

Abstract
Background
Recent evidence has depicted a role of macrophage migration inhibitory factor (MIF) in cardiac homeostasis under pathological conditions. This study was designed to evaluate the role of MIF in doxorubicin‐induced cardiomyopathy and the underlying mechanism involved with a focus on autophagy.
Methods and Results
Wild‐type (WT) and MIF knockout (MIF−/−) mice were given saline or doxorubicin (20 mg/kg cumulative, i.p.). A cohort of WT and MIF−/− mice was given rapamycin (6 mg/kg, i.p.) with or without bafilomycin A1 (BafA1, 3 μmol/kg per day, i.p.) for 1 week prior to doxorubicin challenge. To consolidate a role for MIF in the maintenance of cardiac homeostasis following doxorubicin challenge, recombinant mouse MIF (rmMIF) was given to MIF−/− mice challenged with or without doxorubicin. Echocardiographic, cardiomyocyte function, and intracellular Ca2+ handling were evaluated. Autophagy and apoptosis were examined. Mitochondrial morphology and function were examined using transmission electron microscopy, JC‐1 staining, MitoSOX Red fluorescence, and mitochondrial respiration complex assay. DHE staining was used to evaluate reactive oxygen species (ROS) generation. MIF knockout exacerbated doxorubicin‐induced mortality and cardiomyopathy (compromised fractional shortening, cardiomyocyte and mitochondrial function, apoptosis, and ROS generation). These detrimental effects of doxorubicin were accompanied by defective autophagolysosome formation, the effect of which was exacerbated by MIF knockout. Rapamycin pretreatment rescued doxorubicin‐induced cardiomyopathy in WT and MIF−/− mice. Blocking autophagolysosome formation using BafA1 negated the cardioprotective effect of rapamycin and rmMIF.
Conclusions
Our data suggest that MIF serves as an indispensable cardioprotective factor against doxorubicin‐induced cardiomyopathy with an underlying mechanism through facilitating autophagolysosome formation.
Keywords: autophagolysosome, doxorubicin, heart failure, MIF, rapamycin
Introduction
Doxorubicin has been used extensively as a potent anticancer chemotherapeutic agent since the late 1960s.1 Nonetheless, accumulating studies have depicted that doxorubicin directly triggers cardiotoxicity, thus limiting its clinical application.2 Chronic use of doxorubicin has been shown to prompt cardiotoxicity and congestive heart failure in a dose‐dependent manner.2–4 Although ample studies have been seen with regard to doxorubicin‐induced cardiomyopathy, the precise mechanisms of action behind doxorubicin toxicity still remain elusive.4 A number of signaling molecules have been identified for doxorubicin‐induced cardiomyopathy and resulted cell death.1,3–4 Among the signaling molecules mentioned, oxidative stress derived from subcellular sources, including mitochondria, NOS, NADPH, and ion complexes, appears to play an essential role in doxorubicin‐induced cardiac remodeling and contractile defects.5–9 At the same time, experimental studies have demonstrated a pivotal role for apoptosis and necrosis in doxorubicin‐induced cardiomyocyte death.4
Macrophage migration inhibitory factor (MIF) was initially identified as a proinflammatory cytokine expressed ubiquitously.10 Recent studies also indicated that MIF may be secreted by cardiomyocytes.11 More intriguingly, various studies have demonstrated that MIF is involved in the regulation of cardiac function under different pathological conditions including burn injury,12 diabetes mellitus,13 and ischemia‐reperfusion injury.11,14–15 The cardioprotective effect of MIF is believed to be mainly dependent on the activation of AMPK and inhibition of JNK under ischemia reperfusion injury.11,14–15 However, whether and how MIF is involved in doxorubicin‐induced cardiomyopathy is still unknown.
Autophagy is an evolutionarily conserved pathway responsible for bulk degradation of intracellular components.16 It is accepted that basal autophagy may be cardioprotective and serve as an indispensable factor in maintaining cardiac geometry and function.17–18 Although ample studies have indicated increased cardiac autophagy in response to various stress‐inducers, it is still controversial whether autophagy induction is adaptive or maladaptive.19–22 While certain studies suggest that autophagy induction may be detrimental to pressure overload‐induced cardiac hypertrophy and heart failure,20,22 others indicate that autophagy induction may be cardioprotective in pressure overload‐induced cardiac hypertrophy in experimental and clinical settings of heart failure.18,21,23 Although the role of autophagy in the maintenance of cardiac geometry and function is extensively studied, its role in doxorubicin‐induced cardiomyopathy remains unclear. Recent in vitro studies suggested that autophagy activation is detrimental for cardiomyocyte survival24–25 although the role of autophagy may be different in the in vivo model of doxorubicin‐induced cardiomyopathy.26–27 To this end, this study was designed to examine the role of MIF in the etiology of doxorubicin‐induced cardiomyopathy, and the underlying mechanisms involved with a special focus on autophagy.
Methods
Experimental Animals
All animal procedures performed in this study were approved by the Animal Care and Use Committee at the University of Wyoming (Laramie, WY) and was in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996). In brief, 4‐month‐old adult male Wild‐type (WT) and MIF−/− mice, both with the C57BL/6 background were given doxorubicin (10 mg/kg, i.p., twice at 3‐day intervals, 20 mg/kg cumulative, Sigma, D‐1515) or the vehicle saline.3,28 A cohort of WT and MIF−/− mice was given rapamycin (6 mg/kg, i.p., at 2‐day intervals, 3 injections) for 1 week prior to doxorubicin challenge (Figure 1).29 Mice were housed in a climate‐controlled environment (22.8±2.0°C, 45% to 50% humidity) with a 12/12–light/dark cycle with access to food and water ad libitum until experimentation.
Figure 1.
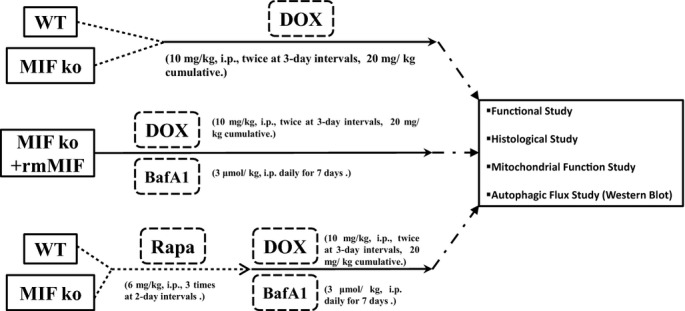
Study flowchart explaining the various groups of mice and treatments. BafA1 indicates bafilomycin A1; DOX, doxorubicin; i.p., intraperitoneal; ko, knockout; MIF, migration inhibitory factor; Rapa, rapamycin; rmMIF, recombinant mouse macrophage migration inhibitory factor; WT, wild type.
Echocardiographic Assessment
Cardiac geometry and function were evaluated in anesthetized (ketamine 80 mg/kg and xylazine 12 mg/kg, i.p.) mice (8 to 9 per group) using the 2‐dimensional guided M‐mode echocardiography (Philips SONOS 5500, Phillips Medical Systems) equipped with a 15 to 6 MHz linear transducer (Phillips Medical Systems). The chests were shaved and mice were placed in a shallow left lateral position on a heating pad. Using the 2‐dimensional (2D) parasternal short‐axis image obtained at a level close to papillary muscles as a guide, a 2D‐guided M‐mode trace crossing the anterior and posterior wall of the LV was obtained at a sweep speed of 50 mm/s. The echocardiographer was blind to treatment. Caution was taken to avoid excessive pressure over the chest, which may induce bradycardia and deformation of the heart. Left ventricular (LV) anterior and posterior wall dimensions during diastole and systole were recorded from 3 consecutive cycles in M‐mode using a method adopted by the American Society of Echocardiography. Fractional shortening was calculated from LV end‐diastolic (EDD) and end‐systolic (ESD) diameters using the equation (EDD−ESD)/EDD×100. The estimated echocardiographically derived LV mass was calculated as ([LVEDD+septal wall thickness+posterior wall thickness]3−LVEDD3)×1.055, where 1.055 (mg/mm3) denotes the density of myocardium. Heart rates were averaged over 10 consecutive cycles.30
Isolation of Murine Cardiomyocytes
Hearts were rapidly removed from anesthetized mice and mounted onto a temperature‐controlled (37°C) Langendorff system. After perfusion with a modified Tyrode's solution (Ca2+ free) for 2 minutes, the heart was digested with a Ca2+‐free KHB buffer containing liberase blendzyme 4 (Hoffmann‐La Roche Inc) for 20 minutes. The modified Tyrode solution (pH 7.4) contained the following (in mmol/L): NaCl 135, KCl 4.0, MgCl2 1.0, HEPES 10, NaH2PO4 0.33, glucose 10, butanedione monoxime 10, and the solution was gassed with 5% CO2–95% O2. The digested heart then was removed from the cannula and the left ventricle was cut into small pieces in the modified Tyrode's solution. Tissue pieces were gently agitated and a pellet of cells was resuspended. Extracellular Ca2+ was added incrementally back to 1.20 mmol/L over 30 minutes. A yield of at least 60% to 70% viable rod‐shaped cardiomyocytes with clear sarcomere striations was achieved. Only rod‐shaped myocytes with clear edges were selected for contractile and intracellular Ca2+ studies.
Cell Shortening/Relengthening
Mechanical properties of cardiomyocytes (100 to 130 cells from 5 mice per group) were assessed using a SoftEdge MyoCam system (IonOptix Corporation). IonOptix SoftEdge software was used to capture changes in cardiomyocyte length during shortening and relengthening. In brief, cardiomyocytes were placed in a Warner chamber mounted on the stage of an inverted microscope (Olympus, IX‐70) and superfused (≈1 mL/minutes at 25°C) with a buffer containing (in mmol/L): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, at pH 7.4. Cells were field stimulated with supra‐threshold voltage at a frequency of 0.5 Hz, 3 ms duration, using a pair of platinum wires placed on opposite sides of the chamber connected to a FHC stimulator (Brunswick, NE). The myocyte being studied was displayed on the computer monitor using an IonOptix MyoCam camera. IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS)—the amplitude myocytes shortened on electrical stimulation, which is indicative of peak ventricular contractility; time‐to‐PS (TPS)—the duration of myocyte shortening, which is indicative of contraction duration; time‐to‐90% re‐lengthening (TR90)—the duration to reach 90% relengthening, which represents cardiomyocyte relaxation duration (90% rather 100% re‐lengthening was used to avoid signal noise at baseline concentration); and maximal velocities of shortening (+dL/dt) and relengthening (−dL/dt)—maximal slope (derivative) of shortening and relengthening phases, which are indications of maximal velocities of ventricular pressure rise/fall.
Intracellular Ca2+ Transient Measurement
Myocytes (100 to 130 cells from 5 mice per group) were loaded with fura‐2/AM (0.5 μmol/L) for 10 minutes and fluorescence measurements were recorded with a dual‐excitation fluorescence photomultiplier system (IonOptix). Cardiomyocytes were placed on an Olympus IX‐70 inverted microscope and imaged through a Fluor ×40 oil objective. Cells were exposed to light emitted by a 75 W lamp and passed through either a 360 or a 380 nm filter, while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after first illuminating the cells at 360 nm for 0.5 seconds then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360 nm excitation scan was repeated at the end of the protocol and qualitative changes in intracellular Ca2+ concentration were inferred from the ratio of fura‐2 fluorescence intensity (FFI) at 2 wavelengths (360/380). Fluorescence decay time was measured as an indication of the intracellular Ca2+ clearing rate.
Western Blot Analysis
Murine (5 to 6 mice per group) hearts were flash‐frozen in liquid nitrogen and stored at −80°C before protein extraction. For protein extraction, heart tissues were homogenized and sonicated in RIPA buffer containing 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 0.1% sodium dodecyl sulfate (SDS), and a protease inhibitor cocktail (Roche Diagnostics). Heart homogenates containing equal amount of proteins were resolved by SDS‐polyacrylamide gels in a mini‐gel apparatus (Mini‐PROTEAN II, Bio‐Rad) and proteins were transferred to nitrocellulose membranes, incubated overnight with primary antibodies at 4°C. After washing 3 times, the membranes were incubated with horseradish peroxidase (HRP)‐coupled secondary antibody for 1 hr at room temperature. The membranes were washed again 3 times for 10 minutes each time, and the signals were quantified with a Bio‐Rad calibrated densitometer. The densitometric intensity of immunoblot bands was normalized to that of GAPDH. For reprobing, membranes were stripped with 50 mmol/L Tris‐HCl, 2% SDS, and 0.1 mol/L β‐mercaptoethanol (pH 6.8). Polyclonal rabbit antibodies against MIF (Santa Cruz, sc‐20121), phosphorylated AMPK (pAMPKα) at Thr172 (Cell Signaling, 2535S), total AMPKα (Cell Signaling, 2532S), Bip (Cell Signaling, 3177S), LC3B (Cell Signaling, 3868S), GAPDH (Cell Signaling, 2118L), p62 (Guinea Pig; Enzo Life Sciences, GP62‐C), and ubiquitin (Abcam, ab7780) were examined by standard Western immunoblotting.
TUNEL Staining
Mouse (9 mice per group) hearts were frozen immediately after euthanasia, and 7‐μm thickness sections were obtained using a Leica, cryomicrotome (Model CM3050S, Leica Microsystems). Sections were stained with in situ terminal dUTP nick end‐labeling (TUNEL) staining kit (Roche Diagnostics Corporation, 11684795910) to detect apoptotic cells according to the manufacturer's instructions.31 To confirm apoptosis for both cardiomyocytes and noncardiomyocytes in the heart, we performed triple immunofluorescence for DESMIN, TUNEL, and DAPI for nuclei.32 Tissue sections were first stained with DESMIN (Cell Signaling, D93F5, 5332) followed by Alexa Fluor 568 (Invitrogen, A‐11011), then stained with TUNEL staining kit (Roche Diagnostics Corporation, 11684795910). Nuclei were stained with DAPI. An Image J software was employed for quantitative analysis. False‐positive TUNEL spots were ascertained by nuclear counterstaining with DAPI and enumeration by Image J analysis.
Immunohistochemical Staining of α‐Smooth Muscle Actin
Frozen left ventricle was sectioned at 7‐μm thickness by a Leica, cryomicrotome (Leica Microsystems) prior to fixation in 4% paraformaldehyde for 10 minutes. Following 3 washes using distilled water, slides were incubated with the anti‐α‐SMA antibody (abcam, ab5694) overnight at room temperature. They were then stained with Alexa Fluor 568 (Invitrogen, A‐11011). Slides were mounted with aqueous mounting media, coverslipped, and visualized using an Olympus BX‐51 light microscope (Olympus America Inc).
H&E Staining
Following anesthesia, hearts were arrested in diastole with saturated KCl, excised and fixed in 10% neutral‐buffered formalin at room temperature for 24 hours. The specimen was processed through graded alcohols, cleared in xylenes, embedded in paraffin, serial sections were cut at 5‐μm and stained H&E, dehydrated, and mounted. Myocardial inflammation was examined under a light microscope (Olympus America Inc).33
MIF Reconstitution
For MIF reconstitution, MIF−/− mice were given recombinant mouse MIF (rmMIF, 2 injections of 10 μg of lipopolysaccharide [LPS]‐free rmMIF at 24‐hour intervals, i.p.). Prior to doxorubicin administration, MIF−/− mice were given the first injection of rmMIF (i.p., 10 μg). To maintain MIF level in vivo, MIF−/− mice were given rmMIF (i.p., 10 μg) daily prior to experimentation (Figure 1).34
Mitochondrial Electron Transport Chain Enzymatic Activity (Cytochrome C Oxidase [CCO], Succinate/Cytochrome C Reductase, and Reduced Nicotinamide Adenine Dinucleotide [NADH]/Succinate Cytochrome C Reductase)
Mitochondrial electron transport chain enzymatic activity was evaluated as previously described.35 Mitochondria were isolated from left ventricles (5 to 6 mice per group) for analysis of mitochondrial respiration. CCO activity was detected by monitoring the change in absorbance at 550 nm (∆A550) resulting from the oxidation of reduced cytochrome c. The CCO activity was monitored until ∆A550 was ≈0.6 U, at which point potassium cyanide (0.17 mmol/L) was added to terminate the CCO reaction. Succinate (12.5 mmol/L) was added to detect succinate: cytochrome c reductase activity by monitoring ∆A550. Next, malonate (17 mmol/L) was added to terminate succinate:cytochrome c reductase. NADH (2.8 nmol/L) was added, and NADH:cytochrome c reductase activity was measured by monitoring ∆A550. The rate of cytochrome c oxidation orreduction (nmole per minute) was calculated using a molar extinction coefficient of 19 600 for cytochrome c.35
Transmission Electron Microscopy
Small cubic pieces ≤1 mm3 were dissected from the left ventricle and fixed with 2.5% glutaraldehyde in 0.1 mol/L sodium phosphate (pH 7.4) overnight at 4°C. After postfixation in 1% OsO4, samples were dehydrated through graded alcohols and embedded in Epon Araldite. Ultrathin sections (50 nm) were cut using an ultramicrotome (Ultracut E, Leica), and stained with uranyl acetate and lead citrate. The specimens were viewed on a Hitachi H‐7000 Electron Microscope (Pleasanton, CA). Images were captured with a Gatan high resolution 4 k×4 k digital camera and Gatan Ditital Micrograph software. In 5 randomly picked regions of each sample, the area of mitochondria was calculated (≈50 mitochondria per group).36–37
Measurement of Mitochondrial Membrane Potential (∆Ψm)
Mitochondrial membrane potential was evaluated using JC‐1 (Invitrogen, T‐3168) as described.38–39 Cardiomyocytes (≈50 cells from 3 mice per group) isolated from WT and MIF−/− mice following doxorubicin or saline treatment were seeded on gelatin‐coated culture chamber slides and stained with JC‐1 (5 μmol/L) at 37°C for 10 minutes. Cells were rinsed with the HEPES‐saline buffer. Fluorescence of each sample was read at excitation wavelength of 490 nm and emission wavelength of 530 and 590 nm using a spectrofluorimeter (Spectra MaxGeminiXS, Spectra Max, Atlanta, GA). In healthy cells, a high concentration of JC‐1 forms aggregates that yield red fluorescence at ≈590 nm. In unhealthy cells, JC‐1 exists as a monomer at low concentration emitting red fluorescence at ≈530 nm. Results in fluorescence intensity were expressed as the ratio of 590‐ to 530‐nm emission.
Detection of Mitochondrial ROS Using MitoSOX Red Fluorescence
Mitochondrial‐derived ROS (O2− in particular) was measured by incubating cardiomyocytes with MitoSOX as previously described.40 Briefly, cardiomyocytes (50 fields per group) isolated from WT and MIF−/− mice with or without doxorubicin treatment were incubated with MitoSOX Red (2 μmol/L, Molecular Probes) at 37°C for 10 minutes. Following incubation for 1 hour at 37°C, cardiomyocytes were rinsed with the perfusion buffer and MitoSOX Red fluorescence intensity was measured at 510/ 580 nm using an Olympus BX51 microscope equipped with a digital cooled charged‐coupled device camera. InSpeck microspheres (Molecular Probes) were used to calibrate MitoSOX Red fluorescence by calculating the ratio of myocyte fluorescent intensities to the fluorescent beads.40
Detection of Reactive Oxygen Species Production
To evaluate tissue production of ROS (O2− in particular), fresh, frozen left ventricular myocardium (7‐μm sections) was incubated with dihydroethidium (2 mmol/L; Molecular Probes, D‐1168) for 1 hour at room temperature, as previously described.41 Following 2 washes by phosphate buffered saline (PBS), the sections were stained with DAPI (1 μg/mL, Sigma, D9564) for 5 minutes at room temperature. Slides were rinsed 3 times with PBS, mounted with prolong gold antifade mounting reagent and coverslipped. Tissue slides were examined using an Olympus BX‐51 epifluorescence microscope. The numbers of DHE‐positive nuclei and the total nuclei were counted (50 fields per group), as described previously.39
Blood Pressure Measurement
Systolic and diastolic blood pressure was examined in conscious mice (8 mice per group) using a CODA semi‐automated non‐invasive blood pressure device (Kent Scientific Co).42
Administration of Rapamycin
Rapamycin was dissolved as previously described.43 One week prior to doxorubicin injection, rapamycin (6 mg/kg, i.p., LC Laboratories, R‐5000) was administered to WT and MIF−/− AAC or saline‐treated mice for 3 times at 2‐day intervals (Figure 1).29
Assessment of Autophagic Flux
To assess the role of autophagy flux in rapamycin‐induced myocardial responses, WT and MIF−/− mice (5 to 6 mice per group) were treated with the lysosomal inhibitor bafilomycin A1 (BafA1, 3 μmol/kg, i.p.) daily for 7 days immediately following the initiation of rapamycin administration.44 To evaluate the role of autophagy flux in rmMIF‐induced beneficial myocardial response, if any, MIF−/− mice were treated Baf A1 (3 μmol/kg, i.p.) daily for 7 days immediately following the injection of rmMIF (Figure 1).
Data Analysis
D'Agostino‐Pearson omnibus test was used to determine the normality of data. Data were expressed as Mean±SEM. Statistical significance (P<0.05) was estimated by one‐way analysis of variation (ANOVA) followed by a Tukey's test for post hoc analysis. Two‐way ANOVA was employed to discern potential interactions among doxorubicin, rapamycin, bafilomycin A1, rmMIF treatment and MIF genotype in Figures 8,9A through 9F, and 10. The log rank test was used for Kaplan‐Meier survival comparison. All statistics were performed with GraphPad Prism 4.0 software (GraphPad).
Figure 8.
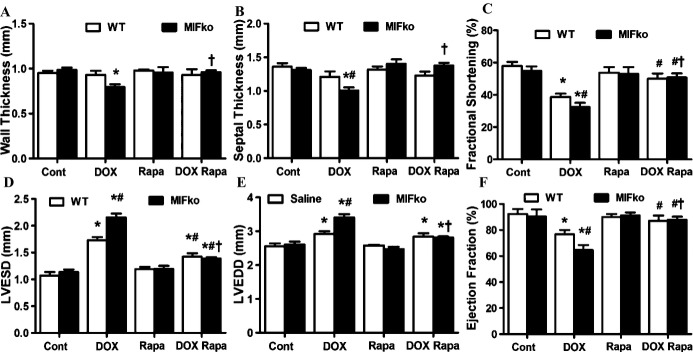
Echocardiographic parameters in WT and MIF−/− mice treated with rapamycin for 7 days prior to doxorubicin (DOX) challenge. A, LV wall thickness; B, septal thickness; C, fractional shortening (FS, %); D, LVEDD; E, LVESD; F, ejection fraction (EF, %). Mean±SEM, n=8 to 9 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group, †P<0.05 vs MIF knockout DOX group. ko indicates knockout; LV, left ventricle; LVEDD, LV end diastolic diameter; LVESD, LV end systolic diameter; MIF, macrophage migration inhibitory factor; Rapa, rapamycin; WT, wild type.
Figure 9.

A through F, Cardiomyocyte contractile properties of WT and MIF−/− mice treated with rmMIF reconstitution or rapamycin in the presence or absence of the lysosomal inhibitor bafilomycin A1 (BafA1, 3 μmol/kg per day, i.p.) for 7 days immediately following initiation of rmMIF or rapamycin treatment. A, Resting cell length; B, peak shortening (PS, normalized to resting cell length); C, +dL/dt; D, −dL/dt; E, TPS; F, TR90. Mean±SEM, n=100 to 130 cells from 5 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group, †P<0.05 vs MIFko DOX group. G through K, Effect of rmMIF reconstitution on myocardial autophagy flux in MIF−/− mice treated with or without DOX. G, Representative gel blots depicting protein levels of LC3BI/II, p62 and GAPDH (loading control) using specific antibodies; H, LC3B I expression; I, LC3B II expression; J, LC3B II‐to‐LC3B I ratio; K, p62 expression. Mean±SEM, n=5 to 6 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group. BafA1 indiactes bafilomycin A1; +dL/dt, maximal velocity of shortening; −dL/dt, maximal velocity of relengthening; DOX, doxorubicin; i.p., intraperitoneal; ko, knockout; MIF, macrophage migration inhibitory factor; Rapa, rapamycin; rmMIF, recombinant mouse macrophage migration inhibitory factor; TPS, time‐to‐PS; TR90, time‐to‐90% relengthening; WT, wild type.
Figure 10.

ROS production in hearts of WT and MIF−/− mice treated with saline or doxorubicin (DOX) in the absence or presence of rapamycin pretreatment. Frozen myocardial tissue sections were stained with DHE (red), and nucleus with DAPI (blue). Data were from 3 independent experiments each with 3 mice per group (for a total of 9 mice per group). The percentage of DHE‐positive nuclei was quantified (≈50 fields from 3 mice per group, scale bar=50 μm). Mean±SEM, n=50 fields per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group, †P<0.05 vs MIF knockout DOX group. DOX indicates doxorubicin; ko, knockout; MIF, macrophage migration inhibitory factor; Rapa, rapamycin; WT, wild type.
Results
MIF Deficiency Accentuates Doxorubicin‐induced Cardiac Remodeling
MIF is known to be expressed constitutively in murine cardiomyocytes and released in response to ischemic injury.11 Seven days following initial injection of doxorubicin, myocardial MIF expression was significantly upregulated (Figure 2A and 2B). There was no mortality in either WT or MIF knockout saline groups (n=12). With doxorubicin challenge, MIF−/− mice displayed a significantly greater mortality (53.8%, P<0.05 versus WT group, n=13 mice) compared with WT mice (16.7%, n=12) (Figure 2C). Little phenotypical changes were observed in MIF−/− mice, as evidenced by physiological parameters such as heart rate and blood pressure (Table 1). Echocardiography was performed to examine doxorubicin‐induced myocardial remodeling in WT and MIF−/− mice. Neither saline nor MIF deletion significantly affected cardiac geometry. Doxorubicin treatment significantly increased LVEDD and LVESD, as well as decreased fractional shortening and ejection fraction without affecting LV wall and septal thickness in WT mice (P<0.05 versus WT mice in the absence of doxorubicin). Interestingly, doxorubicin‐induced cardiac remodeling was exacerbated by MIF knockout as evidenced by more pronounced changes in wall thickness, septal thickness, LVEDD, LVESD, fractional shortening, and ejection fractioning (Figure 2D through 2I). In addition, doxorubicin significantly increased the lung/body weight ratio, with a more pronounced effect in MIF−/− mice (Table 1). These results suggest that MIF knockout may exacerbate doxorubicin‐induced cardiac dysfunction, leading to the development of heart failure and increased mortality.
Figure 2.

A, Representative gel blots depicting protein levels of MIF and GAPDH (loading control) using specific antibodies. B, Quantitative analysis of MIF expression (normalized to GAPDH). C, Kaplan‐Meier survival curves of WT and MIF−/− mice after doxorubicin (DOX) injection (P<0.05 using the Log‐rank test). D, Wall thickness; E, septal thickness; F, fractional shortening (FS); G, LV end diastolic diameter (LVEDD); H, LV end systolic diameter (LVESD); I, ejection fraction (EF); J and K, immunohistochemical staining of α‐SMA in WT and doxorubicin‐treated WT hearts; L and M, H&E staining in WT and doxorubicin‐treated WT hearts. Mean±SEM, n=8 to 9 mice per group, *P<0.05 vs WT, #P<0.05 vs WT DOX group. ko indicates knockout; MIF, macrophage migration inhibitory factor; α‐SMA, α‐smooth muscle actin; WT, wild type.
Table 1.
Hemodynamics of WT and MIF−/− Mice Treated With or Without DOX
| WT | WT DOX | MIFko | MIFko DOX | |
|---|---|---|---|---|
| Lung weight/body weight, mg/g | 5.71±0.26 | 7.63±0.23* | 5.36±0.29 | 9.54±0.44*† |
| Heart rate, bpm | 511.3±6.0 | 506.6±6.5 | ||
| Diastolic blood pressure, mm Hg | 87.4±3.8 | 87.4±3.4 | ||
| Systolic blood pressure, mm Hg | 121.9±4.4 | 115.4±3.2 | ||
| Mean blood pressure, mm Hg | 98.7±3.9 | 96.0±3.3 |
Mean±SEM, n=8 mice per group. DOX indicates doxorubicin; ko, knockout; MIF, migration inhibitory factor; WT, wild type.
*P<0.05, vs the WT group; †P<0.05, vs the WT DOX group.
We also examined the possibility that additional cell populations may contribute to increased myocardial MIF protein. α‐Smooth muscle actin (α‐SMA), primarily expressed in smooth muscle cells, is a widely used indicator for myofibroblast.45 In WT mice, vascular smooth muscle cells were positively stained with α‐SMA while few myofibroblasts were found in myocardial interstitial tissues (Figure 2J). Treatment with doxorubicin did not significantly alter the number of myofibroblasts (Figure 2K). In addition, H&E staining showed that doxorubicin did not overtly increase the number of inflammatory cells in myocardium (Figure 2L and 2M).33 Taken together, these data suggest that the cardiac cellular source of MIF after doxorubicin treatment is predominantly from cardiomyocytes.
MIF Deficiency Augments Doxorubicin‐induced Cardiomyocyte Dysfunction
Consistent with echocardiographic findings, 7 days following the initial doxorubicin challenge, cardiomyocyte contractile function was suppressed in WT mice, as evidenced by decreased peaking shortening (PS) and maximal velocity of shortening/relengthening (±dL/dt), increased TR90 associated with unchanged TPS. Neither saline treatment nor MIF knockout affected cardiomyocyte contractile properties. However, MIF knockout overtly accentuated doxorubicin‐induced cardiomyocyte contractile dysfunction (manifested by further depressed PS and ±dL/dt) (Figure 3A through 3F). Reconstitution of MIF−/− mice with rmMIF partially but significantly rescued doxorubicin‐induced cardiomyocyte contractile anomalies in MIF−/− mice, as evidenced by normalized PS, ±dL/dt and TR90. Cardiomyocyte contractile function in control MIF−/− mice was not affected by rmMIF (Figure 9A through 9F).
Figure 3.

Cardiomyocyte contractile and intracellular Ca2+ handling properties in WT and MIF−/− mice 7 days after doxorubicin (DOX) injection. A, Resting cell length; B, peak shortening (PS, normalized to resting cell length); C, maximal velocity of shortening (+dL/dt); D, maximal velocity of relengthening (−dL/dt); E, time‐to‐PS (TPS); F, time‐to‐90% relengthening (TR90); G, resting fura‐2 fluorescence intensity (FFI); H, electrically stimulated rise in FFI (∆FFI); I, single exponential intracellular Ca2+ decay rate. Mean±SEM, n=100 to 130 cells from 5 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group. ko indicates knockout; MIF, macrophage migration inhibitory factor; WT, wild type.
To further uncover the potential mechanisms behind doxorubicin‐induced cardiomyocyte contractile dysfunction in WT and MIF−/− mice, intracellular Ca2+ handling was monitored in cardiomyocytes from WT and MIF−/− mice with or without doxorubicin treatment. Our data showed that doxorubicin promoted intracellular Ca2+ disturbance in cardiomyocytes as evidenced by a reduced electrical stimulus‐induced rise in intracellular Ca2+ and prolonged intracellular Ca2+ clearance associated with unchanged baseline intracellular Ca2+ levels. Although MIF deficiency did not elicit any notable effect on intracellular Ca2+ properties, it exacerbated doxorubicin‐induced intracellular Ca2+ mishandling (Figure 3G through 3I).
MIF Deficiency Exacerbates Doxorubicin‐induced Cardiomyocyte Apoptosis
Given evidence that apoptosis plays an important role in doxorubicin‐induced cardiomyopathy,4 apoptosis was examined in cardiomyocytes from WT and MIF−/− mice using confocal microscopy. One week after the initial challenge, doxorubicin significantly increased apoptosis in myocardial tissues, reminiscent to previous reports.7 Our triple immunofluorescence staining confirmed that doxorubicin induced apoptosis in cardiomyocytes. More importantly, MIF deficiency exacerbated doxorubicin‐induced cardiomyocyte apoptosis (Figure 4A through 4P and 4Y). In MIF−/− mice, rmMIF did not significantly affect cardiomyocyte viability. However, rmMIF subtly although significantly attenuated doxorubicin‐induced cardiomyocyte cell death (Figure 4Q through 4Y).
Figure 4.

Confocal microscopic images depicting MIF deficiency‐induced accentuation of doxorubicin (DOX)‐induced cardiomyocyte apoptosis. A through X, Frozen myocardial sections from WT and MIF−/− mice treated with either saline or DOX were stained with desmin (red), TUNEL (green), and nucleus with DAPI (blue). Data were from 3 independent experiments each with 3 mice per group (for a total of 9 mice per group). Arrows denote cardiomyocyte apoptosis (the co‐localization of desmin, TUNEL and DAPI staining); Y, Quantitative analysis of apoptosis using TUNEL staining 7 days after DOX injection (≈50 fields from 3 mice per group). Mean±SEM, n=3 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group, †P<0.05 vs MIFko DOX group. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; ko, knockout; MIF, macrophage migration inhibitory factor; rmMIF, recombinant mouse macrophage migration inhibitory factor; TUNEL, terminal dUTP nick end‐labeling; WT, wild type.
MIF Deficiency Augments Doxorubicin‐induced Mitochondrial Dysfunction
Given that mitochondria play a key role in apoptosis induction and that doxorubicin may directly trigger cardiac mitochondrionopathy,4,46 mitochondrial function was evaluated in cardiomyocytes from WT and MIF−/− mice treated with or without doxorubicin. Neither saline nor MIF deficiency affected mitochondrial morphology as evidenced by organized cristae structure and intact morphology (Figure 5A and 5C). As expected, doxorubicin elicited overt cardiac ultrastructural changes characterized by mitochondrial swelling, disorganization of cristae and loss of sarcomere integrity (Figure 5B). MIF deficiency exacerbated doxorubicin‐induced mitochondrial morphological changes (Figure 5D). Mitochondria from MIF−/− mice treated with doxorubicin showed more pronounced swelling and fragmentation of the cristae (Figure 5D). Quantification showed that doxorubicin treatment significantly increased mitochondrial area, the effect of which was exacerbated in MIF−/− mice (Figure 5E). Next, JC‐1 was employed to monitor the mitochondrial membrane potential in cardiomyocytes.38–39 Our results showed a significant reduction in the red, green fluorescence ratio in cardiomyocytes from doxorubicin‐treated WT mice, the effect of which was exacerbated by MIF knockout (Figure 5F through 5I and 5L). Neither saline nor MIF deficiency exerted any significant effect on ∆Ψm (Figure 5F, 5I, and 5L). Although rmMIF did not overtly influence ∆Ψm in cardiomyocytes isolated from untreated MIF−/− mice, it significantly ameliorated doxorubicin‐induced ∆Ψm loss in MIF−/− mice (Figure 5J through 5L). Furthermore, our data demonstrated that MIF knockout significantly exacerbated doxorubicin‐induced changes in mitochondrial respiration (Figure 5M through 5O). These data indicate that doxorubicin treatment dampened mitochondrial function and respiration, the effect of which was exacerbated by deletion of MIF.
Figure 5.

Mitochondrial morphology, function, and respiration in hearts from WT and MIF−/− mice treated with saline or doxorubicin (DOX). Representative transmission electron microscopy (TEM) image of left ventricular tissues (A through D, scale bar=500 nm); E, quantitative analysis of the percentage of mitochondrial area (≈50 mitochondria per group); representative images of JC‐1 staining of cardiomyocytes (F through K, scale bar=30 μm); L, quantitative analysis of the red/green fluorescence ratio (≈50 cardiomyocytes from 3 mice per group); mitochondrial respiration (M, CCO activity/complex IV; N, complex I/III; O, complex II/III). M1: Healthy mitochondria; M3: degenerating mitochondria; Sar: sarcomere; arrows denote double membrane vacuoles. Mean±SEM, n=50 cardiomyocytes per group, *P<0.05 vs saline group, #P<0.05 vs WT DOX group, †P<0.05 vs MIFko DOX group. CCO indicates cytochrome c oxidase; ko, knockout; MIF, macrophage migration inhibitory factor; rmMIF, recombinant mouse macrophage migration inhibitory factor; WT, wild type.
MIF Deficiency Exacerbates Doxorubicin‐induced Cardiac ROS Generation
Next, DHE was used to evaluate ROS (O2− in particular) production in the hearts from WT and MIF−/− mice with or without doxorubicin treatment. Neither saline treatment nor MIF deficiency significantly affected DHE staining. Doxorubicin significantly increased ROS generation, consistent with previous report.39 More importantly, doxorubicin‐induced ROS production was further enhanced by MIF deficiency (Figure 6A through 6L and 6Q), indicating a role of ROS generation in MIF deficiency‐accentuated myocardial response following doxorubicin challenge. Furthermore, MitoSOX Red was employed to detect levels of ROS derived from mitochondria. MitoSOX Red is widely employed for detection of mitochondrial O2−, the primary source of ROS in mitochondria.40 Our data revealed that doxorubicin treatment significantly promoted mitochondria‐derived ROS, the effect of which was exacerbated by MIF deletion (Figure 6M through 6P and 6R).
Figure 6.
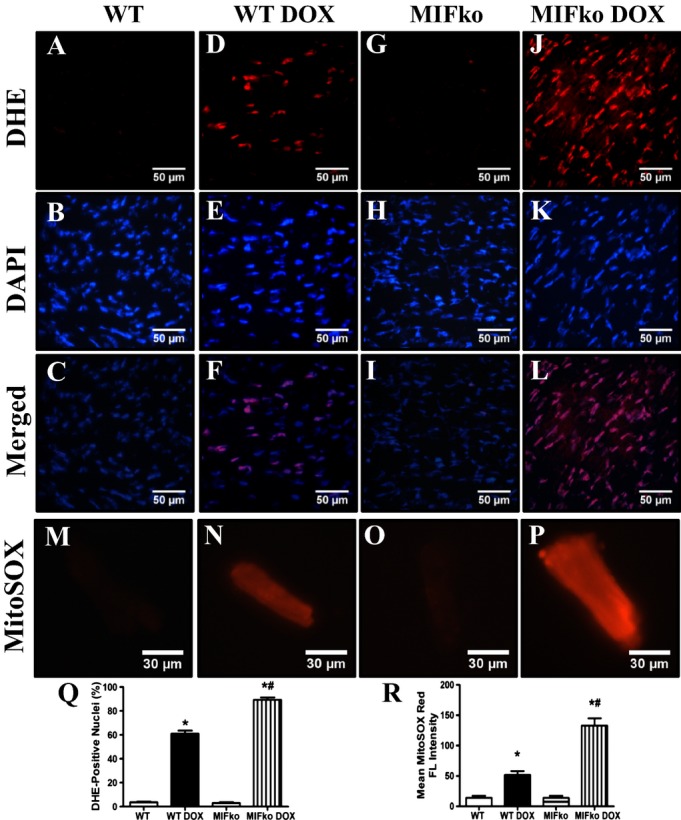
ROS production in hearts and myocardial mitochondria from WT and MIF−/− mice treated with saline or doxorubicin (DOX). A through L, Frozen myocardial tissue sections from WT and MIF−/− mice treated with saline or DOX were stained with DHE (red) and nucleus with DAPI (blue). Representative images of heart sections from WT and MIF−/− mice stained with DHE and DAPI. Oxidized DHE intercalates into DNA and the nuclei appear in red; M through P, Representative images of MitoSOX‐stained cardiomyocytes from WT and MIF−/− mice treated with or without DOX. Data were from 3 independent experiments each with 3 mice per group (for a total of 9 mice per group); Q, Quantitative analysis of DHE‐positive nuclei 7 days after DOX injection (≈50 fields from 3 mice per group); R, MitoSOX red fluorescence intensity. Mean±SEM, n=50 fields per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group. DHE indicates dihydroethidium; DAPI, 4′,6‐diamidino‐2‐phenylindole; ko, knockout; MIF, macrophage migration inhibitory factor; MitoSOX, the brand name of the reagent used for mitochondrial superoxide staining; WT, wild type.
MIF Deficiency Augments Doxorubicin‐caused Autophagolysosome Formation Deficit
By electron microscopic examination, doxorubicin treatment was found to dramatically promote accumulation of double‐membrane vacuoles (Figure 5B and 5D), indicating that doxorubicin leads to possibly reduced formation of autophagolysosomes.27 MIF deficiency further increased doxorubicin‐induced accumulation of these double‐membrane vacuoles in myocardial tissues, suggesting that the absence of MIF exacerbates doxorubicin‐induced reduction of autophagolysosome in the heart. Western blot also was employed to evaluate cardiac autophagolysosome formation. Consistently, our data suggested that doxorubicin treatment suppressed cardiac autophagolysosome formation, as evidenced by the increased LC3BII and p62 (Figure 7A, 7D through 7G). Moreover, MIF deficiency further augmented doxorubicin‐induced interruption of autophagolysosome formation (Figure 7A, 7D through 7G). Although rmMIF itself failed to significantly alter myocardial autophagic flux in control MIF−/− mice, it significantly reconciled doxorubicin‐induced disruption of autophagic flux in MIF−/− mice (Figure 9G through 9K). Furthermore, our data showed that doxorubicin inhibited cardiac AMPK phosphorylation while increasing marker of ER stress and accumulation of ubiquitinated proteins in myocardial tissues, the effects of which were exacerbated by MIF deficiency (Figure 7A through 7C, 7H, 7I). Taken together, these data suggested that doxorubicin treatment inhibits formation of autophagolysosome in the heart, with a more pronounced effect under MIF deficiency.
Figure 7.

Effect of MIF deficiency on doxorubicin (DOX)‐induced changes in autophagy, ER stress and ubiquitination. A, Representative gel blots depicting levels of p‐AMPK, t‐AMPK, LC3BI/II, p62, Bip, ubiquitin, and GAPDH (loading control) using specific antibodies. B, AMPKα phosphorylation (Thr172, pAMPKα‐to‐AMPKα ratio). C, Total AMPK expression. D, LC3B I expression. E, LC3B II expression. F, LC3B II‐to‐I ratio. G, p62 expression. H, Bip expression. I, Ubiquitin expression. Mean±SEM, n = 5 to 6 mice per group, *P<0.05 vs WT group, #P<0.05 vs WT DOX group. ER indicates endoplasmic reticulum; ko, knockout; MIF, macrophage migration inhibitory factor; p‐AMPK, phosphorylated AMP kinase; t‐AMPK, total AMP kinase; WT, wild type.
Rapamycin Pretreatment Alleviated Doxorubicin‐induced Cardiomyopathy in WT and MIF−/− Mice
Since doxorubicin‐induced cardiomyopathy was associated with autophagolysosome formation defect, we next analyzed the effect of autophagy activation using rapamycin, a specific inhibitor of mammalian target of rapamycin (mTOR) to induce autophagy.29,47 One week of rapamycin pretreatment failed to significantly influence cardiac geometry in WT and MIF−/− mice. However, rapamycin pretreatment dramatically protected against doxorubicin‐induced cardiac remodeling in WT mice, as evidenced by improved LVESD, FS and EF (Figure 8C, 8E, and 8F). More importantly, it significantly alleviated the detrimental effect of MIF deficiency in murine hearts following doxorubicin challenge. Cardiac geometry was not significantly affected by rapamycin or MIF deficiency (Figure 8A through 8F).
To further confirm the beneficial effect of rapamycin against doxorubicin‐induced cardiomyopathy, cardiomyocyte contractile function was evaluated. Our data revealed that rapamycin rescued doxorubicin‐induced cardiomyocyte contractile dysfunction as evidenced by normalized peaking shortening (PS) and maximal velocity of shortening/relengthening (±dL/dt). More importantly, rapamycin pretreatment protected against doxorubicin‐induced contractile dysfunction in cardiomyocytes from MIF−/− mice. Neither rapamycin nor MIF deficiency significantly affected cardiomyocyte contractile function (Figure 9A through 9F). To further examine if the beneficial effect of rapamycin relies on autophagy flux, bafilomycin A1 was employed to block autophagolysosome formation.44 Our data showed that inhibiting autophagolysosome formation with bafilomycin A1 negated the cardioprotective effect of rapamycin, favoring the notion that facilitated autophagolysosome formation might be the primary mechanism behind the beneficial effect of rapamycin pretreatment (Figure 9A through 9F).
We went on to further examine ROS generation in myocardial tissues from doxorubicin‐treated WT and MIF−/− mice following a 1‐week pretreatment with rapamycin. Doxorubicin‐induced ROS were dramatically alleviated in myocardial tissues from WT and MIF−/− mice pretreated with rapamycin (Figure 10). These findings implicate that rapamycin pretreatment significantly attenuates doxorubicin‐induced cardiomyopathy in WT and MIF−/− mice.
Discussion
The salient findings from our present work suggest that endogenous MIF may serve as a unique protective factor for doxorubicin‐induced cardiomyopathy. Doxorubicin‐induced cardiac remodeling and dysfunction were accentuated by genetic MIF deficiency, as evidenced by more pronounced adverse effects on mortality, myocardial geometry, and function. In addition, MIF deficiency exacerbated doxorubicin‐induced apoptosis, mitochondrial injury (TEM image, JC‐1 staining and mitochondrial respiration) and ROS generation (global or mitochondria originated O2−). Our TEM and Western blot data indicated that doxorubicin suppressed the degradation of cardiac autophagosomes, and that these effects were exacerbated by MIF deficiency. In MIF−/− mice, reconstitution with recombinant MIF partially although significantly improved doxorubicin‐induced cardiomyocyte contractile dysfunction, cardiomyocyte cell death, mitochondrial dysfunction, and interrupted myocardial autophagic flux. Pretreatment with rapamycin to induce cardiac autophagy dramatically attenuated doxorubicin‐induced cardiomyopathy in WT and MIF−/− mice. More importantly, treatment with bafilomycin A1 confirmed that facilitated autophagolysosome formation was probably the primary mechanism underlying the cardioprotective effect of rmMIF and rapamycin. Taken together, these data indicate that by facilitating autophagolysosome formation, endogenous MIF exerts a cardioprotective action by preventing doxorubicin‐induced exacerbation of cardiomyopathy. Facilitation of cardiac autophagy may be beneficial for the treatment of doxorubicin‐induced cardiomyopathy, especially in those patients who are genetic low producers of MIF.48
Our echocardiographic data showed that doxorubicin treatment increased LVEDD and LVESD, as well as decreased fractional shortening and ejection fraction in WT hearts. These findings are in agreement with the previous findings of cardiac remodeling induced by acute doxorubicin treatment.3 MIF deletion further exacerbated the doxorubicin‐induced rise in the lung/body weight ratio, suggesting that the doxorubicin‐challenged MIF−/− mice might have already developed heart failure prior to death.49 In addition, data from the present study revealed that doxorubicin treatment triggered cardiomyocyte contractile dysfunction, including suppressed peak shortening and maximal velocity of shortening/relengthening. Moreover, intracellular Ca2+ handling was disturbed in response to doxorubicin treatment, as evidenced by a depressed electrically stimulated rise in intracellular Ca2+ (ΔFFI) and prolonged intracellular Ca2+ clearance. MIF deficiency itself was not found to significantly affect cardiac geometry, cardiomyocyte contractile function, and intracellular Ca2+ handling properties. However, the absence of MIF exacerbated doxorubicin‐induced cardiac remodeling, cardiomyocyte contractile dysfunction, and disturbances in intracellular Ca2+ handling. Accumulating evidence indicates a pivotal role of interrupted intracellular Ca2+ homeostasis in cardiac pathological responses,50–52 further suggesting a specific function of MIF in the maintenance of intracellular Ca2+ handling in cardiomyocytes during doxorubicin challenge. Although it is still unclear how MIF‐1 deficiency exacerbates doxorubicin‐induced intracellular Ca2+ handing, myocardial autophagy is speculated to play a role. Our previous report showed that myocardial autophagy activity is involved in the regulation of intracellular Ca2+ handing in cardiomyocytes.36 Moreover, our present finding that reconstitution of rmMIF significantly improved cardiomyocyte function in doxorubicin‐treated MIF−/− mice further confirmed the indispensible role of MIF in the maintenance of cardiac homeostasis in the face of doxorubicin challenge.
As a cytokine involved in innate immunity, MIF plays an essential role in mediating antimicrobial and stress responses.10 There is an increasing recent interest in the role of MIF in the etiology of cardiovascular diseases, including atherosclerosis,53–55 coronary heart disease,56 type 2 diabetes mellitus‐associated cardiac complications,13,57–58 cardiac ischemia reperfusion injury,11,14–15 and pressure overload‐induced cardiac hypertrophy.59 MIF has been shown to be expressed in cardiomyocytes and to be secreted by cardiomyocytes.11,14 In the course of ischemia/reperfusion, type 1 diabetes or pressure overload, endogenous MIF has been demonstrated to be tissue protective against stress‐caused cardiac injuries.11,13–15,59 The mechanism through which MIF exerts its cardioprotective effect is dependent upon binding to and activating its cardiac receptor CD74.11,15 More importantly, the cardioprotective effect of MIF may rely on activation of AMPK11,14 and inhibition of the JNK/MAPK pathway.15 Reminiscent of these previous findings, the current results indicate that MIF deficiency exacerbates cardiac geometric, cardiomyocyte contractile, and intracellular Ca2+ handling defects induced by doxorubicin treatment. Our data showed that MIF deficiency further suppressed cardiac AMPK in response to doxorubicin treatment.
Our in vivo data revealed, for the first time, that MIF deficiency exacerbates doxorubicin‐induced deficit of cardiac autophagolysosome formation. Our TEM data displayed that doxorubicin overtly promoted the accumulation of double‐membrane vacuoles in myocardial tissues, representing an increased cellular content of autophagosomes. In addition, our data depicted a significant rise in LC3B II, a widely accepted marker for autophagosomes, in cardiac tissue following doxorubicin challenge. The increased appearance of LC3B II may be caused by either an increase in its formation (autophagy initiation), or inhibition of its degradation (autophagolysosome formation).36,44,60 p62, also known as SQSTM1/sequestome 1, is an autophagy adaptor protein specifically degraded by autophagy.61–62 Total cellular content of p62 inversely correlates with autophagy function. Our data showed that p62 was increased in cardiac tissues following doxorubicin treatment, the effect of which was further augmented by MIF deficiency. An increase in LC3B II with a concomitant elevation in p62 suggests a deficit in autophagolysosome formation following doxorubicin treatment. However, in vitro data suggests that doxorubicin at low dosages may induce autophagy in cardiomyocytes, which is detrimental to cardiomyocyte survival.24–25 It may be speculated that such discrepancies in doxorubicin‐induced autophagy response may be attributed to different experimental settings. For example, autophagy was deemed detrimental in the in vitro setting of doxorubicin (1 μmol/L) treatment.24–25 On the other hand, our group and others used a 20 mg/kg cumulative dose of doxorubicin in vivo to trigger doxorubicin cardiomyopathy acutely.3,24–25 Although the precise mechanism behind MIF‐regulated autophagolysosome formation remains unknown, our data suggest that MIF deficiency exacerbates doxorubicin‐induced defect in cardiac autophagolysosome formation, which may further exacerbate doxorubicin cardiomyopathy.
Perhaps the most intriguing finding from our current study is that facilitated autophagy using rapamycin rescued doxorubicin‐induced changes in cardiac geometry and contractile function in WT and MIF−/− mice. As a specific inhibitor of mammalian target of rapamycin (mTOR), rapamycin is a potent inducer of autophagy.24,63 Data from other laboratories have shown that starvation‐induced autophagy is beneficial for homeostasis of cardiac geometry in the face of doxorubicin toxicity.26 At the same time, recent studies showed that prior starvation, a widely employed inducer for autophagy, is capable of rescuing doxorubicin‐induced deficit in cardiac autophagolysosome formation.26 Although the precise underlying mechanisms are unclear, it is possible that pretreatment with rapamycin might activate cardiac autophagy to increase myocardial ATP levels to prevent doxorubicin‐induced deficit in autophagolysosome formation.24 Further experiments are warranted to identify the possible mechanisms underlying the beneficial effect of rapamycin pretreatment against doxorubicin‐induced cardiomyopathy.
In conclusion, the findings from our present study provide the first evidence that MIF exerts a permissive role in doxorubicin cardiomyopathy. Our data revealed a pivotal role of interrupted autophagolysosome formation in doxorubicin‐induced cardiac anomalies in WT and MIF−/− mice. Moreover, facilitating autophagy using rapamycin and replenishment of MIF using rmMIF significantly ameliorated doxorubicin‐induced cardiac anomalies. Although it is still premature to discern the precise mechanism through which MIF governs myocardial autophagolysosome formation in the setting of doxorubicin challenge, our current study has shed some light towards a better understanding for the role of MIF and autophagy in the maintenance of myocardial geometry and function under doxorubicin toxicity. Further study is warranted to elucidate the underlying mechanisms as well as the therapeutic potential of pharmacologically augmented autophagy in the management of doxorubicin cardiomyopathy.64
Sources of Funding
This work was supported in part by NIH/NCRR 5P20RR016474, NIH/NIGMS 8P20GM103432, NIH R01AI042310, and the Brookdale Foundation.
Disclosures
None.
References
- 1.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003; 97:2869-2879 [DOI] [PubMed] [Google Scholar]
- 2.Singal PK, Iliskovic N. Doxorubicin‐induced cardiomyopathy. N Engl J Med. 1998; 339:900-905 [DOI] [PubMed] [Google Scholar]
- 3.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ. Acute doxorubicin cardiotoxicity is associated with p53‐induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009; 119:99-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin‐induced cardiotoxicity. Arch Immunol Ther Exp (Warsz). 2009; 57:435-445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L. Gp91phox‐containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol Med. 2007; 42:466-473 [DOI] [PubMed] [Google Scholar]
- 6.Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, Bertrand R, Andrews NC, Santos MM. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003; 102:2574-2580 [DOI] [PubMed] [Google Scholar]
- 7.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin‐induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012; 52:1213-1225 [DOI] [PubMed] [Google Scholar]
- 8.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000; 39:257-288 [DOI] [PubMed] [Google Scholar]
- 9.Vasquez‐Vivar J, Martasek P, Hogg N, Masters BS, Pritchard KA, Jr, Kalyanaraman B. Endothelial nitric oxide synthase‐dependent superoxide generation from adriamycin. Biochemistry. 1997; 36:11293-11297 [DOI] [PubMed] [Google Scholar]
- 10.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003; 3:791-800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP‐activated protein kinase in the ischaemic heart. Nature. 2008; 451:578-582 [DOI] [PubMed] [Google Scholar]
- 12.Willis MS, Carlson DL, Dimaio JM, White MD, White DJ, Adams GA, IV, Horton JW, Giroir BP. Macrophage migration inhibitory factor mediates late cardiac dysfunction after burn injury. Am J Physiol Heart Circ Physiol. 2005; 288:H795-H804 [DOI] [PubMed] [Google Scholar]
- 13.Tong C, Morrison A, Yan X, Zhao P, Yeung ED, Wang J, Xie J, Li J. Macrophage migration inhibitory factor deficiency augments cardiac dysfunction in type 1 diabetic murine cardiomyocytes. J Diabetes. 2010; 2:267-274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, Bucala R, Li J. Impaired macrophage migration inhibitory factor‐AMP‐activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010; 122:282-292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009; 119:3807-3816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010; 140:313-326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004; 432:1032-1036 [DOI] [PubMed] [Google Scholar]
- 18.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007; 13:619-624 [DOI] [PubMed] [Google Scholar]
- 19.Matsui Y, Kyoi S, Takagi H, Hsu CP, Hariharan N, Ago T, Vatner SF, Sadoshima J. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy. 2008; 4:409-415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. 2011; 108:4123-4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009; 120:S191-S197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007; 117:1782-1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito T. Autophagic changes in cardiomyocyte predict good prognosis for dilated cardiomyopathy. Circulation. 2012; 216:A13562 [Google Scholar]
- 24.Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin‐induced autophagy and cardiomyocyte death. J Biol Chem. 2010; 285:793-804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X, Chen K, Kobayashi S, Timm D, Liang Q. Resveratrol attenuates doxorubicin‐induced cardiomyocyte death via inhibition of p70 S6 kinase 1‐mediated autophagy. J Pharmacol Exp Ther. 2012; 341:183-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, Ogino A, Tsujimoto A, Goto K, Maruyama R, Kawasaki M, Mikami A, Fujiwara T, Fujiwara H, Minatoguchi S. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012; 96:456-465 [DOI] [PubMed] [Google Scholar]
- 27.Xu X, Ren J. Macrophage migration inhibitory factor knockout exacerbates doxorubicin‐induced cardiomyopathy: role of autophagy. Circulation. 2012; 126:A13473 [Google Scholar]
- 28.Fingerle‐Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Muller W, Bucala R. The p53‐dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003; 100:9354-9359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski DJ, Yu JJ, Henske EP. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)‐dependent. Proc Natl Acad Sci USA. 2011; 108:12455-12460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hua Y, Zhang Y, Ren J. IGF‐1 deficiency resists cardiac hypertrophy and myocardial contractile dysfunction: role of microRNA‐1 and microRNA‐133a. J Cell Mol Med. 2012; 16:83-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuzman JA, Gerdes AM, Kobayashi S, Liang Q. Thyroid hormone activates Akt and prevents serum starvation‐induced cell death in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005; 39:841-844 [DOI] [PubMed] [Google Scholar]
- 32.Hua Y, Zhang Y, Dolence J, Shi GP, Ren J, Nair S. Cathepsin K knockout mitigates high‐fat diet‐induced cardiac hypertrophy and contractile dysfunction. Diabetes. 2013; 62:498-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, Sonderegger I, Bachmaier K, Kopf M, Penninger JM. Dendritic cell‐induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003; 9:1484-1490 [DOI] [PubMed] [Google Scholar]
- 34.Xu X, Pacheco BD, Leng L, Bucala R, Ren J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res. 2013; 99:412-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Relling DP, Esberg LB, Johnson WT, Murphy EJ, Carlson EC, Lukaski HC, Saari JT, Ren J. Dietary interaction of high fat and marginal copper deficiency on cardiac contractile function. Obesity (Silver Spring). 2007; 15:1242-1257 [DOI] [PubMed] [Google Scholar]
- 36.Xu X, Hua Y, Sreejayan N, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high‐fat diet‐induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. 2013; 5:61-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol. 2012; 14:177-185 [DOI] [PubMed] [Google Scholar]
- 38.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol. 2009; 54:2187-2196 [DOI] [PubMed] [Google Scholar]
- 39.Zhang S, Liu X, Bawa‐Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin‐induced cardiotoxicity. Nat Med. 2012; 18:1639-1642 [DOI] [PubMed] [Google Scholar]
- 40.Guo R, Ren J. Alcohol dehydrogenase accentuates ethanol‐induced myocardial dysfunction and mitochondrial damage in mice: role of mitochondrial death pathway. PLoS One. 2010; 5:e8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS, Furutani E, Perez‐Sanz TM, Graveline A, Janssens SP, Picard MH, Scherrer‐Crosbie M, Bloch KD. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dys‐function, and mortality induced by doxorubicin. Circulation. 2007; 116:506-514 [DOI] [PubMed] [Google Scholar]
- 42.Ceylan‐Isik AF, Guo KK, Carlson EC, Privratsky JR, Liao SJ, Cai L, Chen AF, Ren J. Metallothionein abrogates GTP cyclohydrolase I inhibition‐induced cardiac contractile and morphological defects: role of mitochondrial biogenesis. Hypertension. 2009; 53:1023-1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, Wu X, Lauriol J, Wang B, Bauer M, Bronson R, Franchini KG, Neel BG, Kontaridis MI. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of leopard syndrome‐associated PTPN11 mutation. J Clin Invest. 2011; 121:1026-1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011; 124:2117-2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor‐beta‐induced alpha‐smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003; 29:397-404 [DOI] [PubMed] [Google Scholar]
- 46.Wallace KB. Doxorubicin‐induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003; 93:105-115 [DOI] [PubMed] [Google Scholar]
- 47.Xu X, Hueckstaedt LK, Ren J. Deficiency of insulin‐like growth factor 1 attenuates aging‐induced changes in hepatic function: role of autophagy. J Hepatol. 2013; 59:308-317 [DOI] [PubMed] [Google Scholar]
- 48.Bucala R. Mif, mif alleles, and prospects for therapeutic intervention in auto‐immunity. J Clin Immunol. 2013; 33:S72-S78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meguro T, Hong C, Asai K, Takagi G, McKinsey TA, Olson EN, Vatner SF. Cyclosporine attenuates pressure‐overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circ Res. 1999; 84:735-740 [DOI] [PubMed] [Google Scholar]
- 50.Dong F, Li Q, Sreejayan N, Nunn JM, Ren J. Metallothionein prevents high‐fat diet induced cardiac contractile dysfunction: role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes. 2007; 56:2201-2212 [DOI] [PubMed] [Google Scholar]
- 51.Fang CX, Dong F, Thomas DP, Ma H, He L, Ren J. Hypertrophic cardiomyopathy in high‐fat diet‐induced obesity: role of suppression of forkhead transcription factor and atrophy gene transcription. Am J Physiol Heart Circ Physiol. 2008; 295:H1206-H1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Li L, Hua Y, Nunn JM, Dong F, Yanagisawa M, Ren J. Cardiac‐specific knockout of et(a) receptor mitigates low ambient temperature‐induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Biol. 2012; 4:97-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burger‐Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002; 105:1561-1566 [DOI] [PubMed] [Google Scholar]
- 54.Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R, Lau CP, Lan HY. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res. 2000; 87:1202-1208 [DOI] [PubMed] [Google Scholar]
- 55.Verschuren L, Kooistra T, Bernhagen J, Voshol PJ, Ouwens DM, van Erk M, de Vries‐van der Weij J, Leng L, van Bockel JH, van Dijk KW, Fingerle‐Rowson G, Bucala R, Kleemann R. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res. 2009; 105:99-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herder C, Illig T, Baumert J, Muller M, Klopp N, Khuseyinova N, Meisinger C, Martin S, Thorand B, Koenig W. Macrophage migration inhibitory factor (MIF) and risk for coronary heart disease: results from the MONICA/KORA Augsburg case‐cohort study, 1984–2002. Atherosclerosis. 2008; 200:380-388 [DOI] [PubMed] [Google Scholar]
- 57.Atsumi T, Cho YR, Leng L, McDonald C, Yu T, Danton C, Hong EG, Mitchell RA, Metz C, Niwa H, Takeuchi J, Onodera S, Umino T, Yoshioka N, Koike T, Kim JK, Bucala R. The proinflammatory cytokine macrophage migration inhibitory factor regulates glucose metabolism during systemic inflammation. J Immunol. 2007; 179:5399-5406 [DOI] [PubMed] [Google Scholar]
- 58.Yabunaka N, Nishihira J, Mizue Y, Tsuji M, Kumagai M, Ohtsuka Y, Imamura M, Asaka M. Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care. 2000; 23:256-258 [DOI] [PubMed] [Google Scholar]
- 59.Koga K, Kenessey A, Ojamaa K. Macrophage migration inhibitory factor antagonizes pressure overload‐induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013; 304:H282-H293 [DOI] [PubMed] [Google Scholar]
- 60.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation‐induced autophagy in cardiac myocytes. Circ Res. 2010; 107:1470-1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on Huntingtin‐induced cell death. J Cell Biol. 2005; 171:603-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post‐abscission event of cytokinesis. Nat Cell Biol. 2009; 11:65-70 [DOI] [PubMed] [Google Scholar]
- 63.Hua Y, Zhang Y, Ceylan‐Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging‐induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011; 106:1173-1191 [DOI] [PubMed] [Google Scholar]
- 64.Jorgensen WL, Gandavadi S, Du X, Hare AA, Trofimov A, Leng L, Bucala R. Receptor agonists of macrophage migration inhibitory factor. Bioorg Med Chem Lett. 2010; 20:7033-7036 [DOI] [PMC free article] [PubMed] [Google Scholar]