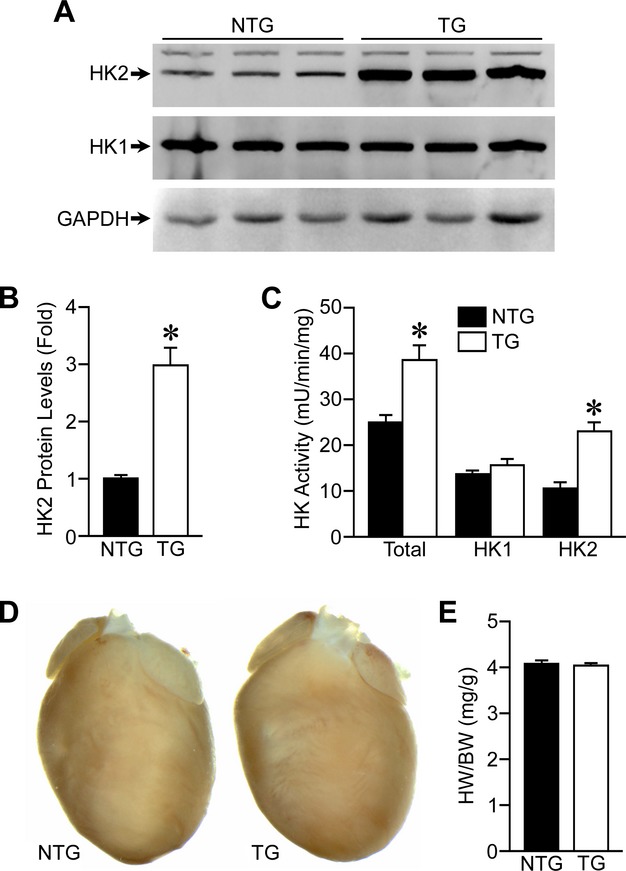
Characterization of αMHC‐HK2 transgenic mice. A, Western blotting for HK2 and HK1 protein levels in αMHC‐HK2 non‐transgenic (NTG) and transgenic (TG) whole‐heart lysates. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as a loading control. B, Quantification of HK2 protein expression in NTG and TG hearts (n=6). C, Total (HK1 and HK2), HK1, and HK2 enzymatic activities in whole heart lysates from NTG and TG mice (n=4). D, Representative heart images from NTG and TG mice. E, Gravimetric analysis of heart weight/body weight ratio (HW/BW) for NTG and TG mice (n=26 to 30). Error bars represent SEM with *P<0.05 vs NTG. αMHC‐HK2 indicates α‐myosin heavy chain hexokinase‐2; SEM, standard error of the mean.
Table 1.
Echocardiographic Data From NTG and HK2 TG Mice Treated With Isoproterenol
| Vehicle | Isoproterenol | |||
|---|---|---|---|---|
| NTG | HK2 TG | NTG | HK2 TG | |
| LVEDD, mm | 3.34±0.07 | 3.30±0.09 | 3.42±0.07 | 3.47±0.11 |
| LVESD, mm | 2.06±0.08 | 1.98±0.07 | 1.98±0.08 | 2.11±0.10 |
| FS, % | 38.6±1.2 | 40.1±1.2 | 42.2±1.7 | 39.2±1.5 |
| EF, % | 75.5±1.3 | 77.2±1.2 | 79.2±1.8 | 75.9±1.7 |
| HR, bpm | 404±23 | 368±19 | 393±24 | 435±25 |
EF indicates ejection fraction; FS, fractional shortening; HK2, hexokinase‐2; HR, heart rate; LVEDD, left‐ventricular end diastolic dimension; LVESD, left‐ventricular end systolic dimension; NTG, non‐transgenic; TG, transgenic.
As HK2 can be localized to both the cytosol and mitochondria, we subfractionated NTG and TG hearts into cytosolic and mitochondrial fractions to assess the localization of the overexpressed HK2. Surprisingly, the majority of the overexpressed HK2 was found in the cytosolic fraction (Figure 2A and 2B). However, there was a small but significant increase in mitochondrial HK2 expression as well (Figure 2A and 2B). Total HK and HK2 activity were correspondingly increased mainly in the cytosolic fraction (Figure 2C), but also in the mitochondrial fraction (Figure 2D). Oxygen consumption was then measured in isolated cardiac mitochondria to assess functional differences. TG heart mitochondria exhibited a significant reduction in State3 respiration when stimulated with complex I substrates (glutamate/malate), but not with complex II substrate (succinate) (Figure 2E).
Figure 2.

HK2 overexpression is mostly cytosolic. A, Cytosolic and mitochondrial HK2 protein expression in subfractionated hearts of NTG and TG mice. Voltage‐dependent anion channel (VDAC) demarcates mitochondrial lysates and lactate dehydrogenase (LDH) labels cytosolic lysates. B, Quantification of cytosolic and mitochondrial HK2 expression in NTG and TG mouse hearts (n=6). C, Total (HK1 and HK2), HK1, and HK2 enzymatic activities from cytosolic lysates of NTG and TG mouse hearts (n=4). D, Total, HK1, and HK2 enzymatic activities from mitochondrial lysates of NTG and TG mouse hearts (n=4). E, Ratio of ADP‐stimulated (State3) to nonstimulated (State2) respiration measured in isolated mitochondria from NTG and TG hearts (n=10 to 12). F, Average swelling traces for de‐energized NTG and TG cardiac mitochondria treated with 250 μmol/L Ca2+ (n=11). G, Average swelling traces for glutamate/malate energized NTG and TG cardiac mitochondria treated with 100 μmol/L Ca2+ (n=11 to 12). H, Maximal change in absorbance from mitochondrial swelling experiments shown in (F and G). Error bars represent SEM with *P<0.05 vs NTG. Deenerg indicates de‐energized; Energ, energized; HK2, hexokinase‐2; NTG, non‐transgenic; SEM, standard error of the mean; TG, transgenic.
To determine whether mitochondrial permeability transition was inhibited in the TG mice, heart mitochondria were isolated and subjected to mitochondrial swelling induced by calcium. Despite a small yet significant HK2 overexpression on the mitochondria (Figure 2A and 2B), αMHC‐HK2 hearts showed no protection against mitochondrial swelling under either de‐energized (Figure 2F and 2H) or energized (Figure 2G and 2H) conditions.
αMHC‐HK2 Mice Display Decreased Cardiac Hypertrophy in Response to Isoproterenol
We next investigated the effects of HK2 overexpression on the hypertrophic response. Isoproterenol infusion resulted in increased heart size (Figure 3A), but not body weight (data not shown), which significantly increased the HW/BW ratio (Figure 3B). HK2 overexpression attenuated the hypertrophic response to isoproterenol (Figure 3A and 3B). Wheat‐germ agglutinin staining of LV heart sections indicated increased cardiomyocyte cross‐sectional area with isoproterenol, and HK2 overexpression decreased this response (Figure 3C and 3D). Cardiomyocyte cross‐sectional area trends were similar for septal wall cardiomyocytes, however, RV cardiomyocytes showed only ≈15% increase in cell size with isoproterenol and HK2 overexpression had no effect (data not shown).
Figure 3.

HK2 overexpression attenuates cardiac hypertrophy. A, Representative heart images from NTG and TG mice treated with 0.9% saline (vehicle) or 60 mg/kg per day isoproterenol (ISO) for 2 weeks. B, Gravimetric analysis of heart weight/body weight ratio (HW/BW) for Vehicle‐ or ISO‐treated NTG and TG mice (n=10 to 12). D, Representative wheat‐germ agglutinin (WGA) images from left‐ventricular (LV) sections of vehicle‐ or ISO‐treated NTG and TG mouse hearts. D, LV cardiomyocyte cross‐sectional area measured from WGA images as in (C) (n=4 to 6). E, Myocyte apoptosis, as determined by TUNEL staining, in vehicle‐ or ISO‐treated NTG and TG mouse hearts (n=4 to 6). F through H, mRNA expression measured by qRT‐PCR for (F), atrial natriuretic peptide (ANP), (G), B‐type natriuretic peptide (BNP), and (H), β‐Myosin heavy chain (βMHC) in vehicle‐ or ISO‐treated NTG and TG mouse hearts. The 18s ribosomal subunit was used as loading control (n=6). Error bars represent SEM with *P<0.05 vs Vehicle and †P<0.05 vs NTG. HK2 indicates hexokinase‐2; NTG, non‐transgenic; qRT‐PCR, quantitative reverse transcription‐polymerase chain reaction; SEM, standard error of the mean; TG, transgenic; TUNEL, terminal deoxynucleotidyl transferase‐mediated dUTP nick end labeling.
We then assessed the effects of HK2 overexpression on cardiomyocyte survival and cardiac function after isoproterenol infusion. In general, there were no signs of heart failure induced by the isoproterenol infusion. LV ejection fraction and fractional shortening remained normal with isoproterenol, and was unaltered by HK2 overexpression (Table). Overt fibrosis was not seen in either group as well (data not shown). Isoproterenol infusion did result in a small, yet significant increase in cardiomyocyte apoptosis, as measured by TUNEL staining of heart sections, and HK2 overexpression attenuated this increase in cardiomyocyte death (Figure 3E). Altogether, these results suggest that isoproterenol infusion induces cardiac hypertrophy, but not heart failure, and that HK2 overexpression reduces the cardiomyocyte hypertrophy.
Isoproterenol infusion also increased transcripts for ANP, BNP, and βMHC, common markers of the “fetal gene program” (Figure 3F through 3H). As HK2 overexpression limited hypertrophy, it was expected that the αMHC‐HK2 hearts would exhibit decreased transcripts for these markers. In fact, HK2 overexpression resulted in non‐significantly increased ANP, BNP, and βMHC transcript expression (Figure 3F through 3H). However, these transcripts were also slightly elevated in vehicle‐treated TG mice compared to NTG littermates. Increased levels of these “hypertrophy” markers have also been shown in other models of increased glucose utilization.34
Overexpression of HK2 in vitro Attenuates Cardiomyocyte Hypertrophy
To better study the mechanisms of HK2's anti‐hypertrophic effect, we wanted to recapitulate our in vivo findings in a cell culture system. NRVMs were infected with either control β‐galactosidase adenovirus or a mouse HK2 adenovirus (Figure 4A and 4B), and treated with phenylephrine (PE) to induce hypertrophy. PE (25 μmol/L) resulted in a 37% increase in NRVM area, and HK2 overexpression reduced this hypertrophic response to only 16% (Figure 4C and 4D). To confirm that this was not specific to PE we also tested angiotensin‐II (AngII). AngII (2 μmol/L) increased NRVM area by 27%, while HK2 overexpression limited the AngII‐induced hypertrophy to only 10% (Figure 4E). These results confirm our in vivo findings in that HK2 overexpression attenuates cardiomyocyte hypertrophy. We also investigated the effect of reduced HK2 expression by transfecting NRVMs with a HK2 siRNA, which reduced HK2 protein expression 60%, but did not affect HK1 (Figure 5A and 5B). In agreement with our HK2 overexpression results, decreased HK2 expression led to significantly increased NRVM hypertrophy in response to 25 μmol/L PE (Figure 5C and 5D). These results further indicate that HK2 expression modulates cardiomyocyte hypertrophy.
Figure 4.
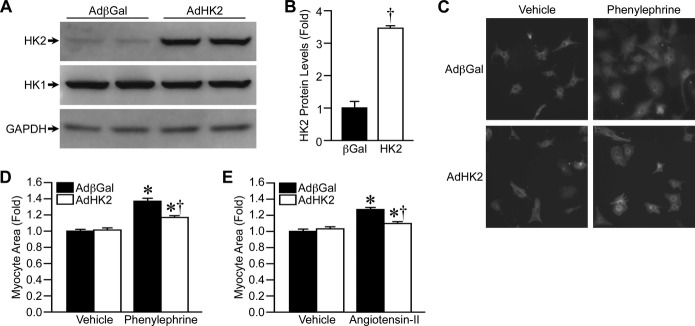
HK2 overexpression decreases cardiomyocyte hypertrophy in vitro. A, Western blotting for HK2 and HK1 protein levels in isolated neonatal rat ventricular cardiomyocytes (NRVMs) infected with adenovirus expressing either β‐galactosidase (AdβGal) or HK2 (AdHK2). GAPDH was used as a loading control. B, Quantification of HK2 protein expression in AdβGal‐ and AdHK2‐infected NRVM (n=4). C, Representative images of AdβGal‐ and AdHK2‐infected NRVMs treated with either vehicle or 25 μmol/L phenylephrine for 48 hours, and immunostained for tropomyosin. D, Mean cell area for AdβGal‐ and AdHK2‐infected NRVMs treated with either vehicle or 25 μmol/L phenylephrine for 48 hours (n=4). E, Mean cell area for AdβGal‐ and AdHK2‐infected NRVMs treated with either vehicle or 2 μmol/L angiotensin‐II (AngII) for 48 hours (n=4). Error bars represent SEM with *P<0.05 vs Vehicle and †P<0.05 vs AdβGal. HK1 indicates hexokinase‐1; HK2, hexokinase‐2; SEM, standard error of the mean.
Figure 5.
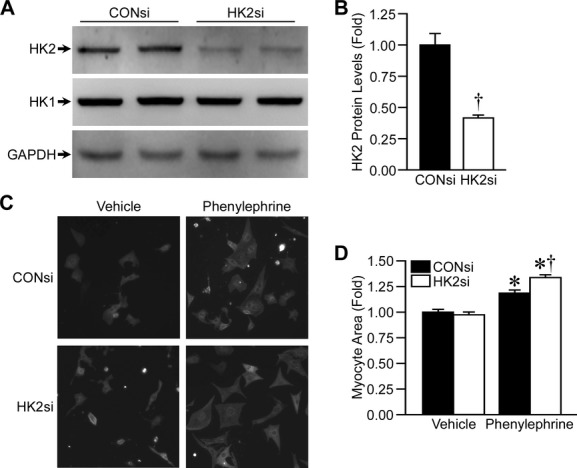
HK2 knockdown increases hypertrophy in vitro. A, Western blotting for HK2 and HK1 protein levels in NRVMs transfected with a control (CONsi) or anti‐HK2 siRNA (HK2si). B, Quantification of HK2 protein expression in CONsi‐ and HK2‐transfected NRVM (n=4). C, Representative images of CONsi‐ and HK2‐transfected NRVM treated with either vehicle or 25 μmol/L phenylephrine for 48 hours, and immunostained for tropomyosin. D, Mean cell area for CONsi‐ and HK2si‐transfected NRVMs treated with either vehicle or 25 μmol/L phenylephrine for 48 hours (n=4). Error bars represent SEM with *P<0.05 vs Vehicle and †P<0.05 vs CONsi. HK1 indicates hexokinase‐1; HK2, hexokinase‐2; NRVM, neonatal rat ventricular myocytes; SEM, standard error of the mean.
HK2 Modulates ROS During Cardiomyocyte Hypertrophy
As ROS levels have been shown to be elevated during hypertrophy,35–37 and HK2 is known to be protective against ROS‐induced death,12–16 we decided to investigate whether HK2 expression affected ROS levels during cardiomyocyte hypertrophy. PE treatment increased NRVM ROS levels by 65% as measured by DCF fluorescence (Figure 6A). HK2 overexpression reduced the ROS accumulation during hypertrophy, to an increase of only 24% (Figure 6A). HK2 depletion by siRNA exacerbated the accumulation of ROS during PE‐induced hypertrophy (Figure 6B). This modulation of ROS levels during hypertrophy led us to question whether HK2 could decrease generalized oxidative stress; therefore we measured ROS levels in NRVMs overexpressing HK2 after treatment with 50 μmol/L H2O2. ROS levels were increased nearly 3‐fold with H2O2 treatment, and HK2 overexpression significantly reduced this ROS accumulation (Figure 6C). HK2 overexpression also significantly reduced H2O2‐induced NRVM death (Figure 6D). These results suggest that HK2 expression mediates ROS levels during cardiomyocyte hypertrophy and can protect against ROS‐induced cardiomyocyte death.
Figure 6.
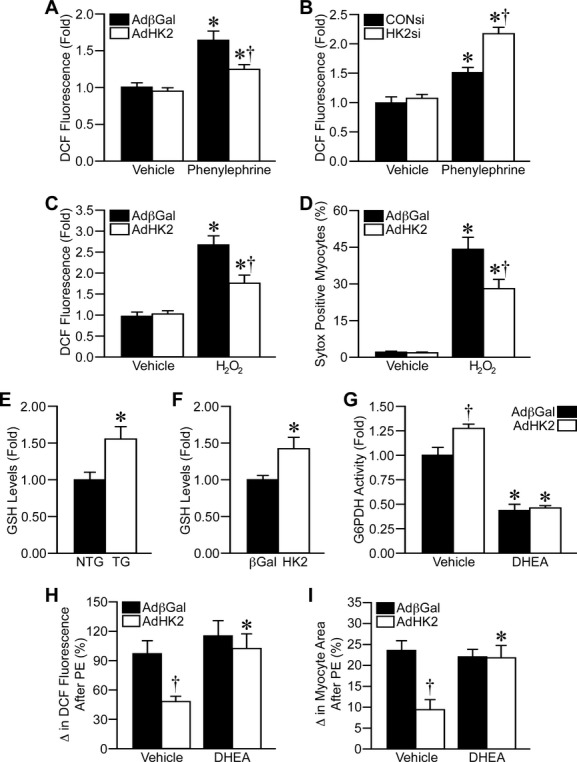
HK2 modulates reactive oxygen species (ROS) levels by increasing pentose phosphate pathway flux. A, Mean ROS levels as measured by 2′,7′‐di‐chlorofluorescein (DCF) fluorescence in AdβGal‐ and AdHK2‐infected NRVMs treated with either vehicle or 25 μmol/L phenylephrine for 48 hours (n=4). B, Mean ROS levels as measured by DCF fluorescence in CONsi‐ and HK2‐transfected NRVM treated with either vehicle or 25 μmol/L phenylephrine for 48 hours (n=4). C, Mean ROS levels as measured by DCF fluorescence in AdβGal‐ and AdHK2‐infected NRVMs treated with vehicle or 50 μmol/L H2O2 for 1 hour (n=4). D, Cell death, as measured by Sytox Green, in AdβGal‐ and AdHK2‐infected NRVMs treated with vehicle or 50 μmol/L H2O2 for 1 hour (n=6). E, Reduced glutathione (GSH) levels in cardiac lysates from NTG and TG mice (n=6). F, GSH levels in AdβGal‐ and AdHK2‐infected NRVMs (n=3). G, Glucose‐6‐phosphate dehydrogenase (G6PDH) enzymatic activity in AdβGal‐ and AdHK2‐infected NRVMs treated with vehicle or 25 μmol/L of the G6PDH inhibitor dehydroepiandrosterone (DHEA, n=4). H, Mean phenylephrine (PE)‐induced increase in ROS levels in AdβGal‐ and AdHK2‐infected NRVMs treated with vehicle or 25 μmol/L DHEA (n=4). I, Mean PE‐induced increase in NRVM cell area in AdβGal‐ and AdHK2‐infected NRVMs treated with vehicle or 25 μmol/L DHEA (n=5). Error bars represent SEM with *P<0.05 vs Vehicle and †P<0.05 vs AdβGal or CONsi. AdβGal indicates adenovirus expressing β‐galactosidase; HK2, hexokinase‐2; NRVM, neonatal rat ventricular myocytes; NTG, non‐transgenic; SEM, standard error of the mean; TG, transgenic.
HK2 Modulates ROS via the Pentose Phosphate Pathway
Lastly, we wanted to determine how HK2 controlled ROS levels during cardiomyocyte hypertrophy. One possible connection between glucose utilization and ROS is the pentose phosphate pathway. The initial step in this pathway is the conversion of glucose‐6‐phosphate into 6‐phosphogluconolactone by G6PDH, which involves the reduction of NADP+ to NADPH. NADPH can then be utilized to reduce glutathione and control ROS levels.38 We therefore decided to test whether HK2 overexpression was controlling ROS levels by increasing flux through the pentose phosphate pathway. One of the end points of the pentose phosphate pathway is the reduction of glutathione. Consistent with increased flux we found that GSH levels were significantly increased in both HK2 TG hearts and HK2‐overexpressing NRVMs compared to the respective controls (Figure 6E and 6F). We next treated NRVMs with dehydroepiandrosterone (DHEA), an inhibitor of G6PDH 39 to decrease pentose phosphate pathway flux. DHEA (25 μmol/L) treatment significantly decreased G6PDH activity by 55% (Figure 6G). HK2 overexpression resulted in significantly increased G6PDH activity (27% increase); however, DHEA maintained the ability to inhibit G6PDH activity with HK2 overexpression (Figure 6G). Similar to what was shown before, HK2 overexpression attenuated the increase in ROS accumulation induced by PE in vehicle treated NRVMs; however, DHEA cotreatment abolished the ability of HK2 to decrease ROS accumulation (Figure 6H). DHEA treatment also blocked the ability of HK2 to attenuate the PE‐induced increase in cardiomyocyte size (Figure 6I). Together, these results suggest that HK2 overexpression attenuates ROS levels and hypertrophy by increasing glucose flux through the pentose phosphate pathway.
Discussion
Cardiac hypertrophy is characterized by a transition into the “fetal metabolic program”, involving decreased fatty acid metabolism and increased glucose utilization. Recent evidence from genetic models suggests that altered fatty acid metabolism is the driving force of pathologic hypertrophy, and increased glucose utilization may be a beneficial compensation. Therefore, we hypothesized that a mouse model of increased cardiac HK2 expression would result in a decrease in hypertrophy in response to chronic β‐adrenergic stimulation. Indeed, αMHC‐HK2 mice displayed decreased cardiac and cardiomyocyte hypertrophy in response to isoproterenol. HK2 overexpression also attenuated the hypertrophic response in vitro in NRVMs treated with PE or AngII. Oppositely, HK2 depletion by treatment of NRVMs with a HK2 siRNA exacerbated PE‐induced hypertrophy. These results are in full agreement with most other studies investigating cardiac hypertrophy in models of altered glucose utilization. Cardiac‐specific deletion of the GLUT4 transporter was shown to induce cardiac hypertrophy at basal conditions.20 Antithetically, cardiac‐specific GLUT1 overexpression was shown to decrease hypertrophy, attenuate the transition to heart failure, and increase survival in response to pressure overload.19 Cardiac‐specific PPARα overexpression has also been shown to increase fatty acid metabolism, decrease glucose utilization, and result in cardiac hypertrophy at basal conditions.17 Most recently, Hk2 heterozygous mice were shown to have increased hypertrophy and exacerbated heart failure in response to pressure overload.22 We also found that HK2 overexpression could reduce the TUNEL staining associated with isoproterenol‐induced hypertrophy, suggesting that HK2 may also have anti‐apoptotic properties in this context. We also found that HK2 could attenuate oxidant‐induced necrosis in NRVMs. These data are consistent with previous studies demonstrating that reduced HK2 levels enhanced pressure‐overload‐ and ischemia/reperfusion‐induced myocyte apoptosis,22,40 and that overexpression of HK2 in NRVMs protects against oxidative‐stress‐induced death.41
ROS levels have been shown to increase during hypertrophy,22,35–37 where they are thought to mediate the hypertrophic response by activating pro‐hypertrophic signaling enzymes such as Ca2+/Calmodulin‐dependent kinase,42 calcineurin/NFAT,43 and mitogen‐activated protein kinase pathways.44 HK2 is known to be protective against ROS‐induced death.13–16 Consistent with these data we found that treatment of NRVMs with PE resulted in increased ROS accumulation, and HK2 overexpression attenuated this increase in ROS levels. Conversely, HK2 knockdown further increased the ROS accumulation during PE‐induced hypertrophy. The increased hypertrophy observed with decreased HK2 expression was suggested to be due to increased ROS accumulation as a result of mitochondrial permeability transition.22 Indeed mitochondrial permeability transition has been reported to contribute to aging‐related hypertrophy,45 and overexpression of the mitochondrial pore subunit cyclophilin‐D induces hypertrophy.31 However, isolated mitochondria from αMHC‐HK2 hearts showed no difference in mitochondrial swelling experiments, suggesting that inhibition of permeability transition may not play a role in the anti‐oxidant and anti‐hypertrophic effects of HK2. We cannot fully rule out a mitochondrial mechanism, as there is a small but significant increase in HK2 mitochondrial expression in our overexpression model. Nevertheless, the much greater increase in HK2 expression in the cytosolic fraction in our model leads us to surmise that the anti‐oxidant effect of HK2 is likely due to a more cytosolic or glucose utilization‐based mechanism, rather than a mitochondrial mechanism.
We investigated how HK2 reduced ROS levels, and tested the theory that increased HK2 expression could increase pentose phosphate pathway flux, leading to reduced ROS. The pentose phosphate pathway is important for the formation of NADPH reducing equivalents that are necessary for glutathione peroxidase activity and production of reduced glutathione.38 Thus, the pentose phosphate pathway is vitally important in controlling oxidative stress and, therefore, could represent a non‐mitochondrial mechanism by which HK2 suppresses ROS. Glucose‐6‐phosphate, the product of the reaction catalyzed by HK2, is the substrate for the initial reaction of the pentose phosphate pathway performed by G6PDH. Increased G6PDH activity,24–25 and increased pentose phosphate pathway flux 46–48 have been observed during cardiac hypertrophy. Genetic models of altered pentose phosphate pathway activity also suggest a prominent connection between this pathway and hypertrophy. Mice that are deficient in G6PDH develop hypertrophy ≈9 months of age,49 and display increased hypertrophy after myocardial infarction or pressure overload.50 In the present study, DHEA treatment was utilized to decrease G6PDH activity by ≈55%. This level of inhibition did not result in significantly increased ROS accumulation or cardiomyocyte hypertrophy. However, inhibition of G6PDH did abolish the beneficial effects of HK2 overexpression against α‐adrenergic stimulation‐induced ROS production and cardiomyocyte hypertrophy. Interestingly, our ≈3‐fold HK2 overexpression increased G6PDH activity ≈30%, which fits nicely with the estimation that 10% to 20% of glucose oxidation occurs through the pentose phosphate pathway.51 Consistent with this was our finding that GSH levels are elevated when HK2 is increased in cardiac myocytes. Thus, these results indicate that increased pentose phosphate pathway flux is an important component of the anti‐hypertrophic effect of HK2.
Another possible fate of glucose‐6‐phosphate is the formation of O‐linked β‐N‐acetylglucosamine residues, also known as protein GlcNAcylation, by the hexosamine biosynthetic pathway. Flux through this pathway also appears to be increased during cardiac hypertrophy as GlcNAc levels are increased in hypertrophied hearts.26–27 Hypertrophy also increases the expression of O‐linked β‐N‐acetylglucosamine transferase, the enzyme responsible for adding GlcNAc residues to proteins, and deletion of this enzyme increases cardiomyocyte hypertrophy, apoptosis, and dysfunction.27 We did not thoroughly investigate the hexosamine biosynthetic pathway in this study; however, the wheat germ agglutinin used in this study is a marker of N‐acetylglucosamine residues on cell plasma membranes. Analysis of the fluorescence intensity of these images does suggest that both HK2 overexpression and hypertrophy increase N‐acetylglucosamine levels (Figure 7A), and blotting demonstrated a modest increase in protein O‐GlcNAcylation in HK2 overexpressing hearts (Figure 7B). Therefore, increased protein O‐GlcNAcylation could potentially be another mechanism decreasing both cardiomyocyte hypertrophy and apoptosis in our HK2 overexpression model. Further study is warranted to thoroughly investigate the response of the hexosamine biosynthetic pathway to altered HK2 expression.
Figure 7.
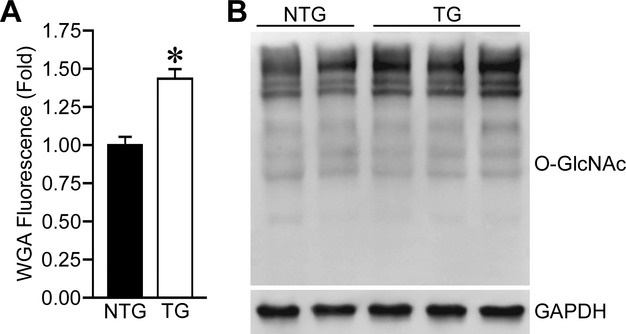
HK2 overexpression increases O‐GlcNAcylation. A, Quantification of wheat germ agglutinin (WGA) fluorescence intensity from left‐ventricular sections of NTG and TG mouse hearts (n=3). B, Western blotting for protein O‐GlcNAcylation in NTG and TG cardiac lysates. GAPDH was used as a loading control. Blots representative of n=5 to 6 in each group. Error bars represent SEM with *P<0.05 vs NTG. HK2 indicates hexokinase‐2; NTG, non‐transgenic; SEM, standard error of the mean; TG, transgenic.
A consistent finding was that overexpression of HK2 only partially abrogated the hypertrophy, increases in ROS, and cell death, whether in the mice or the cultured myocytes. This most likely stems from the fact that HK2 did not completely abolish the increases in ROS, which in turn would drive the hypertrophy and cell death. This could be due to the levels of HK2 and it is feasible that a greater level of HK2 could overcome this, totally prevent ROS production, and therefore fully restore a normal phenotype. However, it is likely that other ROS‐independent pathways are also at play here. For example, the antioxidant tempol was only able to partially reverse the hypertrophy seen in GLUT4‐deficient hearts.52 Similarly, n‐acetylcysteine was only partially effective in blocking AngII‐induced hypertrophy in vivo.53 Therefore, other HK2‐insensitive signaling may contribute to the residual hypertrophy and cell death exhibited in the cells and mice.
To conclude, our results show that HK2 overexpression attenuates cardiomyocyte hypertrophy and death in response to adrenergic stimulation. Our in vitro studies suggest that HK2 modulates hypertrophy by controlling cellular ROS levels by increasing pentose phosphate pathway flux. Thus, therapeutic approaches to increase HK2 expression or pentose phosphate pathway flux may assist in the treatment of pathologic cardiac hypertrophy.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL094404 (C.P.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. K.S.M. was supported by an American Heart Association Predoctoral Fellowship (11PRE6930000).
Disclosures
Dr Baines receives research support from National Institutes of Health (>$10K R01HL094404). Dr McCommis received research support from the American Heart Association (>$10K 11PRE6930000).
Acknowledgments
The technical expertise of Kathryn Crombie is gratefully acknowledged.
References
- 1.Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. 1972; 15:289-329 [DOI] [PubMed] [Google Scholar]
- 2.Taegtmeyer H, Overturf ML. Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension. 1988; 11:416-426 [DOI] [PubMed] [Google Scholar]
- 3.Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. 1999; 318:36-42 [DOI] [PubMed] [Google Scholar]
- 4.Dávila‐Román V, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002; 40:271-277 [DOI] [PubMed] [Google Scholar]
- 5.Katzen HM. The multiple forms of mammalian hexokinase and their significance to the action of insulin. Adv Enzyme Regul. 1967; 5:335-336 [DOI] [PubMed] [Google Scholar]
- 6.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003; 206:2049-2057 [DOI] [PubMed] [Google Scholar]
- 7.Osawa H, Printz RL, Whitesell RR, Granner DK. Regulation of hexokinase II gene transcription and glucose phosphorylation by catecholamines, cyclic AMP, and insulin. Diabetes. 1995; 44:1426-1432 [DOI] [PubMed] [Google Scholar]
- 8.Printz RL, Osawa H, Ardehali H, Koch S, Granner DK. Hexokinase II gene: structure, regulation and promoter organization. Biochem Soc Trans. 1997; 25:107-112 [DOI] [PubMed] [Google Scholar]
- 9.Fiek C, Benz R, Roos N, Brdiczka D. Evidence for identity between the hexokinase‐binding protein and the mitochondrial porin in the outer membrane of rat liver mitochondria. Biochim Biophys Acta. 1982; 688:429-440 [DOI] [PubMed] [Google Scholar]
- 10.Linden M, Gellerfors P, Nelson BD. Pore protein and the hexokinase binding protein from the outer membrane of rat liver mitochondria are identical. FEBS Lett. 1982; 141:189-192 [DOI] [PubMed] [Google Scholar]
- 11.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax‐induced cytochrome c release and apoptosis. J Biol Chem. 2002; 277:7610-7618 [DOI] [PubMed] [Google Scholar]
- 12.Majewski N, Nogueira V, Bhasker P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase‐mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004; 16:819-830 [DOI] [PubMed] [Google Scholar]
- 13.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase‐II. Cell Death Differ. 2008; 15:521-529 [DOI] [PubMed] [Google Scholar]
- 14.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008; 28:1007-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, Klawitter H, Przesdzing I, Neeb L, Freyer D, Priller J, Collins TJ, Megow D, Dirnagl U, Andrews DW, Meisel A. Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cell fate depending on the metabolic state. Proc Natl Acad Sci USA. 2012; 109:1518-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Da‐Silva WS, Gomez‐Puyou A, de Gomez‐Puyou MT, Moreno‐Sanchez R, De Felice FG, de Meis L, Oliviera MF, Galina A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady‐state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem. 2004; 279:39846-39855 [DOI] [PubMed] [Google Scholar]
- 17.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002; 109:121-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR‐gamma coactivator 1alpha. Proc Natl Acad Sci USA. 2006; 103:10086-10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac‐specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002; 106:2125-2131 [DOI] [PubMed] [Google Scholar]
- 20.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste‐Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved ejection contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. 1999; 104:1703-1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, Doenst T, Medina‐Gomez G, Litwin SE, Lelliott CJ, Vidal‐Puig A, Abel ED. PGC‐1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011; 109:783-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, Epting CL, Ardehali H. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012; 4:1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolwicz SC, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011; 90:194-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allard MF, Henning SL, Walbolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation. 1997; 96:676-682 [DOI] [PubMed] [Google Scholar]
- 25.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose‐6‐phosphate dehydrogenase‐derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006; 41:340-349 [DOI] [PubMed] [Google Scholar]
- 26.Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, Jones SP. O‐GlcNAc signaling is essential for NFAT‐mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol. 2012; 302:H2122-H2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O‐linked β‐N‐acetylglucosamine transferase is indispensible in the failing heart. Proc Natl Acad Sci USA. 2010; 107:17797-17802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baines CP, Kaiser RA, Sheiko T, Craigen WJ. Voltage‐dependent anion channels are dispensable for mitochondrial‐dependent cell death. Nat Cell Biol. 2007; 9:550-555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson JE. Rapid purification of mitochondrial hexokinase from rat brain by a single affinity chromatography step on AffiGel Blue. Prep Biochem. 1989; 19:13-21 [DOI] [PubMed] [Google Scholar]
- 30.Wilson JE. Distinguishing the type I and type II isozymes of hexokinase: the need for a reexamination of past practice. Diabetes. 1998; 47:1544-1548 [DOI] [PubMed] [Google Scholar]
- 31.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005; 434:658-662 [DOI] [PubMed] [Google Scholar]
- 32.Schramm C, Fine DM, Edwards MA, Reeb AN, Krenz M. The PTPN11 loss‐of‐function mutation Q510E‐Shp2 causes hypertrophic cardiomyopathy by dysregulating mTOR signaling. Am J Physiol Heart Circ Physiol. 2012; 302:H231-H243 [DOI] [PubMed] [Google Scholar]
- 33.Tian WN, Pignatare JN, Stanton RC. Signal transduction proteins that associate with the platelet‐derived growth factor (PDGF) receptor mediate the PDGF‐induced release of glucose‐6‐phosphate dehydrogenase from permeabilized cells. J Biol Chem. 1994; 269:14798-14805 [PubMed] [Google Scholar]
- 34.Young ME, Yan J, Razeghi P, Cooksey RC, Guthrie PH, Stepkowski SM, McClain DA, Tian R, Taegtmeyer H. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul Syst Bio. 2007; 1:251-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal. 2003; 5:731-740 [DOI] [PubMed] [Google Scholar]
- 36.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007; 93:903-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007; 49:241-248 [DOI] [PubMed] [Google Scholar]
- 38.Robey RB, Hay N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene. 2006; 25:4683-4696 [DOI] [PubMed] [Google Scholar]
- 39.Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC. Importance of glucose‐6‐phosphate dehydrogenase activity for cell growth. J Biol Chem. 1998; 273:10609-10617 [DOI] [PubMed] [Google Scholar]
- 40.Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L, Chawla K, Hollmann MW, Nagpal V, Heikkinen S, Laakso M, Jujo K, Wasserstrom JA, Zuurbier CJ, Ardehali H. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ Res. 2011; 1:60-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts DJ, Tan‐Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK‐II at Thr‐473 and increases mitochondrial HK‐II association to protect cardiomyocytes. J Biol Chem. 2013; 288:23798-23806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin‐Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell. 2008; 133:462-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adiga IK, Nair RR. Multiple signaling pathways coordinately mediate reactive oxygen species dependent cardiomyocyte hypertrophy. Cell Biochem Funct. 2008; 26:346-351 [DOI] [PubMed] [Google Scholar]
- 44.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves‐Cintrón M, Chen T, Marcinek DJ, Dorn GW, II, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II‐induced cardiac hypertrophy and Gαq overexpression‐induced heart failure. Circ Res. 2011; 108:837-846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3‐mediated deacetylation of CypD at lysine 166 suppresses age‐related cardiac hypertrophy. Aging. 2010; 2:914-923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meerson FZ, Spiritchev VB, Pshennikova MG, Djachkova LV. The role of the pentose‐phosphate pathway in adjustment of the heart to a high load and the development of myocardial hypertrophy. Experientia. 1967; 23:530-532 [DOI] [PubMed] [Google Scholar]
- 47.Zimmer HG, Ibel H, Steinkopff G. Studies on the hexose monophosphate shunt in the myocardium during development of hypertrophy. Adv Myocardiol. 1980; 1:487-492 [PubMed] [Google Scholar]
- 48.Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 2010; 3:420-430 [DOI] [PubMed] [Google Scholar]
- 49.Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, Koh S, Sawyer DB, Leopold JA, Handy DE, Loscalzo J, Apstein CS, Liao R. Glucose‐6‐phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003; 93:e9-e16 [DOI] [PubMed] [Google Scholar]
- 50.Hecker PA, Lionetti V, Ribeiro RF, Rastogi S, Brown BH, O'Connell KA, Cox JW, Shekar KC, Gamble DM, Sabbah HN, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Glucose‐6‐phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ Heart Fail. 2013; 6:118-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wamelink MMC, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008; 31:703-717 [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Wende AR, Nunthakungwan O, Huang Y, Hu E, Jin H, Boudina S, Abel ED, Jalili T. Cytosolic, but not mitochondrial, oxidative stress is a likely contributor to cardiac hypertrophy resulting from cardiac specific GLUT4 deletion in mice. FEBS J. 2012; 279:599-611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakagami H, Takemoto M, Liao JK. NADPH oxidase‐derived superoxide anion mediates angiotensin II‐induced cardiac hypertrophy. J Mol Cell Cardiol. 2003; 35:851-859 [DOI] [PubMed] [Google Scholar]