

Abstract
Background
Rheumatoid arthritis (RA) is associated with heightened mortality due to atherosclerotic cardiovascular disease (CVD). Inflammatory pathways in RA negatively affect vascular physiology and promote metabolic disturbances that contribute to CVD. We hypothesized that the peroxisome proliferator activated receptor‐γ (PPAR‐γ) pioglitazone could promote potent vasculoprotective and anti‐inflammatory effects in RA.
Methods and Results
One hundred forty‐three non‐diabetic adult RA patients (76.2% female, age 55.2±12.1 [mean±SD]) on stable RA standard of care treatment were enrolled in a randomized, double‐blind placebo controlled crossover trial of 45 mg daily pioglitazone versus placebo, with a 3‐month duration/arm and a 2‐month washout period. Pulse wave velocity of the aorta (PWV), brachial artery flow mediated dilatation (FMD), nitroglycerin mediated dilatation (NMD), microvascular endothelial function (reactive hyperemia index [RHI]), and circulating biomarkers of inflammation, insulin resistance, and atherosclerosis risk all were quantified. RA disease activity was assessed with the 28‐Joint Count Disease Activity Score (DAS‐28) C‐reactive protein (CRP) and the Short Form (36) Health Survey quality of life questionnaire. When added to standard of care RA treatment, pioglitazone significantly decreased pulse wave velocity (ie, aortic stiffness) (P=0.01), while FMD and RHI remained unchanged when compared to treatment with placebo. Further, pioglitazone significantly reduced RA disease activity (P=0.02) and CRP levels (P=0.001), while improving lipid profiles. The drug was well tolerated.
Conclusions
Addition of pioglitazone to RA standard of care significantly improves aortic elasticity and decreases inflammation and disease activity with minimal safety issues. The clinical implications of these findings remain to be established.
Clinical Trial Registration
URL: ClinicalTrials.gov Unique Identifier: NCT00554853.
Keywords: drugs, inflammation, rheumatoid arthritis, vasodilation
Introduction
Rheumatoid arthritis (RA), a chronic inflammatory disease that affects ≈1% of the population in the United States, is associated with significant levels of functional disability, joint destruction, and loss of productivity. RA is also associated with enhanced mortality, which is predominantly due to accelerated atherosclerotic cardiovascular disease (CVD).1–4 Indeed, women with RA are twice as likely to suffer myocardial infarction (MI) compared to women without RA, and the risk is considered equivalent to the one observed in diabetes mellitus (DM).2 Largely due to this heightened CVD risk, the overall life expectancy in RA is significantly reduced.2,5–6 The Framingham risk equation does not fully account for the enhanced CVD risk in RA,7 which appears to convey a risk beyond that attributable to standard risk factors. As such, it has been proposed that immune dysregulation, inflammation, and metabolic disturbances characteristic of RA play an important role in the development of endothelial dysfunction, plaque progression, and acute coronary syndromes (ACS).8 Indeed, chronic inflammation has been firmly associated with endothelial cell (EC) activation and vascular dysfunction, leading to noncompliance of vessels and development of atheroma. However, the reasons for the dramatic increase in CVD in RA are not totally understood and appear to be complex.
While growing evidence suggests that disease modifying antirheumatic drugs (DMARDs) and various biologic agents may decrease CV risk in RA,9–11 these drugs can also pose significant risks to selected RA patients, are not accessible to all patients, and their long‐term protective effects on the endothelium are uncertain.12 Therefore, it is important to identify therapeutic targets that are antiatherogenic without causing significant immunosuppression.
The thiazolidinediones (TZD) are a class of drugs used in the treatment of type 2 diabetes mellitus (DM). These drugs are potent peroxisome proliferator‐activated receptor‐γ (PPAR‐γ) agonists with significant pleiotropic effects on the vasculature and on inflammatory pathways that are both antiatherogenic and anti‐inflammatory.13 We hypothesized that TZDs could significantly improve endothelial function and CV risk in RA and might represent an important preventive strategy for this potentially fatal complication in individuals with this disease. Given the anti‐inflammatory properties reported previously by PPAR‐γ agonists both in vitro and in vivo,13–18 we also proposed that TZDs could ameliorate RA disease activity.
This study assessed the efficacy of TZDs with regards to modification of vascular function, biomarkers of CV risk and disease activity in a cohort of patients with stable RA.
Methods
Subjects
The study protocol complied with the Declaration of Helsinki and was approved by the University of Michigan Medical Institutional Review Board. All participants signed informed consent. Subjects fulfilling the 1987 American College of Rheumatology diagnostic criteria for RA19 were recruited from the Rheumatology clinic at the University of Michigan and by advertisement. For inclusion, stable doses (>3 months prior to enrollment) of corticosteroids, DMARDs and/or biological agents were required. Subjects were excluded if they were current smokers or had smoked within 6 months prior to enrollment, had significant hepatic or renal impairment, diabetes mellitus, congestive heart failure (NYHA class III or IV), or acute infection. Subjects treated with lipid‐lowering drugs were required to be on stable doses for at least 6 months prior to enrollment. Women of reproductive age were excluded if pregnant or lactating, and were required to be on adequate contraception to be enrolled.
Study Protocol
The study was a single‐center, randomized, double‐blind, placebo‐controlled, crossover treatment trial of 45 mg daily of pioglitazone. Pioglitazone and placebo tablets were provided by Takeda Pharmaceutical Company. Patients were treated for 3 months with pioglitazone or placebo, underwent a washout period of 8 weeks, and then crossed over to the other arm for an additional 3 months. Patients typically took 30 mg daily of pioglitazone for 2 weeks, followed by an increase to 45 mg daily for the rest of the treatment arm. Functional and serum biomarkers of vascular damage, atherosclerosis risk, and RA disease activity were assessed at baseline and at 3, 5, and 8 months after enrollment using the 28‐Joint Count Disease Activity Score C‐reactive protein (DAS28‐CRP). Safety visits occurred at 1 and 6 months postenrollment (see Figure 1 for detailed protocol).
Figure 1.
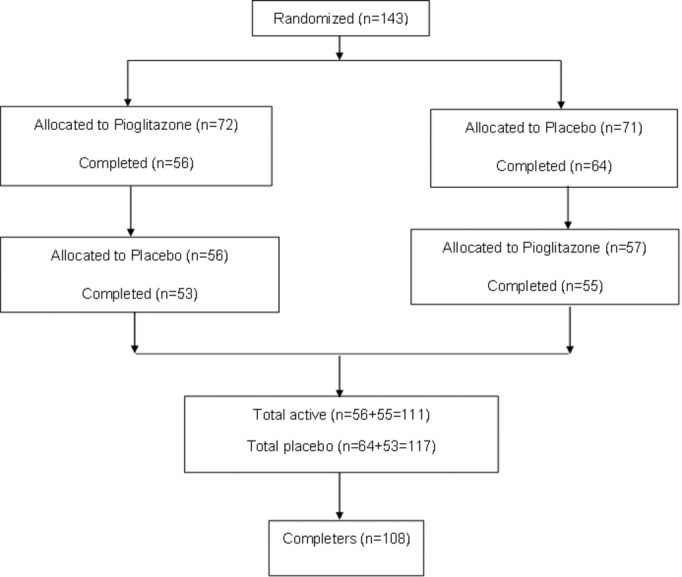
Study population recruitment summary.
Vascular function assessment
Studies were conducted at the University of Michigan Research Vascular Laboratory in a temperature‐controlled room. Prior to testing, patients were instructed to fast and to hold vasoactive drugs for at least 12 h. Detailed methodology has been previously described by our group.20–21
Pulse wave analyses and arterial pulse wave velocity
After subjects had rested supine for 10 minutes, applanation tonometry was measured once at the right carotid and femoral arteries for 10 seconds each, following manufacturer's guidelines (Sphygmocor device; Atcor). Three‐lead ECG recordings were obtained simultaneously with the tonometry to calculate aortic pulse wave velocity (PWV) (a direct measure of arterial stiffness). The operator quality index was 80% in all accepted values used for the study.
Simultaneous microvascular and conduit brachial endothelial function quantification, assessment of vascular smooth muscle function and endothelium‐independent nitric oxide responsiveness
Simultaneous measurement of conduit artery endothelial‐dependent and independent vasodilatation by brachial flow mediated dilatation (FMD) and nitroglycerin mediated dilatation (NMD), respectively, and of microvascular endothelial‐dependent vasodilatation using the Endo‐Pat2000 device (Itamar) was performed on the dominant arm as previously described by us.20–24 The end points of these measurements were the percentage of change in mean brachial and radial diameter in response to reactive hyperemia (FMD) or to nitroglycerin (NMD). The reproducibility of these techniques has been previously reported by our group.22,24
Quantification of circulating markers of CV risk and inflammation
Quantification of serum inflammatory cytokines, adhesion molecules and chemokines
Enzyme‐linked immunosorbent assay (ELISAs) were performed following manufacturer's instructions and using a Biotek plate reader (BioTek Instruments Inc) to quantify serum levels of the following molecules: monocyte chemoattractant protein‐1 (MCP‐1) (BD Biosystems), soluble intercellular adhesion molecule‐1 (sICAM‐1), soluble vascular cellular adhesion molecule (sVCAM‐1), and interleukin‐6 (IL‐6) (all from R&D systems). For circulating IL‐17 quantification by ELISA, we used a protocol previously reported by our group.20
Other laboratory measurements
Total cholesterol, HDL and LDL cholesterol, triglycerides, insulin, glucose, and high sensitivity C‐reactive protein (hsCRP) were quantified in the Michigan Diabetes Research and Training Center. Complete blood count (CBC), liver function tests (LFTs), Westergren sedimentation rate (ESR), pregnancy test, rheumatoid factor (RF), and anti‐CCP Abs (ACPA) were measured at the central laboratories at the University of Michigan.
Quantification of insulin resistance
The Homeostasis Model of Assessment‐Insulin Resistance 2 (HOMA‐2) was used to quantify insulin resistance25 as follows: HOMA‐IR=(Glucose [mg/dL]×Insulin [μU/mL])/405. Values >2.5 were considered consistent with insulin resistance.
Assessment of RA disease activity
Disease Activity Score 28 based on C‐reactive protein (DAS‐28 CRP)26 was used to quantify RA disease activity as previously described. The SF‐36 quality of life (QOL) questionnaire27 was also applied to patients at each visit.
Statistical considerations and analysis
The randomization for treatment sequence was conducted using computer‐generated lists stratified for baseline DAS‐28 scores and patients' age and managed through the University of Michigan pharmacy. The primary outcome of the study was modifications in pulse wave velocity (PWV) of the aorta. Secondary outcomes were changes in other vascular function assessments (FMD, NMD, RHI), as well as RA disease activity. Sample size formula for a 2‐period, 2‐treatment crossover design was used to estimate the detectable treatment effect with the α level fixed at 0.05 and power at 80%. Standard deviations were determined from the literature at the time the study was planned and from pilot studies on similar populations. For FMD 0.5% change was used and for NMD 1.4% change was used. In addition, the sample size calculation assumed different possible correlations between observations over time. We powered the study for differences of 0.26 to 0.36 times the assumed standard deviations. A 1 in 6 (17%) subject dropout rate was assumed, so the planned sample size was a total of 120 subjects completing the study. We conducted a repeated measures analysis of variance including treatment, sequence, and period effects and incorporated baseline measures on the primary measures and on QOL. If the tests for sequence and period effect were not significant they were dropped from the model. The analysis was conducted on the complete dataset and restricted to those subjects who completed all assessments (n=108). Continuous variables were summarized using means and standard deviations (SD) and categorical variables by counts and percentages. To assess carryover effects, the 2 baseline measures of all outcome variables were compared using paired t test for normal data or Wilcoxon sign rank test for non‐normal data.
Two sample t tests or Wilcoxon rank sum tests were used to compare the first period outcomes between the 2 groups. Because no carryover effect was detected, a paired t test or Wilcoxon signed rank test was used to compare the change from baseline to end of treatment for additional known CV and inflammation markers. A chi square or Fisher's exact test was used to compare the rate of various adverse events between the placebo and treatment groups. An exact binomial test was used for comparing the dropout rates between those starting treatment with pioglitazone versus those starting with placebo. Due to the pilot and hypothesis‐generating nature of this study, no adjustment was used for multiple statistical testing and multiplicity of outcomes in the sample size calculation. A P<0.05 was considered significant for all hypothesis tests. The above procedures were done in SAS 9.3 (SAS Institute Inc).
Results
Patients' Characteristics and Recruitment
A total of 143 nondiabetic RA subjects were randomized to participate in the study. Baseline demographics and clinical characteristics and measurements of vascular function and inflammation are presented in Tables 1 and 2 and include information on all subjects enrolled, as well as on completers. Of patients enrolled, 95% were seropositive for rheumatoid factor (RF) and 57.3% for anti‐CCP antibodies. At baseline, treatment groups did not significantly differ with respect to RA disease activity (P=0.4), RA duration (P=0.2), or medications used to treat RA, including biologics, DMARDs, or corticosteroids (P=0.1). There were also no differences between groups in the Framingham risk equation (P=0.2) or use of vasoactive medications. Overall, subjects demonstrated high prevalence of insulin resistance at baseline (113/140 patients [81%]; HOMA‐IR2=5.11, ±3.44, mean±SD). At baseline, 68.5% of patients were on DMARDs, 46.8% were on biologics, and 49.6% were on steroids. In addition, 37.7% of patients were taking a vasoactive drug, 12.5% were on lipid‐lowering agents, and 16.7% took daily aspirin.
Table 1.
Baseline Demographic, Clinical Characteristics, and Medications of Study Participants
| Completers (n=108), n (%) | All Subjects (n=143), n (%) | |
|---|---|---|
| Gender (females), n (%) | 84 (77.8) | 109 (76.2) |
| Race, n (%) | ||
| White | 98 (90.7) | 124 (86.7) |
| African American | 6 (5.6) | 13 (9.1) |
| Asian | 2 (1.9) | 4 (2.8) |
| American Indian/Alaska Native | 1 (0.9) | 1 (0.7) |
| Other | 1 (0.9) | 1 (0.7) |
| Ethnicity (Hispanic), n (%) | 5 (4.6) | 10 (7.0) |
| RF (positive), n (%) | 105 (97.2) | 137 (95.8) |
| Anti‐CCP (positive), n (%) | 62 (57.4) | 82 (57.3) |
| Mean (SD) | Mean (SD) | |
| Age, y | 54.9 (12.5) | 55.2 (12.1) |
| Anti‐CCP*, U/mL | 31.1 (95) | 31.6 (95) |
| RF*, IU/mL | 30.5 (97) | 29.0 (81) |
| DAS28‐CRP* | 2.8 (1.98) | 3.0 (1.91) |
| Years since RA diagnosis | 11.2 (11.2) | 11.0 (10.5) |
| ESR*, mm/h | 9.0 (12) | 10.0 (12) |
| Medications, n (%) | ||
| Biologics | 53 (49.1) | 67 (46.85) |
| Biologics only | 2 (3.8) | 3 (4.48) |
| Biologics and DMARD | 4 (7.5) | 4 (5.97) |
| Biologics and other* | 12 (22.6) | 19 (28.36) |
| Biologics and DMARD and other* | 35 (66.0) | 41 (61.19) |
| DMARD | 77 (71.3) | 98 (68.53) |
| DMARD only | 5 (6.5) | 6 (6.12) |
| DMARD and biologics | 4 (5.2) | 4 (4.08) |
| DMARD and others | 33 (42.9) | 47 (47.96) |
| DMARD and biologics and other* | 35 (45.5) | 41 (41.84) |
| Vasoactive | 41 (37.96) | 54 (37.76) |
| Lipid lowering | 14 (12.96) | 18 (12.58) |
| Aspirin | 18 (16.66) | 24 (16.78) |
| Steroid | 53 (49.07) | 71 (49.65) |
| NSAID | 58 (53.7) | 78 (54.54) |
CCP indicates cyclic citrullinated peptide; DAS28‐CRP, Disease Activity Score of 28 Joints‐C reactive protein; DMARD, disease modifying anti‐rheumatic drug; ESR, erythrocyte sedimentation rate; IQR, interquartile range; NSAID, nonsteroidal antiinflammatory drugs; RA, rheumatoid arthritis; RF, rheumatoid factor; SD, standard deviation.
Median (IQR).
Other includes vasoactive and lipid‐lowering drugs, aspirin, steroids, and NSAIDs.
Table 2.
Baseline CV Measurements, Risk Factors and Inflammatory Markers
| Completers (n=108), Mean (SD) | All Subjects (n=143), Mean (SD) | |
|---|---|---|
| Brachial SBP, mm Hg | 131.5 (17.6) | 131.9 (17.7) |
| Brachial DBP, mm Hg | 78.7 (10.7) | 78.6 (11.0) |
| Aortic PP, mm Hg | 46.0 (14.9) | 45.1 (14.3) |
| Aortic DP, mm Hg | 124.4 (18.4) | 123.5 (18.4) |
| Augmentation index | 28.6 (12.0) | 28.3 (11.5) |
| BMI | 29.5 (6.7) | 29.8 (6.8) |
| hsCRP, mg/dL | 4.2 (6.2) | 5.3 (10.5) |
| Framingham composite score, % | 3.9 (5.3) | 3.7 (4.9) |
| MCP‐1*, pg/mL | 473.2 (250.77) | 471.9 (294.96) |
| IL‐17*, pg/mL | 60.5 (642.6) | 17.1 (642.3) |
| IL‐6*, pg/mL | 1.0 (6.4) | 1.4 (6.2) |
| Total cholesterol, mg/dL | 186.8 (41.7) | 186.0 (41.7) |
| Triglycerides, mg/dL | 110.4 (59.8) | 109.6 (59.5) |
| HDL, mg/dL | 65.3 (18.0) | 65.2 (17.7) |
| LDL, mg/dL | 111.3 (35.5) | 111.1 (35.2) |
| Fasting serum insulin, μU/mL | 20.3 (10.9) | 21.2 (12.3) |
| Fasting glucose, mg/dL | 95.6 (11.6) | 95.0 (11.0) |
| HOMA‐IR, mg/dL×μU/mL | 5.0 (3.4) | 5.1 (3.4) |
| FMD, % | 8.8 (4.2, 48%*) | 8.3 (4.0, 48%*) |
| NMD, % | 1.9 (1.6, 84%*) | 2.0 (1.6, 80%*) |
| PWV, m/s | 9.0 (2.6, 29%*) | 9.2 (2.8, 30%*) |
| RHI, % | 2.1 (0.7, 33%*) | 2.1 (0.7, 33%*) |
BMI indicates body mass index; CV, cardiovascular; DBP, diastolic blood pressure; FMD, flow‐mediated dilatation; HDL, high‐density lipoprotein; HOMA‐IR, homeostatic model assessment‐insulin resistance; hsCRP, high‐sensitivity C‐reactive protein; DP, delta pressure; IL, interleukin; LDL, low‐density lipoprotein; MCP‐1, monocyte chemotactic protein‐1; NMD, nitroglycerin‐mediated dilatation; PP, pulse pressure; PWV, pulse wave velocity; RHI, reactive hyperemia index; SBP, systolic blood pressure.
Median(IQ).
Coefficient of variation.
There was >90% compliance in enrolled patients, as measured by pill counts returned at study visits. The dropout rates were similar between both initially randomized groups: treatment (19 of 72; 26%) versus placebo (16 of 71; 23%). Overall, 108 patients completed the study. Figure 1 shows the flow diagram of the study.
Effect of Pioglitazone on Aortic Compliance
Aortic stiffness, quantified as PWV, significantly decreased from baseline after 3 months on the pioglitazone treatment arm, when compared to the placebo arm (−0.48±2.10 m/s for treatment arm versus 0.27±1.46 m/s for placebo arm; P<0.0001). Overall, aortic PWV significantly decreased from baseline in the pioglitazone arm (P<0.0001), while mean brachial diastolic BP increased from baseline during pioglitazone treatment when compared to the placebo arm (−3.89±9.78 during pioglitazone versus −9.08±86 during placebo; P=0.02). No significant changes form baseline occurred in systolic BP. Both mean aortic systolic pressure and mean aortic pulse pressure significantly improved from baseline with treatment (−5.75±13.84 during pioglitazone versus 0.82±13.93 during placebo; P=0.0001 and −4.16±12.95 during pioglitazone versus 1.30±13.33 during placebo; P=0.0003), respectively.
Conduit vessel endothelial‐dependent vasorelaxation (brachial artery FMD) showed a trend to improve during pioglitazone treatment but this did not reach statistical significance, nor did microvascular endothelial function (RHI) significantly improve during treatment with active drug. Brachial artery vascular smooth muscle function, as assessed by NMD significantly increased from baseline during pioglitazone treatment when compared to the placebo arm (0.32±1.45% pioglitazone arm versus −0.20±1.25% placebo arm; P<0.0001) (Figure 2).
Figure 2.

Change in various CV measures and inflammatory markers during study. Mean change is plotted over time between groups. The dash line represents the mean change for individuals who started the treatment with pioglitazone; the solid line represents the mean change of individuals who started the treatment with placebo. CRP indicates C‐reactive protein; CV, cardiovascular; DAS28, 28‐Joint Disease Activity Scale; HDL, high‐density lipoprotein; HOMA IR, homeostasis model of assessment‐insulin resistance; NMD, nitroglycerin mediated dilatation; Pl/Tx, placebo/treatment group; PWV, pulse wave velocity.
Several markers associated with metabolic disturbances and CV risk were also significantly modified during the pioglitazone arm when compared to the placebo arm. Indeed, HDL levels significantly increased from baseline during pioglitazone treatment (5.07±13.86 mg/dL pioglitazone versus −0.64±13.98 mg/dL placebo; P=0.01), while triglycerides and hsCRP decreased (−16.13±44.51 mg/dL pioglitazone versus 2.06±46.79 mg/dL; P=0.01; −1.31±5.96 mg/dL pioglitazone versus 0.54±11.70 mg/dL placebo; P=0.001, respectively). Total cholesterol, LDL, and ESR were not significantly modified during pioglitazone treatment when compared to the placebo arm. As expected, the mean changes in HOMA‐IR values from baseline were significantly different between pioglitazone and placebo (−0.07±0.18 during pioglitazone arm versus 0.03±0.18 during placebo arm; P≤0.0001). Similarly, mean insulin levels significantly decreased from baseline while in the pioglitazone arm (−4.36±9.76 μU/mL on pioglitazone versus 2.21±10.87 μU/mL on placebo; P≤0.0001). No significant changes in adhesion molecules and chemokines were observed in the pioglitazone arm when compared to the placebo arm.
Overall, these observations indicate that short‐term treatment with pioglitazone significantly improves vascular function, particularly large vessel compliance and conduit vessel smooth muscle function, while also promoting beneficial changes in inflammatory markers, lipid profile and insulin resistance.
Pioglitazone and RA Disease Activity
When added to standard of care treatment of RA, disease activity measured by the DAS‐28‐CRP28 significantly decreased from baseline measurements on the pioglitazone arm when compared to placebo arm (mean=0.31±1.2 pioglitazone versus 0.15±1.2, P=0.02) after 3 months of treatment. QOL, as assessed by SF‐36, did not differ between treatment arms. Levels of IL‐17 and IL‐6 and ESR, were not differentially modified between treatment arms, while CRP significantly decreased during pioglitazone treatment (Figure 2). Overall, these observations indicate that short‐term treatment with pioglitazone significantly improves RA disease activity and decreases inflammatory burden when compared to placebo.
Pioglitazone Adverse Events
Patients with RA often present with various comorbidities, which can make treatments challenging, due to drug interactions and potential adverse effects. In this cohort of patients with overall low Framingham risk factors and preserved heart function, markers of target organ function, including LFTs and CBC were not different between treatment versus placebo groups at the end of the study. Overall, pioglitazone was well tolerated. There were more adverse events while on the active treatment group, and these consisted primarily of expected side effects related to this drug class such as weight gain, lower extremity edema, and dyspnea (Table 3). Of the 16 serious adverse events (SAEs) in the trial, only one was probably related to the study drug. This was a case of lower‐extremity edema and chest pain that resolved with discontinuation of the study drug. There were 5 other SAEs considered to be not related to the study drug including one case of dyspnea and chest pain, one case of tachycardia and elevated blood pressure, one case of low potassium and sodium, one hip fracture, and one intracranial bleed. The study drug was discontinued only for the case of dyspnea with chest pain, and all resolved. The remainder of SAEs were considered “definitely not” related to the study drug.
Table 3.
Adverse Event (AE) by Body System and Treatment
| N=143 | |||
|---|---|---|---|
| Body System | Placebo (n=127) | Treatment (n=129) | P Value |
| Body as a whole | 3 (2.36%) | 5 (3.87%) | 0.72 |
| Cardiovascular | 4 (3.15%) | 8 (6.25%) | 0.25 |
| Edema | 0 (0.00%) | 6 (4.65%) | NA |
| Hemic and lymphatic | 0 (0.00%) | 1 (3.87%) | NA |
| Infection | 5 (3.94%) | 5 (3.87%) | 1.00 |
| Metabolic and nutritional | 3 (2.36%) | 4 (3.10%) | 1.00 |
| Musculoskeletal | 5 (3.94%) | 8 (6.25%) | 0.57 |
| Nervous system | 2 (1.57%) | 4 (3.1%) | 0.68 |
| Respiratory | 1 (0.79%) | 6 (4.65%) | 0.12 |
| Skin and appendages | 4 (3.15%) | 3 (2.33%) | 0.72 |
| Total number of AEs | 27 (21.26%) | 50 (38.75%) | 0.003 |
| Total number of subjects with at least one AE | 23 (18.11%) | 39 (30.23%) | 0.028 |
Values in bold are statistically significant.
Discussion
As CVD now represents one of the most important causes of morbidity and mortality among patients with RA, it has become a priority in this and other chronic inflammatory diseases to identify novel adjuvant therapies that can improve the aortic compliance and arterial endothelial dysfunction common among those with RA, while also improving RA disease activity.
The results of this proof of concept study suggest that adding pioglitazone to RA standard of care treatment produces a small but statistically significant improvement in aortic compliance, decreases RA disease activity, and positively modulates several markers of CV risk and inflammation. While there were no improvements in conduit or microvascular endothelial‐dependent vasodilatation (ie, unchanged FMD and RHI), there was a small, but significant increase in conduit vascular smooth muscle vasodilatory function (NMD). Previous studies have indicated that RA is associated with endothelial dysfunction and increased arterial stiffness, in association with enhanced circulating levels of CRP and proinflammatory cytokines. These observations have implicated chronic inflammation as an important mediator CVD in RA.29–31 While the mechanisms for the PWV reduction in our trial remain speculative, previous meta‐analyses indicate that a 1 m/s increase in PWV corresponds to an age and risk factor adjusted 14% increase in total CVD events.32 Whether lowering PWV with treatment directly translates into event reduction has never been specifically tested; however, the PWV difference from baseline in our 2 groups was roughly one half to two thirds (0.75). We acknowledge that the clinical significance of the reduction in PWV observed in our trial remains uncertain, and while it represents a favorable modification in a surrogate marker of CVD health, other markers (eg, FMD) were not similarly improved. Therefore, the importance of this isolated reduction in PWV, albeit likely a reflection of beneficial vascular alterations, on hard clinical outcomes remains unclear. Extrapolation from the meta‐analysis results suggests it is plausible that the reduced PWV alone by pioglitazone could produce a 10% reduction in CVD (in addition to the other vascular and metabolic changes). Supporting the hemodynamic significance and health relevance of the reduction in PWV, central aortic systolic BP and pulse pressure (parameters that are also independently associated with CV risk33) were reduced by treatment. We recognize that a formal clinical outcome trial testing the efficacy of pioglitazone would be required to confirm the clinical importance of these findings.
While the implications of abnormal vascular smooth muscle function (ie, direct vasodilatory response to NTG, meaning NMD) in conduit arteries have been less commonly evaluated or discussed than those for abnormal endothelium‐dependent vasorelaxation, recent evidence in specific patient populations indicates that NMD adds important information to conventional CV risk stratification. Indeed, aberrant NMD has recently been proposed to be an independent predictor of future CV events, particularly in patients with long‐term exposure to traditional and nontraditional CV risk factors.34–36 Maruhashi et al have provided recent further evidence that NMD may be an important physiological parameter that is often overlooked as compared to FMD. They showed in 436 patients that NMD is significantly impaired among patients with underlying CV disease and is an equal predictor to the presence of atherosclerosis compared with FMD.37 The mechanisms responsible for the lack of change in FMD concomitant with the small increase in NMD in our study remain unclear. We can speculate, as outlined by Maruhashi et al, that potential causes could be a small reduction in vascular oxidative stress that yields an improvement in NMD but not FMD (as endogenous nitric oxide from endothelial cells is still overwhelmed by the remaining reactive oxygen species while the high dose of exogenous NO derived from NTG can now overcome the oxidative stress). Other possibilities include augmented function of the cCMP cascade within the smooth muscle cells, or favorable structural remodeling of the conduit vessels. The biological importance of the NMD increase also remains speculative. Nonetheless, when taken together with the reduced PWV and aortic BP values, the improved NMD suggests that vascular compliance and perhaps smooth muscle function were enhanced. We acknowledge that our study was aiming to evaluate a change in FMD, not NMD. Hence, these results may be chance findings. FMD is a more widely accepted vascular marker of CVD risk compared with NMD. Nevertheless, we consider that the studies showing a relation between reduced NMD and CVD supports the conjecture that our findings of improved NMD with pioglitazone may reflect true positive changes on vascular health. As arterial stiffness and vascular smooth muscle function (NMD) predict future CV damage, our observations support the notion that TZDs can have dual beneficial effects on RA disease activity and its associated vascular risk.32 Given the nature of our findings, we consider these hypothesis‐generating results and a future trial is merited to evaluate the impact of TZD therapy directly on smooth muscle functionality (eg, NMD) in order to corroborate these observations.
Our results are in agreement with observations that PPAR‐γ signaling pathways play critical roles in the regulation of a variety of biological processes within the CV system.38–40 Furthermore, TZDs have pleiotropic vasculoprotective roles in diabetics as well as in other patient populations, including modulation of CV risk factors, independent of glucose control. PPAR‐γ agonists also exert significant anti‐inflammatory effects in experimental arthritis, with reduction in clinical, radiographic, and histopathologic indicators.18,41 Indeed, PPAR‐γ ligands inhibit the growth of RA synoviocytes in vitro and suppress IL‐1β‐induced prostaglandin E2 synthesis in RA synovial fibroblasts.42 A recent preliminary study in a small sample of RA patients found a significant improvement in disease activity and inflammatory markers when 30 mg of pioglitazone were combined with methotrexate after 12 weeks of treatment.43 All these observations support a potential role for TZDs as adjuvant therapy in RA and its associated CV risk.
One of the reasons pioglitazone was selected as the TZD to test in this trial, was the previous evidence that it promotes a more antiatherogenic lipid profile than rosiglitazone.44 Indeed, during the pioglitazone treatment arm patients showed a significant increase in levels of HDL and significant decreases in triglycerides. These changes may further contribute to vasculoprotective effects of pioglitazone in this patient population.
It is important to notice that recent safety issues regarding the use of TZDs have been raised, due to outcomes of large placebo‐controlled and open‐label clinical trials in type 2 diabetics. Both pioglitazone and rosiglitazone have been associated with fluid retention and heart failure in diabetics, despite reduced CV morbidity and mortality.45–46 Since RA patients are at increased risk for heart failure,47 the careful selection of patients is warranted if these drugs are selected for use in this disease. Additionally, a concern for malignancy associated with these medications exists, as a dose‐effect relationship was observed between their use for ≥2 years with bladder cancer in patients with diabetes.48 Finally, future studies would need to address how TZDs affect bone health in RA patients, a group already at heightened risk for osteoporosis due to disease activity and corticosteroid use.49 PPAR agonist subtypes with selectivity for PPAR‐γ have been associated with loss of bone density and strength in animal models and in clinical trials, due to unfavorable balance between bone formation and reduction,50 while other PPAR subtypes have been associated with a protective effect on bone mineral density in animal models,51–52 However, there is recent evidence that, in animal models of arthritis, pioglitazone actually prevents inflammatory bone loss.53 As such, further investigation is required into how these drugs modulate bone biology in patients with chronic inflammatory diseases. Given these potential safety concerns, it will be important to establish whether PPAR‐γ modulators (SPPARγMs) or dual PPAR γ/α agonists may offer antiatherogenic and anti‐inflammatory effects without the associated risks of the current generation of TZDs. In recognition of the potential side effects and health risks of TZDs and the mixed benefits in vascular health observed in this trial, future studies are required to discern the risk versus benefit ratio of pioglitazone treatment in regards to overall morbidity and mortality among high‐risk RA patients.
Limitations of this study include the relatively small sample size, the short duration of the trial, and finally an unexpected finding related to blood pressure changes observed between groups: while no significant increases in blood pressure were observed with pioglitazone treatment, there was a small decrease in blood pressure in the placebo‐treated group, which may be a potential limitation of treatment. However, we do not believe it outweighs the other potential benefits of pioglitazone, as aortic SBP, PP, and PWV all improved, and DBP did not increase, but simply remained stable. The crossover study design was a strength of this study, as patients served as their own controls and we found no evidence for a carryover effect. In general, pioglitazone was well tolerated in a chronically ill population with a complex inflammatory disease.
The mechanisms involved in the improvement of vascular function and large and medium vessels and in RA disease activity by pioglitazone are likely multifactorial, and associated with modulation of inflammatory pathways, metabolic abnormalities, and function of endothelial and vascular smooth muscle cells. Recent evidence suggests that TZDs decrease vasoconstriction by inhibiting PI3K/Akt pathway.54 Further, in prediabetes, pioglitazone slows progression of carotid intima media thickness, independent of improvement in hyperglycemia, insulin resistance, dyslipidemia, and systemic, suggesting a possible direct vascular benefit.55 It is important to note that resting brachial BP determined by basal arteriole tone was not decreased by active treatment. Thus, the reduction in PWV (ie, improved aortic compliance) cannot be explained as a consequence of BP‐lowering per se or by a direct hemodynamic action.
Overall, pioglitazone therapy in this cohort of RA patients improved disease activity, as evidenced by improvement in DAS score and decreases in markers of inflammation. However, pioglitazone use was associated with some significant cardiovascular side effects, and the positive effects on macrovascular and conduit artery function were modest. While at this stage the study does not support the addition of PPAR‐γ agonists to standard of care therapy in patients with RA, it will hopefully add to the growing body of work supporting a paradigm shift toward prevention of endothelial injury that leads to atherosclerosis and CV events in RA, and in other populations of patients with inflammatory diseases.
Sources of Funding
This work was supported by the National Institutes of Health through PHS grant RO1 HL086553 (to Kaplan) and by the Michigan Diabetes Research and Training Center, University of Michigan. Marder was supported by K12HD001438 from National Institutes of Health. Additional financial support for statistical analysis was provided by NIH grant UL1TR000433. Takeda Pharmaceuticals provided pioglitazone and placebo tablets.
Disclosures
Dr Kaplan has received pioglitazone from Takeda for investigator‐initiated studies. This article was prepared while Mariana Kaplan was employed at the University of Michigan. The opinions expressed in this article are the author's own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
References
- 1.Wållberg‐Jonsson S, Johansson H, Ohman ML, Rantapää‐Dahlqvist S. Extent of inflammation predicts cardiovascular disease and overall mortality in seropositive rheumatoid arthritis. A retrospective cohort study from disease onset. J Rheumatol. 1999; 26:2562-2571 [PubMed] [Google Scholar]
- 2.Solomon DH. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003; 107:1303-1307 [DOI] [PubMed] [Google Scholar]
- 3.Goodson NJ, Wiles NJ, Lunt M, Barrett EM, Silman AJ, Symmons DPM. Mortality in early inflammatory polyarthritis: cardiovascular mortality is increased in seropositive patients. Arthritis Rheum. 2002; 46:2010-2019 [DOI] [PubMed] [Google Scholar]
- 4.Goodson N, Symmons D. Rheumatoid arthritis in women: still associated with an increased mortality. Ann Rheum Dis. 2002; 61:955-956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sattar N, McCarey DW, Capell H, McInnes IB. Explaining how “high‐grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003; 108:2957-2963 [DOI] [PubMed] [Google Scholar]
- 6.Watson DJ, Rhodes T, Guess HA. All‐cause mortality and vascular events among patients with rheumatoid arthritis, osteoarthritis, or no arthritis in the UK General Practice Research Database. J Rheumatol. 2003; 30:1196-1202 [PubMed] [Google Scholar]
- 7.del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001; 44:2737-2745 [DOI] [PubMed] [Google Scholar]
- 8.Yasojima K, Schwab C, McGeer EG, McGeer PL. Generation of C‐reactive protein and complement components in atherosclerotic plaques. Am J Pathol. 2001; 158:1039-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg JD, Kremer JM, Curtis JR, Hochberg MC, Reed G, Tsao P, Farkouh ME, Nasir A, Setoguchi S, Solomon DH. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011; 70:576-582 [DOI] [PubMed] [Google Scholar]
- 10.Choi HK, Hernán MA, Seeger JD, Robins JM, Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. Lancet. 2002; 359:1173-1177 [DOI] [PubMed] [Google Scholar]
- 11.Hurlimann D, Forster A, Noll G, Enseleit F, Chenevard R, Distler O, Béchir M, Spieker LE, Neidhart M, Michel BA, Gay RE, Lüscher TF, Gay S, Ruschitzka F. Anti‐tumor necrosis factor‐alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation. 2002; 106:2184-2187 [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez‐Juanatey C, Testa A, Garcia‐Castelo A, Garcia‐Porrua C, Llorca J, Gonzalez‐Gay MA. Active but transient improvement of endothelial function in rheumatoid arthritis patients undergoing long‐term treatment with anti‐tumor necrosis factor alpha antibody. Arthritis Rheum. 2004; 51:447-450 [DOI] [PubMed] [Google Scholar]
- 13.Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR‐gamma ligands in ischemia‐reperfusion injury, inflammation and shock. Cardiovasc Res. 2005; 65:772-781 [DOI] [PubMed] [Google Scholar]
- 14.Shiojiri T, Wada K, Nakajima A, Katayama K, Shibuya A, Kudo C, Kadowaki T, Mayumi T, Yura Y, Kamisaki Y. PPAR gamma ligands inhibit nitrotyrosine formation and inflammatory mediator expressions in adjuvant‐induced rheumatoid arthritis mice. Eur J Pharmacol. 2002; 448:231-238 [DOI] [PubMed] [Google Scholar]
- 15.Meier CA, Chicheportiche R, Juge‐Aubry CE, Dreyer MG, Dayer JM. Regulation of the interleukin‐1 receptor antagonist in THP‐1 cells by ligands of the peroxisome proliferator‐activated receptor. Cytokine. 2002; 18:320-328 [DOI] [PubMed] [Google Scholar]
- 16.Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop‐Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H. 15‐deoxy‐delta(12,14)‐PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant‐induced arthritis in rats. J Clin Investig. 2000; 106:189-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji JD, Cheon H, Jun JB, Choi SJ, Kim YR, Lee YH, Kim TH, Chae IJ, Song GG, Yoo DH, Kim SY, Sohn J. Effects of peroxisome proliferator‐activated receptor‐gamma (PPAR‐gamma) on the expression of inflammatory cytokines and apoptosis induction in rheumatoid synovial fibroblasts and monocytes. J Autoimmun. 2001; 17:215-221 [DOI] [PubMed] [Google Scholar]
- 18.Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Britti D, Patel NSA, Di Paola R, Genovese T, Di Rosa M, Caputi AP, Thiemermann C. Rosiglitazone, a ligand of the peroxisome proliferator‐activated receptor‐gamma, reduces acute pancreatitis induced by cerulein. Intensive Care Med. 2004; 30:951-956 [DOI] [PubMed] [Google Scholar]
- 19.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988; 31:315-324 [DOI] [PubMed] [Google Scholar]
- 20.Marder W, Khalatbari S, Myles JD, Hench R, Yalavarthi S, Lustig S, Brook R, Kaplan MJ. Interleukin 17 as a novel predictor of vascular function in rheumatoid arthritis. Ann Rheum Dis. 2011; 70:1550-1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brook RD, Yalavarthi S, Myles JD, Khalatbari S, Hench R, Lustig S, Marder W, Neidert A, Kaplan MJ. Determinants of vascular function in patients with chronic gout. J Clin Hypertens (Greenwich). 2011; 13:178-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopalan S, Somers EC, Brook RD, Kehrer C, Pfenninger D, Lewis E, Chakrabarti A, Richardson BC, Shelden E, McCune WJ, Kaplan MJ. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood. 2004; 103:3677-3683 [DOI] [PubMed] [Google Scholar]
- 23.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard‐Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial‐dependent flow‐mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002; 39:257-265 [DOI] [PubMed] [Google Scholar]
- 24.Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F. Inhalation of fine particulate air pollution and ozone causes acute arterial vasoconstriction in healthy adults. Circulation. 2002; 105:1534-1536 [DOI] [PubMed] [Google Scholar]
- 25.Heine RJ, Home PD, Poncher M, Orskov H, Hammond V, McCulloch AJ, Hanning I, Alberti KG. A comparison of 3 methods for assessing insulin sensitivity in subjects with normal and abnormal glucose tolerance. Diabetes Res. 1985; 2:113-120 [PubMed] [Google Scholar]
- 26.Wells G, Becker J‐C, Teng J, Dougados M, Schiff M, Smolen J, Aletaha D, van Riel PLCM. Validation of the 28‐joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C‐reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis. 2009; 68:954-960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ware JE, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Med Care. 1992; 30:473-483 [PubMed] [Google Scholar]
- 28.Fransen J, Welsing PMJ, de Keijzer RMH, van Riel P. Disease activity scores using C‐reactive protein: CRP may replace ESR in the assessment of RA disease activity. Ann Rheum Dis. 2003; 62suppl 1:15112525385 [Google Scholar]
- 29.Klocke R, Cockcroft JR, Taylor GJ, Hall IR, Blake DR. Arterial stiffness and central blood pressure, as determined by pulse wave analysis, in rheumatoid arthritis. Ann Rheum Dis. 2003; 62:414-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong M, Toh L, Wilson A, Rowley K, Karschimkus C, Prior D, Romas E, Clemens L, Dragicevic G, Harianto H, Wicks I, McColl G, Best J, Jenkins A. Reduced arterial elasticity in rheumatoid arthritis and the relationship to vascular disease risk factors and inflammation. Arthritis Rheum. 2003; 48:81-89 [DOI] [PubMed] [Google Scholar]
- 31.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all‐cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001; 37:1236-1241 [DOI] [PubMed] [Google Scholar]
- 32.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all‐cause mortality with arterial stiffness: a systematic review and meta‐analysis. J Am Coll Cardiol. 2010; 55:1318-1327 [DOI] [PubMed] [Google Scholar]
- 33.O'Rourke MF, Adji A. Noninvasive studies of central aortic pressure. Curr Hypertens Rep. 2012; 14:8-20 [DOI] [PubMed] [Google Scholar]
- 34.Kawano N, Emoto M, Mori K, Yamazaki Y, Urata H, Tsuchikura S, Motoyama K, Morioka T, Fukumoto S, Shoji T, Koyama H, Okuno Y, Nishizawa Y, Inaba M. Association of endothelial and vascular smooth muscle dysfunction with cardiovascular risk factors, vascular complications, and subclinical carotid atherosclerosis in type 2 diabetic patients. J Atheroscler Thromb. 2012; 19:276-284 [DOI] [PubMed] [Google Scholar]
- 35.Akamatsu D, Sato A, Goto H, Watanabe T, Hashimoto M, Shimizu T, Sugawara H, Sato H, Nakano Y, Miura T, Zukeran T, Serizawa F, Hamada Y, Tsuchida K, Tsuji I, Satomi S. Nitroglycerin‐mediated vasodilatation of the brachial artery may predict long‐term cardiovascular events irrespective of the presence of atherosclerotic disease. J Atheroscler Thromb. 2010; 17:1266-1274 [DOI] [PubMed] [Google Scholar]
- 36.Kullo IJ, Malik AR, Bielak LF, Sheedy PF, Turner ST, Peyser PA. Brachial artery diameter and vasodilator response to nitroglycerine, but not flow‐mediated dilatation, are associated with the presence and quantity of coronary artery calcium in asymptomatic adults. Clin Sci. 2007; 112:175-182 [DOI] [PubMed] [Google Scholar]
- 37.Maruhashi T, Soga J, Fujimura N, Idei N, Mikami S, Iwamoto Y, Kajikawa M, Matsumoto T, Hidaka T, Kihara Y, Chayama K, Noma K, Nakashima A, Goto C, Higashi Y. Nitroglycerine‐induced vasodilation for assessment of vascular function: a comparison with flow‐mediated vasodilation. Arterioscler Thromb Vasc Biol. 2013; 33:1401-1408 [DOI] [PubMed] [Google Scholar]
- 38.Bishop‐Bailey D. Peroxisome proliferator‐activated receptors in the cardiovascular system. Br J Pharmacol. 2000; 129:823-834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Law RE, Meehan WP, Xi XP, Graf K, Wuthrich DA, Coats W, Faxon D, Hsueh WA. Troglitazone inhibits vascular smooth muscle cell growth and intimal hyperplasia. J Clin Investig. 1996; 98:1897-1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marx N, Kehrle B, Kohlhammer K, Grüb M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation‐associated arteriosclerosis. Circ Res. 2002; 90:703-710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuzzocrea S, Wayman NS, Mazzon E, Dugo L, Di Paola R, Serraino I, Britti D, Chatterjee PK, Caputi AP, Thiemermann C. The cyclopentenone prostaglandin 15‐deoxy‐Delta(12,14)‐prostaglandin J(2) attenuates the development of acute and chronic inflammation. Mol Pharmacol. 2002; 61:997-1007 [DOI] [PubMed] [Google Scholar]
- 42.Tsubouchi Y, Kawahito Y, Kohno M, Inoue K, Hla T, Sano H. Feedback control of the arachidonate cascade in rheumatoid synoviocytes by 15‐deoxy‐Delta(12,14)‐prostaglandin J2. Biochem Biophys Res Commun. 2001; 283:750-755 [DOI] [PubMed] [Google Scholar]
- 43.Shahin D, Toraby EE, Abdel‐Malek H, Boshra V, Elsamanoudy AZ, Shaheen D. Effect of peroxisome proliferator‐activated receptor gamma agonist (pioglitazone) and methotrexate on disease activity in rheumatoid arthritis (experimental and clinical study). Clin Med Insights Arthritis Musculoskelet Disord. 2011; 4:1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, Tan MH, Khan MA, Perez AT, Jacober SJ. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2005; 28:1547-1554 [DOI] [PubMed] [Google Scholar]
- 45.Komajda M, McMurray JJV, Beck‐Nielsen H, Gomis R, Hanefeld M, Pocock SJ, Curtis PS, Jones NP, Home PD. Heart failure events with rosiglitazone in type 2 diabetes: data from the RECORD clinical trial. Eur Heart J. 2010; 31:824-831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dormandy JA, Charbonnel B, Eckland DJA, Erdmann E, Massi‐Benedetti M, Moules IK, Skene AM, Tan MH, Lefèbvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Korányi L, Laakso M, Mokán M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005; 366:1279-1289 [DOI] [PubMed] [Google Scholar]
- 47.Nicola PJ, Maradit‐Kremers H, Roger VL, Jacobsen SJ, Crowson CS, Ballman KV, Gabriel SE. The risk of congestive heart failure in rheumatoid arthritis: a population‐based study over 46 years. Arthritis Rheum. 2005; 52:412-420 [DOI] [PubMed] [Google Scholar]
- 48.Neumann A, Weill A, Ricordeau P, Fagot JP, Alla F, Allemand H. Pioglitazone and risk of bladder cancer among diabetic patients in France: a population‐based cohort study. Diabetologia. 2012; 55:1953-1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Staa TP, Geusens P, Bijlsma JWJ, Leufkens HGM, Cooper C. Clinical assessment of the long‐term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum. 2006; 54:3104-3112 [DOI] [PubMed] [Google Scholar]
- 50.Glintborg D, Andersen M, Hagen C, Heickendorff L, Hermann AP. Association of pioglitazone treatment with decreased bone mineral density in obese premenopausal patients with polycystic ovary syndrome: a randomized, placebo‐controlled trial. J Clin Endocrinol Metab. 2008; 93:1696-1701 [DOI] [PubMed] [Google Scholar]
- 51.Syversen U, Stunes AK, Gustafsson BI, Obrant KJ, Nordsletten L, Berge R, Thommesen L, Reseland JE. Different skeletal effects of the peroxisome proliferator activated receptor (PPAR)alpha agonist fenofibrate and the PPARgamma agonist pioglitazone. BMC Endocr Disord. 2009; 9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Still K, Grabowski P, Mackie I, Perry M, Bishop N. The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif Tissue Int. 2008; 83:285-292 [DOI] [PubMed] [Google Scholar]
- 53.Koufany M, Moulin D, Bianchi A, Muresan M, Sebillaud S, Netter P, Weryha G, Jouzeau J‐Y. Anti‐inflammatory effect of antidiabetic thiazolidinediones prevents bone resorption rather than cartilage changes in experimental polyarthritis. Arthritis Res Ther. 2008; 10:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinagra T, Tamburella A, Urso V, Siarkos I, Drago F, Bucolo C, Salomone S. Reversible inhibition of vasoconstriction by thiazolidinediones related to PI3K/Akt inhibition in vascular smooth muscle cells. Biochem Pharmacol. 2013; 85:551-559 [DOI] [PubMed] [Google Scholar]
- 55.Saremi A, Schwenke DC, Buchanan TA, Hodis HN, Mack WJ, Banerji M, Bray GA, Clement SC, Henry RR, Kitabchi AE, Mudaliar S, Ratner RE, Stentz FB, Musi N, Tripathy D, DeFronzo RA, Reaven PD. Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2013; 33:393-399 [DOI] [PMC free article] [PubMed] [Google Scholar]