

Abstract
Background
The cysteine protease cathepsin K (CatK) has been implicated in the pathogenesis of cardiovascular disease. We sought to determine the link between atrial fibrillation (AF) and plasma CatK levels and to investigate the expression of and therapeutic target for CatK in vivo and in vitro.
Methods and Results
Plasma CatK and extracellular matrix protein peptides (intact procollagen type I of N‐terminal propeptide; carboxyl‐terminal telopeptide of type I collagen [ICTP]) were measured in 209 consecutive patients with AF (paroxysmal AF, 146; persistent AF, 63) and 112 control subjects. In addition, the regulation of CatK expression was investigated in vivo and vitro. Patients with AF had higher plasma CatK and ICTP levels than did control subjects. Patients with persistent AF had higher levels of plasma CatK and ICTP than did patients with paroxysmal AF. CatK was correlated with ICTP concentration and left atrial diameter in all subjects. In rabbits, superoxide production, CatK activity, fibrosis, and the levels of atrial tissue angiotensin II, angiotensin type 1 receptor, gp91phox, phospho‐p38 mitogen‐activated protein kinase, and CatK were greater in those with tachypacing‐induced AF than in controls, and these changes were reversed with angiotensin type 1 receptor antagonist. Olmesartan and mitogen‐activated protein kinase inhibitor decreased the CatK expression induced by angiotensin II in rat neonatal myocytes.
Conclusions
These data indicated that increased plasma CatK levels are linked with the presence of AF. Angiotensin type 1 receptor antagonist appears to be effective in alleviating atrial fibrosis in a rabbit AF model, partly reducing angiotensin type 1 receptor‐p38mitogen‐activated protein kinase‐dependent and ‐independent CatK activation, thus preventing AF.
Keywords: angiotensin type 1 receptor, atrial fibrillation, cathepsin K, extracellular matrix, mitogen‐activated protein kinase
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia in clinical practice. AF itself has been shown to cause changes in the function and structure of the atria, providing a possible explanation for the progressive nature of this arrhythmia.1–3 In the atria, the extracellular matrix provides supportive scaffolding for cardiomyocytes, maintains the structural integrity of cardiac tissue, and is necessary for electrical conduction via cardiomyocytes.4 Growing evidence supports the concept that structural remodeling of the extracellular matrix may be the key event leading to the development of AF and atrial mechanical dysfunction. Lysosmal protease cathepsins (Cats) traditionally have been known to degrade unwanted intracellular or endocytosed proteins.5 However, the recent recognition of the inducible CatK and CatS has revealed their proteolytic functions in inflammatory disease, including atherosclerosis‐based vascular disease processes.6–8 More recently, several studies have reported that Cats play a functional role in intracellular and extracellular protein degradation in cardiac myocytes by contributing to matrix turnover, chamber dilation, and structural remodeling.9–12 A few reports suggest that circulating Cats have a predictive value for proteolysis‐associated disease, and related research has focused on vascular disease (including coronary artery diseases and aortic aneurysm).13–14 To date, no studies have examined atrial Cat expression and plasma levels as potential biomarkers for atrial remodeling in atrial disease.
Recently, activation of the renin–angiotensin system has been implicated as part of the mechanism of AF.15 Cardiac‐specific overexpression of angiotensin‐converting enzyme in mice results in excessive levels of cardiac angiotensin II (Ang II), and the mice develop spontaneous AF.16 Ang II has numerous cardiovascular effects that might lead to cardiac arrhythmia, including the induction of fibrosis and the proliferation of cardiac fibroblasts, the increased synthesis of collagen, and the promotion of reactive oxygen species generation.17 Ang II has been shown to induce p38mitogen‐activated protein kinase (p38MAPK)/extracellular signal‐regulated kinase activation and atrial interstitial fibrosis.18–19 Several recent studies have demonstrated that angiotensin inhibition prevents left ventricular fibrosis by decreasing the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase–dependent Cat activation.11,20 Although the inhibition of Ang II through the use of Ang II type 1 receptor (AT1R) antagonists or angiotensin‐converting enzyme inhibitors has prevented AF in animal models and a diverse human population with AF or at risk of developing AF,21–24 the underlying mechanism is poorly understood.
In this study, we sought to determine whether circulating CatK levels are closely linked to the presence of AF and increased levels of the collagen type I degradation marker. In addition, we explored the possible mechanisms by which Ang II inhibition mitigates atrial remodeling and AF in a rabbit tachypacing model.
Methods
Study Population
We recruited 209 consecutive patients with paroxysmal AF (PAF; n=146) or persistent AF (PeAF; n=63) who were admitted to Nagoya University Hospital between March 2009 and December 2010 for scheduled radiofrequency catheter ablation with coronary angiography. AF in these patients had been diagnosed in light of symptoms, 12‐lead electrocardiogram, and Holter electrocardiogram. As described previously,25 PAF was defined on the basis of a history of 1 or more episodes of AF that self‐resolved or were terminated medically within 7 days, and PeAF was defined according to a history of 1 or more episodes of AF over 7 days that required pharmacological or electrical cardioversion to establish normal sinus rhythm. None of the patients in this study had permanent AF. We excluded patients with dilated or hypertrophic cardiomyopathy, myocardial infarction, congenital heart disease, congestive heart failure, or valvular heart diseases and those receiving hemodialysis. AF patients were receiving standard therapy with antiarrhythmic drugs, β‐blockers, angiotensin‐converting enzyme inhibitors or AT1R blockers, and statins at the time they underwent ablation. We also assessed 112 subjects with and without paroxysmal atrial arrhythmia (no previously documented AF), who were considered to represent the control group. The study protocol was approved by the ethics committee of the Nagoya University School of Medicine, and written informed consent was obtained from all patients.
Laboratory Assay
Laboratory measurements were performed under blinded conditions. Human and rabbit plasma CatK levels were determined by using ELISA kits (Biomedica Gruppe, Biomedica Medizinprodukte). Serum interleukin‐1β levels were measured by using commercially available kits. Serum levels of intact procollagen type I N‐terminal propeptide (I‐PINP), carboxyl‐terminal telopeptide of collagen type I (ICTP), atrial natriuretic peptide, cystatin C, high‐sensitivity C‐reactive protein, hemoglobin A1c, and atrial tissue Ang II were measured at a commercial laboratory (SRL [Tokyo, Japan]). Plasma CatK values were expressed as ng/mL, and interassay and intraassay coefficients of variation were <8% (n=20).
Echocardiography
Two‐dimensional and Doppler echocardiography was performed by an experienced sonographer using a Vivid4 System (GE Healthcare Bio‐Sciences). The images were recorded on a DVD recorder and analyzed offline. Left atrial diameter (LAD) was obtained by using standard M‐mode measurements, as recommended by the American Society of Echocardiography. The left ventricular ejection fraction was calculated using the modified Simpson's rule.
Animal Model and Treatment
A rabbit AF model was induced by ventricular tachypacing as described previously.26 The study protocol was approved by the Institutional Animal Care and Use Committee of Nagoya University School of Medicine. Eleven‐week‐old male New Zealand White rabbits (Kitayama Labs) underwent surgery with or without the implantation of right ventricular pacemakers (Medtronic) under anesthesia with ketamine hydrochloride 35 mg/kg and xylazine 3 mg/kg. After the animals recovered from surgery, the pacemakers were programmed to pace at 380 beats/min for 4 weeks. Rabbits were divided into 3 groups as follows: nonpaced control rabbits (control rabbits; n=7), rabbits subjected to ventricular tachypacing treated with vehicle (0.5% carboxymethylcellulose, AF rabbits; n=7), and rabbits subjected to tachypacing with olmesartan treatment (Olm rabbits; n=5). Daily oral administration of olmesartan (1 mg/kg; Daichi‐Sankyo) via gastric tube was initiated 1 week before surgery and continued throughout the study period. Electrocardiograms were monitored once per week to adjust the pacing rate to the maximum rate, thus allowing for 1:1 capture in each rabbit. At the end of 4 weeks of ventricular tachypacing, open chest electrophysiological studies were performed under anesthesia and ventilated mechanically with isoflurance‐containing room air (0.5% per 2 L/min) as described previously.26 After electrophysiological evaluation, both atria were harvested for biologic and histological analyses.
Western Blotting
On postoperative day, tissue samples obtained on operative day 28 were homogenized lysis buffer containing 20 mmol/L Tris‐HCl, pH 7.4, 1% Nonidet P‐40, 150 mmol/L NaCl, 0.5% deoxycholic acid, 1 mmol/L sodium orthovanadate, and protease inhibitor mixture (Sigma). The protein content was determined by using the Bradford method. The same amount of protein (40 μg) was loaded per lane and separated using denaturing 10% polyacrylamide gels. The membranes were probed with antibodies to CatK,27 to AT1R (Santa Cruz Biotechnology), to gp91phox (BD Biosciences), and to total p38MAPK and phosphorylated p38MAPK (p‐p38MAPK) (Cell Signaling Technology, Inc). Bands were visualized using chemiluminescence (ECL Western Blotting Detection Kit, Amersham Biosciences).
Immunohistochemistry
On postoperative day 28, transverse sections (5‐μm thickness) of atrial tissues were stained with rabbit polyclonal antibody to CatK (dilution, 1:100) as described previously.27 As a negative control, the primary antibody was replaced by nonimmune immunoglobulin G (Vector). Azan Mallory staining was applied to evaluate fibrotic deposition in rabbit atria. We set a threshold to automatically compute the blue pixel area for the histochemical stain and then computed the ratio (percent) of the positively stained area to the total cross‐sectional atrial free‐wall area using BZ‐II Analyzer, Exe 1.42 software (Keyence). Three random microscopic fields (×400) from 8 independent cross sections of atrial free‐wall (24 fields) in each animal were quantified and averaged for each animal.
Assay of Superoxide Production
Specific NADPH oxidase activity of total homogenates of fresh left atrial tissue was measured with the use of a lucigenin‐based enhanced chemiluminescence assay as described.20 A low lucigenin concentration (5 mmol/L) was used to minimize artifactual O2− production attributable to redox cycling. In brief, homogenate protein (1 mg) lysis buffer (1 mL; 20 mmol/L Tris‐HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L ethylene glycol tetraacetic acid, and 1% Triton X‐100) was transferred to an assay tube, and NADPH and dark‐adapted lucigenin were added to final concentrations of 100 and 5 mmol/L, respectively, immediately before the measurement of chemiluminescence. All assays were performed in triplicate. The chemiluminescence signal was sampled every minute for 12 minutes using a tube luminometer (20/20; Turner Biosystems), and the respective background counts were subtracted from the experimental values.
Cell Culture and Simulation
Neonatal rat atrial myocytes from 1‐day‐old Wistar rats were isolated as described previously28 and cultured in a mixture (50:50, v/v) of Dulbecco's modified Eagle's medium and Ham's F‐12 (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics.11 After 24 hours of culture, the cells (5×104/well in 12‐well plates) were pretreated with or without various reagents for appropriate amounts of time, and the cells were subjected to the related biologic assays.11
Gene Expression Assay
Total RNA was isolated cultured cells and was subjected to reverse transcription with a PCR Core kit (Applied Biosystems). The resulting cDNA was subjected to quantitative real‐time PCR analysis with targeted gene primers and with use of the ABI 7300 Real‐Time PCR System under the following conditions: 50°C (2 minutes) for UNG incubation, 94°C (10 minutes) for AmpliTaq Gold activation, 95°C (15 seconds), and 59°C (1 minute) for 40 cycles as previous described.27 The amount of each mRNA was normalized against the corresponding amount of glyceraldehyde‐3‐phosphate dehydrogenase mRNA.
Assay of Collagenolytic Activity
Total protein (100 μg) from the extracts of cells and atrial tissues was incubated with 500 μg/mL fluorescein‐labeled type I collagen (Molecular Probes Inc) for 6 hours. Reactions were performed in the absence or presence of several protease inhibitors at indicated concentrations as described previously.29
Statistical Analysis
Summary descriptive statistics for continuous parameters are presented as mean±SD values. Categorical variables were compared among study groups by using the χ2 test. Student's t test (for comparison of continuous parameters between 2 groups) or 1‐way ANOVA (for comparison of continuous parameters among 3 or more groups), followed by Tukey's post‐hoc test, was used to test significant differences. Cystatin C and high‐sensitive C‐reaction protein concentrations were logarithmically transformed because the data showed a skewed distribution. If the homogeneity of the variance assumption was violated, the nonparametric Kruskal–Wallis test was used instead. The factors that related at the P<0.1 level were selected as independent variable candidates for multiple logistic regression analysis and were used to evaluate the independent contribution of clinical parameters to AF. Correlation coefficients were calculated using linear regression analysis. In animal and in cell experiments, we performed the text related to ensuring normality for the tests with very small sample sizes. StatFlex (version 6.0; Artech) was used for all statistical analysis. P values of <0.05 were considered statistically significant.
Results
All Patients
The baseline clinical and demographic features of the study population are shown in Table 1. The AF group was older and had more men than did the control group (P<0.001). There were no differences between the AF and control groups in potential causal factors. AF patients had taken more β‐blockers than had the control subjects (P=0.02). Patients with AF had significantly (P<0.05) larger LADs and worse ejection fractions than did control subjects.
Table 1.
Patient Characteristics
| AF (n=209) | Control (n=112) | P Value | |
|---|---|---|---|
| Demographic Characteristics | |||
| Age, y | 60.1±10.9 | 54.8±10.7 | <0.001 |
| Female, % | 23.0 | 42.9 | <0.001 |
| Body mass index, kg/m2 | 23.7±3.4 | 23.0±2.8 | 0.09 |
| Smokers, % | 30.1 | 23.2 | 0.25 |
| Causal factors | |||
| Hypertension, % | 45.5 | 38.4 | 0.26 |
| Diabetes mellitus, % | 20.1 | 12.5 | 0.09 |
| Ischemic heart disease, % | 8.1 | 3.6 | 0.12 |
| Chronic heart failure, % | 7.2 | 4.5 | 0.34 |
| Echocardiography | |||
| Left atrial diameter, mm | 39.0±6.9 | 32.3±4.9 | <0.001 |
| LV ejection fraction, % | 61.8±8.4 | 77.1±6.9 | 0.002 |
| Blood examination | |||
| Potassium, mEq/L | 4.3±0.4 | 4.2±0.4 | 0.48 |
| LDL, mg/dL | 118.4±27.8 | 121.2±29.3 | 0.40 |
| HDL, mg/dL | 55.7±14.2 | 57.4±13.3 | 0.32 |
| Hemoglobin A1c, % | 5.6±0.7 | 5.2±0.7 | <0.001 |
| ANP, pg/mL | 70.9±70.9 | 27.3±29.9 | <0.001 |
| hs‐CRP, mg/dL | 0.14±0.4 | 0.06±0.1 | 0.04 |
| IL‐1β, pg/mL | 14.3±7.0 | 10.9±7.9 | <0.001 |
| I‐PINP:ICTP | 13.7±7.7 | 25.1±19.2 | <0.001 |
| ICTP, ng/mL | 3.8±2.7 | 1.5±1.3 | <0.001 |
| CatK, ng/mL | 13.1±6.7 | 6.1±3.9 | <0.001 |
| Medications | |||
| ACEIs, % | 4.3 | 8.0 | 0.17 |
| ARBs, % | 30.1 | 26.8 | 0.53 |
| CCBs, % | 20.1 | 17.0 | 0.50 |
| β‐Blockers, % | 29.7 | 8.9 | 0.02 |
| Statins, % | 20.1 | 22.3 | 0.64 |
| Antiarrhythmic drugs, % | 100 | 0 | |
Values are expressed as mean±SD. AF indicates atrial fibrillation; LV, left ventricular; LDL, low‐density lipoprotein; HDL, high‐density lipoprotein; ANP, atrial natriuretic peptide; hs‐CRP, high‐sensitivity C‐reactive protein; IL, interleukin; I‐PINP, intact procollagen type I N‐terminal propeptide; ICTP, carboxyl‐terminal telopeptide of type I collagen; CatK, cathepsin K; ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; CCB, calcium channel blocker.
Compared with controls, patients with AF had significantly (P<0.001) higher plasma CatK, interleukin‐1β, ICTP, and I‐PINP levels and lower I‐PINP:ICTP ratios than did control subjects. Compared with controls, AF patients had higher levels of hemoglobin A1c (P<0.001), high‐sensitive C‐reactive protein (P=0.04), and atrial natriuretic peptide (P<0.001), whereas there were no significant differences in potassium or in low‐density and high‐density lipoproteins. In all subjects, univariate regression analysis revealed that there was a positive correlation between CatK and ICTP (r=0.3, P<0.0001; Figure 1A) and LAD (r=0.4, P<0.0001; Figure 1B).
Figure 1.
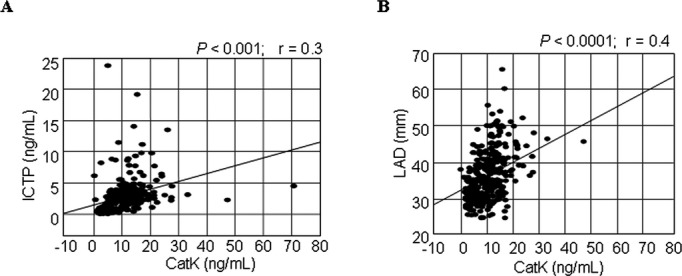
Correlations between plasma levels of CatK and ICTP (A) and (B) LAD. In all patients, there were positive correlations between plasma levels of cathepsin (CatK) and carboxyl‐terminal telopeptide of type I collagen (ICTP) and left atrial diameter (LAD).
PAF Versus PeAF
The baseline characteristics of the PAF and PeAF groups are presented in Table 2. Patients with PeAF had larger LADs and worse ejection fractions than did PAF patients (P<0.05). With the exception of age, the percentage of women, chronic heart failure, and use of statin treatments, there were no significant differences between the 2 groups. As expected, the PeAF group had increased levels of plasma high‐sensitive C‐reactive protein and atrial natriuretic peptide (P<0.05 for both) compared with patients with PAF. In addition, the levels of CatK, ICTP, and interleukin‐1β were higher and the I‐PINP:ICTP ratio lower among subjects in the PeAF group compared with those in the PAF group (P<0.05 for all comparisons; Figure 2).
Table 2.
Patient Characteristics
| Control (n=112) | PAF (n=146) | PeAF (n=63) | ANOVA (P Value) | |
|---|---|---|---|---|
| Demographic characteristics | ||||
| Age, y | 54.8±10.7 | 61.1±10.6* | 57.8±11.3** | 0.04 |
| Female, % | 42.9 | 27.4* | 12.7** | <0.001 |
| Body mass index, kg/m2 | 23.0±2.8 | 3.5±3.5 | 24.0±3.1 | 0.21 |
| Smoker, % | 23.2 | 28.1 | 34.9 | 0.31 |
| Causal factors | ||||
| Hypertension, % | 38.4 | 46.6 | 42.9 | 0.48 |
| Diabetes mellitus, % | 12.5 | 21.9 | 15.9 | 0.47 |
| Ischemic heart disease, % | 3.6 | 7.5 | 9.5 | 0.25 |
| Chronic heart failure, % | 4.5 | 3.4 | 15.9** | 0.002 |
| Echocardiography | ||||
| Left atrial diameter, mm | 32.3±4.9 | 37.2±6.1 | 43.1±7.0** | <0.001 |
| LV ejection fraction, % | 77.1±6.9 | 63.5±7.6* | 58.0±9.0** | <0.001 |
| Blood examination | ||||
| Potassium, mEq/L | 4.6±0.4 | 4.2±1.5 | 4.3±1.3 | 0.06 |
| LDL, mg/dL | 121.3±29.3 | 120.0±26.5 | 114.7±30.4 | 0.24 |
| HDL, mg/dL | 57.4±13.3 | 55.9±15.1 | 55.3±11.8 | <0.001 |
| Hemoglobin A1c, % | 5.2±0.7 | 5.6±0.6* | 5.6±0.8* | 0.89 |
| hs‐CRP, mg/dL | 0.06±0.1 | 0.14±0.1* | 0.16±0.2** | 0.02 |
| ANP, pg/mL | 27.3±29.9 | 59.5±69.9* | 97.3±66.4** | <0.001 |
| Cystatine C, mg/L | 0.9±0.3 | 1.0±0.2 | 1.1±0.9 | 0.23 |
| Medications | ||||
| ARBs or ACEIs, % | 34.8 | 37.7 | 27.0 | 0.08 |
| CCBs, % | 17.0 | 19.9 | 20.6 | 0.05 |
| β‐Blockers, % | 8.9 | 30.8* | 27.0* | 0.01 |
| Statins, % | 22.3 | 25.3 | 7.9** | 0.01 |
Values are expressed as mean±SD. PAF indicates paroxysmal atrial fibrillation; PeAF, persistent atrial fibrillation; LV, left ventricular; LDL, low‐density lipoprotein; HDL, high‐density lipoprotein; hs‐CRP, high‐sensitivity C‐reactive protein; ANP, atrial natriuretic peptide; ARB, angiotensin II receptor blocker; ACEI, angiotensin‐converting enzyme inhibitor; CCB, calcium channel blocker.
P<0.05 compared with control value.
P<0.01 compared with control values.
P<0.05 compared with value for PAF vs PsAF patients.
Figure 2.
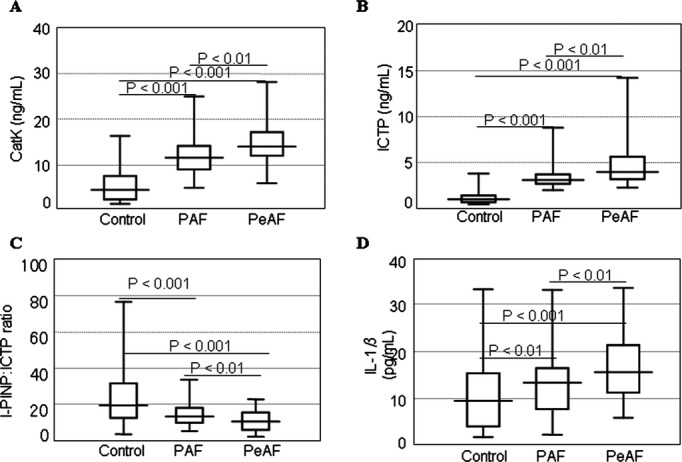
Box plot depiction of the differences in the levels of plasma CatK, I‐PINP, ICTP, and IL‐1β. Plasma levels of CatK, I‐PINP, ICTP, and IL‐1β. Levels of (A) CatK and (B) ICTP gradually increased, whereas (C) the I‐PINP:ICTP ratios decreased from control subjects to patients with PAF to subjects with PeAF. D, IL‐1β levels in control subjects differed from those in both PAF and PeAF patients. Boxes represent the median (black line), 25th percentile, and 75th percentile of observed data; whiskers show the 5th and 95th percentiles of each group. Values are expressed as mean±SD. *P<0.01 vs control; †P<0.01 vs PAF. CatK indicates cathepsin K; ICTP, carboxyl‐terminal telopeptide of type I collagen; IL, interleukin; I‐PINP, intact procollagen type I of N‐terminal propeptide; PAF, paroxysmal atrial fibrillation; PeAF, persistent atrial fibrillation.
Biomarkers for Prediction of AF
Table 3 shows the results of the multiple logistic regression analyses to assess the factors (included at the P<0.1 level in Table 1) that independently contributed to AF. According to the multiple logistic regression analyses, only LAD (odds ratio, 1.19; 95% CI, 1.06 to 1.36; P<0.05), CatK (odds ratio, 1.27; 95% CI, 1.10 to 1.39; P<0.01), and I‐PINP:ICTP (odds ratio, 0.79; 95% CI, 0.69 to 0.90; P<0.001) were significantly associated with AF.
Table 3.
Independent Predictors of AF According to Multivariable Logistic Regression Analysis
| Odds Ratio Estimate | 95% CI | P Value | |
|---|---|---|---|
| LAD | 1.19 | 1.06 to 1.36 | <0.05 |
| CatK | 1.27 | 1.10 to 1.39 | <0.01 |
| I‐PINP/ICTP ratio | 0.79 | 0.69 to 0.90 | <0.001 |
AF indicates atrial fibrillation; LAD, left atrial diameter; CatK, cathepsin K; I‐PINP, intact procollagen type I N‐terminal propeptide; ICTP, carboxyl‐terminal telopeptide of type I collagen.
Inhibitory Effect of AT1R Antagonism on CatK Expression and Atrial Remodeling With AF
Immunostaining showed that the expression of CatK was markedly increased throughout the atrial tissue of AF rabbits, with apparent expression in the myocytes, and this change was inhibited by olmesartan (Figure 3A). As shown in Figure 3B, collagenolytic activity was higher in AF rabbits than in controls, and this change was more sensitive to a specific CatK inhibitor (CatK‐II) and the broad‐spectrum cysteine protease E64 than to GM6001, an inhibitor of matrix metalloproteinases (Figure 3B). Olmesartan reduced the amount of increased collagenolytic activity in the atrial extract of AF rabbits (0.56±0.09 versus 0.94±0.19 fluorescence intensity, P<0.01).
Figure 3.
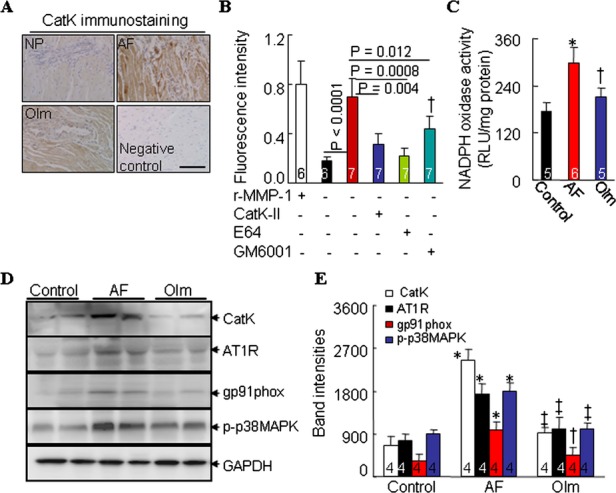
CatK protein expression and NADPH oxidase activity in nonpaced control (NP), ventricular tachypacing (VTP), and ventricular tachypacing plus administration of olmesartan (Olm) rabbits. A, Representative images for CatK immunostaining in the atrial tissues of NP, VTP, Olm rabbits and negative controls (without primary antibody). B, ELISAs of collagenolytic activity in untreated atrial tissues or in those treated with a broad‐spectrum inhibitor of Cats (trans‐epoxysuccinyl‐l‐leucylamido‐(4‐guanidino)butane [E64], 20 μmol/L; Molecular Probes), a CatK‐specific inhibitor (CatK‐II, 10 μmol/L), and an inhibitor of matrix metalloproteinases (GM6001, 10 μmol/L; both from Calbiochem). Recombinant matrix metalloproteinase 1 (rMMP‐1) was included as a positive control. C, Chemiluminescence showing NADPH oxidase activity in atrial tissues from 3 groups. D, Representative Western blots and (E) quantitative data showing the levels of CatK, AT1R, gp91phox, p‐p38MAPK, and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) in left atrial tissue from rabbits. Analyzed animal numbers indicated on related bars. Scale bars indicate 50 μm. Values are expressed as mean±SEM. *P<0.01 vs NP; †P<0.01, ‡P<0.001 vs VTP. AF indicates atrial fibrillation; AT1R, angiotensin type 1 receptor; CatK, cathepsin K; NADPH, nicotinamide adenine dinucleotide phosphate.
AF rabbits had increased levels of NADPH oxidase activity (Figure 3C) and of an NADPH oxidase subunit, gp91phox protein (Figure 3D; P<0.01). As shown in Figure 3D and 3E, Western blotting revealed that the levels of CatK, AT1R, and p‐p38MAPK proteins were greater in AF rabbits than in controls (P<0.01). All of these changes were reversed with olmesartan (P<0.01). In addition, rabbits with AF had significantly higher atrial tissue Ang II levels than did control rabbits (78.3±6.0 versus 36.2±4.8 pg/100 mg, P<0.01), and this effect was also reduced by olmesartan (45.1±3.7 versus 78.3±6.0, P<0.05). The duration of AF was 11.8±0.2 seconds in ventricular‐tachypaced rabbits, whereas AF was not induced in nonpaced controls (Figure 4A). The duration of AF was significantly reduced in olmesartan‐treated rabbits (3.2±0.1 seconds) compared with ventricular tachypacing–only rabbits (P<0.01). Furthermore, olmesartan significantly suppressed atrial fibrosis compared with the control group (3.9±0.6 versus 9.8±1.6%, P<0.05) (Figure 4B and 4C).
Figure 4.
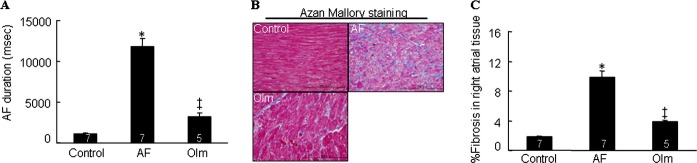
A, Duration of AF in NP, VTP, and olmesartan (Olm)‐treated rabbits. B, Representative images and quantitative data show interstitial fibrosis in the right atrial tissues of 3 experimental groups. Analyzed animal numbers indicated on related bars. Scale bars indicate 100 μm. Values are expressed as mean±SEM. *P<0.01 vs control; †P<0.01 vs VTP. AF indicates atrial fibrillation; NP, nonpaced control; VTP, ventricular tachypacing.
Quantitative real‐time PCR demonstrated that both H2O2 and Ang II significantly (P<0.05) enhanced CatK mRNA expression and that olmesartan reduced CatK expression in response to Ang II in cultured rat neonatal atrial myocytes (Figure 5A). Ang II enhanced the collagenolytic activity in cells extracts, and this effect was sensitive to CatK‐II and E64 (Figure 5B). Western blots showed olmesartan‐mediated reductions in the level of Ang II–induced CatK, gp91phox, and p‐p38MAPK (Figure 5C). Furthermore, Ang II–induced CatK gene expression was suppressed (P<0.001) by olmesartan as well as by NADPH oxidase inhibitor apocynin and p38 inhibitor SB202190 (Figure 5D). Furthermore, apocynin markedly enhanced MAPK inhibitor–mediated action.
Figure 5.
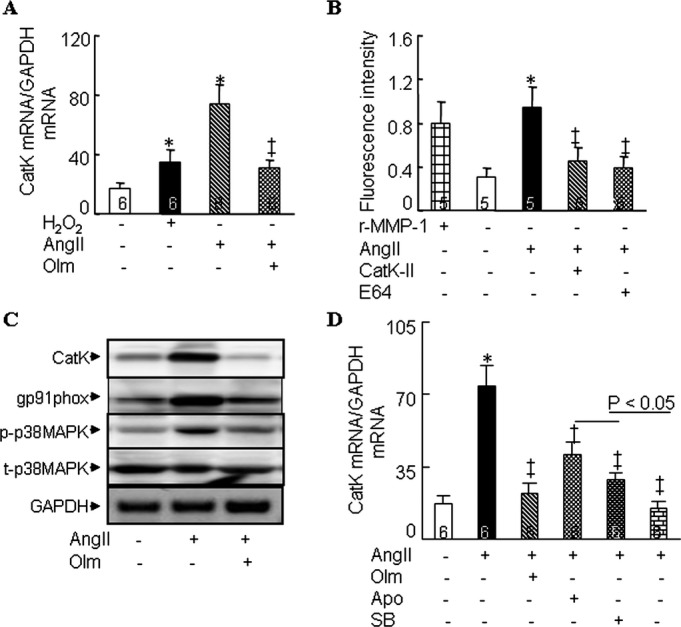
Regulation of CatK expression in cultured rat neonatal atrial myocytes. A, Quantitative real‐time PCR assays showing the expression of CatK mRNA levels in cultured cells treated with and without olmesartan (Olm, 1 μmol/L) in the presence of Ang II (1 μmol/L) or H2O2 (100 μmol/L) for 24 hours. B, Immunofluorescence shows the collagenolytic activity induced by Ang II in cells left untreated or treated with CatK‐II (10 μmol/L) and E64 (10 μmol/L). C, Representative Western blots showing the levels of CatK, gp91phox, p‐p38, and t‐p38 induced by Ang II in cultured neonatal atrial myocytes untreated or treated with Olm (p‐p38MAPK and t‐p38MAPK levels for 30 minutes; CatK and gp91phox levels for 24 hours). D, Quantitative PCR shows Ang II–mediated CatK mRNA expression in cells left untreated or treated with Olm (1 μmol/L), apocynin (Apo, 100 μmol/L), SB202190 (SB, 10 μmol/L), or Apo+SB for 24 hours. Analyzed numbers indicated on related bars. Scale bars indicate 50 μm. Values are expressed as mean±SEM. *P<0.05 vs control; †P<0.01, ‡P<0.001 vs corresponding control. Ang indicates angiotensin II; CatK, cathepsin K; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; rMMP1, recombinant matrix metalloproteinase 1.
Discussion
Plasma Biomarkers of CatK and Collagen Turnover and AF
Several studies have examined the effect of myocardial collagen turnover on the pathogenesis of AF and the outcome of AF ablation.26,30–31 Multiple lines of evidence indicate that CatK is the most abundant and important cysteinyl enzyme synthesized by the cardiovascular system and that it is relevant to cardiovascular disorders, including atherosclerosis,6–8 osteoarthritis,32 and heart failure.27 To the best of our knowledge, this is the first study to show that patients with AF had higher levels of plasma CatK than did control subjects. In agreement with this observation, CatK levels and CatK‐related collagenolytic activity were increased substantially in the atrial tissue of rabbits with tachypacing‐induced AF. Univariate regression analysis showed a positive correlation between plasma ICTP and CatK levels. Moreover, multivariable logistic regression analysis clearly showed that CatK and the I‐PINP:ICTP ratio were independent predictors of AF. Coupled with several recent studies showing increased serum levels of several Cats (S and L) in association with ischemic heart disease,8,14 our findings indicate that elevated plasma levels of CatK with a collagen metabolism–related index (I‐PINP:ICTP ratio) can serve as a novel marker of AF and a noninvasive method of documenting the mechanisms of atrial fibrosis in AF.
Patients initially presenting with PAF often exhibit disease progression and eventually develop PeAF.1 Although the exact pathophysiological mechanisms remain unclear, the persistence of AF is thought to result from atrial remodeling.3,30 Increasing evidence suggests that atrial fibrosis, which has a slower time course than does AF, may be involved in the development and recurrence of AF.1,30 In the current study, CatK and ICTP (a marker of collagen degradation) levels were higher, but the I‐PINP:ICTP ratio was lower, in the PeAF group than in the PAF group. Interestingly, CatK levels were positively correlated with LAD, and patients with PeAF had larger LADs than did those with PAF. These findings imply that—in addition to the duration of arrhythmia—impaired balance between collagen synthesis and metabolism may be one of the initiating factors for AF. However, it should be noted that there are also other initiating factors such as the pulmonary veins for the initiation and maintenance of AF.30
Ang II Inhibition Alleviates Structural Remodeling Related to AF
Many of the Ang II–induced actions in intracellular signaling transduction pathways that regulate gene expression are mediated by the activation and nuclear translocation of MAPKs.33–34 We have shown that Ang II promotes p38MAPK phosphorylation and CatK expression in cultured rat neonatal atrial myocytes and that these effects are reversed with olmesartan. An inhibitor of p38MAPK inhibited Ang II–induced CatK gene expression. In vivo studies, AF rabbits exhibited substantial protein expression of CatK accompanied by increased atrial levels of Ang II, AT1R, and phosphorylated p38MAPK proteins, and these changes were reversed by olmesartan. Thus, AT1R antagonism appears to attenuate CatK expression through the AT1R‐p38MAPK–dependent signaling pathway in the atrial tissues of AF rabbits. Recently, it was reported that the Cat inhibitor E64d prevented hypertensive cardiac remodeling and dysfunction in a Dahl rat model.11 In this study, olmesartan attenuated the duration of AF and atrial fibrosis in tachypacing rabbits. Furthermore, olmesartan reduced the increase in collagenolytic activity in the atrial tissue of AF rabbits. The data from enzyme assays demonstrated that increased collagenolytic activity in both atrial tissues and neonatal atrial myocyte–conditioned medium was attenuated by a CatK‐specific inhibitor as well as a broad‐spectrum Cat inhibitor. Coupled with several recent clinical trials showing the prevention of AF with Ang II inhibition,22,24 these findings indicate that the attenuation of AT1R/p38MAPK‐dependent CatK expression and activity by Ang II inhibition could represent a novel mechanism for the protection of structural remodeling‐related AF, at least in an animal model. In addition, the data from our study and the findings of other researchers35–36 suggest that the inhibition of matrix metalloproteinases also contributes to collagen metabolism in atrial fibrosis. To attain a deeper understanding of the importance of CatK participation in this context, additional experiments using animals with conditional gene knockout may be necessary.
Cross‐talk Between NADPH Oxidase and Ang II/AT1R Signaling Pathway
NADPH oxidase has been implicated in the pathogenesis of cardiovascular disease.37–38 Our data show that the abundance and activity of the NADPH oxidase and its subunit gp91phox markedly increased in the atria from rabbits with AF. In vitro, we observed that both Ang II and H2O2 enhanced CatK expression and activity in cultured neonatal atrial myocytes. Furthermore, NADPH oxidase inhibition with apocynin showed an inhibitory effect on CatK expression in response to Ang II. Because Ang II inhibition attenuated CatS expression and activation via the reduction of NADPH oxidase activity in cultured macrophages,6 we propose that superoxide generation by NADPH oxidase, through “cross‐talk” with the Ang II signaling pathway, can regulate the proteolytic activity of CatK as well as contribute to the pathophysiology of AF. This notion is supported by current and previous findings that AT1R antagonism attenuated atrial and ventricular remodeling and fibrosis associated with NADPH oxidase–derived superoxide production and CatK expression and activation.11,20 It is noteworthy that apocynin enhanced SB202190‐mediated inhibitory effects on CatK expression in neonatal atrial myocytes. This effect raises the possibility that NADPH‐oxidase–derived O2− signaling directly affects CatK expression independent of the AT1R/p38MAPK signaling pathway.
Study Limitations
Several limitations of the present study should be pointed out. First, the small number of participants with and without AF limited the power of this study to prove relationships and differences and limited our power to conduct subgroup analysis by PAF and PeAF. Second, the plasma markers of CatK and collagen turnover are not atrial tissue specific. In addition, patients with cardiomyopathy, myocardial infarction, congenital heart disease, congestive heart failure, or valvular heart diseases and those receiving hemodialysis were excluded. It is unclear how their exclusion may have influenced the present results. Third, despite our efforts to match the control and AF groups, the AF patients were older and included more men. However, the levels of CatK and collagen‐turnover markers were higher in patients with PeAF than in those with PAF, even though the PeAF group was younger and included more men, indicating that age‐ and sex‐associated differences did not influence the interpretation of our findings. In addition, LAD size and the frequency of β‐blocker intake were higher in the AF group than in the control group. Furthermore, a potential impact of these variations on CatK expression was excluded by using multivariable and stepwise logistic models. Fourth, it will also be necessary to investigate cardiovascular events as clinical outcomes in future follow‐up studies. Fifth, further study will be also necessary to evaluate dose‐dependent beneficial effects of olmesartan on CatK expression and atrial remodeling in rabbit model.
Clinical Implication and Conclusions
Our observations show that increased plasma CatK levels are closely linked to the presence of AF and to increased levels of collagen turnover. Therefore, measurement of the circulating CatK level could provide useful information for the evaluation of atrial remodeling in patients with AF. In addition, our findings confirm and extend earlier work19 linking the AT1R/p38MAPK signaling pathway and the activation of NADPH oxidase with AF. We showed that increases in CatK expression and activity were accompanied by local atrial changes in Ang II/AT1R signaling pathway activation in the atrium with AF. The antagonism of AT1R‐mediated beneficial effects on atrial fibrotic remodeling–related AF are likely attributable, at least in part, to the attenuation of CatK expression and activation induced by the AT1R/p38MAPK‐dependent and ‐independent signaling pathway activations.
Sources of Funding
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (24659385 to Dr Cheng; 23390208 to Dr Murohara), by the International Natural Science Foundation of China (grant 81260068), and by the Ministry of Education, Science and Technology of Korea (2012M3A9C6050507).
Disclosures
None.
References
- 1.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011; 91:265-325 [DOI] [PubMed] [Google Scholar]
- 2.Corradi D, Maestri R, Macchi E, Callegari S. The atria: from morphology to function. J Cardiovasc Electrophysiol. 2011; 22:223-235 [DOI] [PubMed] [Google Scholar]
- 3.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002; 54:230-246 [DOI] [PubMed] [Google Scholar]
- 4.Fomovsky GM, Thomopoulos S, Holmes JW. Contribution of extracellular matrix to the mechanical properties of the heart. J Mol Cell Cardiol. 2010; 48:490-496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta. 2000; 1477:98-111 [DOI] [PubMed] [Google Scholar]
- 6.Sasaki T, Kuzuya M, Nakamura K, Cheng XW, Hayashi T, Song H, Hu L, Okumura K, Murohara T, Iguchi A, Sato K. AT1 blockade attenuates atherosclerotic plaque destabilization accompanied by the suppression of cathepsin S activity in apoE‐deficient mice. Atherosclerosis. 2010; 210:430-437 [DOI] [PubMed] [Google Scholar]
- 7.Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation. 2006; 113:98-107 [DOI] [PubMed] [Google Scholar]
- 8.Cheng XW, Huang Z, Kuzuya M, Okumura K, Murohara T. Cysteine protease cathepsins in atherosclerosis‐based vascular disease and its complications. Hypertension. 2011; 58:978-986 [DOI] [PubMed] [Google Scholar]
- 9.Spira D, Stypmann J, Tobin DJ, Petermann I, Mayer C, Hagemann S, Vasiljeva O, Gunther T, Schule R, Peters C, Reinheckel T. Cell type‐specific functions of the lysosomal protease cathepsin L in the heart. J Biol Chem. 2007; 282:37045-37052 [DOI] [PubMed] [Google Scholar]
- 10.Sun M, Chen M, Liu Y, Fukuoka M, Zhou K, Li G, Dawood F, Gramolini A, Liu PP. Cathepsin‐L contributes to cardiac repair and remodelling post‐infarction. Cardiovasc Res. 2010; 89:374-383 [DOI] [PubMed] [Google Scholar]
- 11.Cheng XW, Murohara T, Kuzuya M, Izawa H, Sasaki T, Obata K, Nagata K, Nishizawa T, Kobayashi M, Yamada T, Kim W, Sato K, Shi GP, Okumura K, Yokota M. Superoxide‐dependent cathepsin activation is associated with hypertensive myocardial remodeling and represents a target for angiotensin II type 1 receptor blocker treatment. Am J Pathol. 2008; 173:358-369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation. 2012; 125:1551-1562 [DOI] [PubMed] [Google Scholar]
- 13.Lindholt JS, Erlandsen EJ, Henneberg EW. Cystatin C deficiency is associated with the progression of small abdominal aortic aneurysms. Br J Surg. 2001; 88:1472-1475 [DOI] [PubMed] [Google Scholar]
- 14.Cheng XW, Kikuchi R, Ishii H, Yoshikawa D, Hu L, Takahashi R, Shibata R, Ikeda N, Kuzuya M, Okumura K, Murohara T. Circulating cathepsin K as a potential novel biomarker of coronary artery disease. Atherosclerosis. 2013; 228:211-216 [DOI] [PubMed] [Google Scholar]
- 15.Tsai CT, Lai LP, Lin JL, Chiang FT, Hwang JJ, Ritchie MD, Moore JH, Hsu KL, Tseng CD, Liau CS, Tseng YZ. Renin–angiotensin system gene polymorphisms and atrial fibrillation. Circulation. 2004; 109:1640-1646 [DOI] [PubMed] [Google Scholar]
- 16.Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC, Jr, Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE. Mice with cardiac‐restricted angiotensin‐converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol. 2004; 165:1019-1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsutsumi Y, Matsubara H, Ohkubo N, Mori Y, Nozawa Y, Murasawa S, Kijima K, Maruyama K, Masaki H, Moriguchi Y, Shibasaki Y, Kamihata H, Inada M, Iwasaka T. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circ Res. 1998; 83:1035-1046 [DOI] [PubMed] [Google Scholar]
- 18.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin‐converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing‐induced congestive heart failure. Circulation. 2001; 104:2608-2614 [DOI] [PubMed] [Google Scholar]
- 19.Goette A, Staack T, Rocken C, Arndt M, Geller JC, Huth C, Ansorge S, Klein HU, Lendeckel U. Increased expression of extracellular signal‐regulated kinase and angiotensin‐converting enzyme in human atria during atrial fibrillation. J Am Coll Cardiol. 2000; 35:1669-1677 [DOI] [PubMed] [Google Scholar]
- 20.Cheng XW, Okumura K, Kuzuya M, Jin Z, Nagata K, Obata K, Inoue A, Hirashiki A, Takeshita K, Unno K, Harada K, Shi GP, Yokota M, Murohara T. Mechanism of diastolic stiffening of the failing myocardium and its prevention by angiotensin receptor and calcium channel blockers. J Cardiovasc Pharmacol. 2009; 54:47-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakashima H, Kumagai K, Urata H, Gondo N, Ideishi M, Arakawa K. Angiotensin II antagonist prevents electrical remodeling in atrial fibrillation. Circulation. 2000; 101:2612-2617 [DOI] [PubMed] [Google Scholar]
- 22.Ueng KC, Tsai TP, Yu WC, Tsai CF, Lin MC, Chan KC, Chen CY, Wu DJ, Lin CS, Chen SA. Use of enalapril to facilitate sinus rhythm maintenance after external cardioversion of long‐standing persistent atrial fibrillation. Results of a prospective and controlled study. Eur Heart J. 2003; 24:2090-2098 [DOI] [PubMed] [Google Scholar]
- 23.Arndt M, Lendeckel U, Rocken C, Nepple K, Wolke C, Spiess A, Huth C, Ansorge S, Klein HU, Goette A. Altered expression of ADAMs (A Disintegrin And Metalloproteinase) in fibrillating human atria. Circulation. 2002; 105:720-725 [DOI] [PubMed] [Google Scholar]
- 24.Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, Yusuf S, Connolly SJ. Prevention of atrial fibrillation with angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers: a meta‐analysis. J Am Coll Cardiol. 2005; 45:1832-1839 [DOI] [PubMed] [Google Scholar]
- 25.McNamara RL, Brass LM, Drozda JP, Jr, Go AS, Halperin JL, Kerr CR, Levy S, Malenka DJ, Mittal S, Pelosi F, Jr, Rosenberg Y, Stryer D, Wyse DG, Radford MJ, Goff DC, Jr, Grover FL, Heidenreich PA, Peterson ED, Redberg RF. ACC/AHA key data elements and definitions for measuring the clinical management and outcomes of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Commitee to Develop Data Standards on Atrial Fibrillation). J Am Coll Cardiol. 2004; 44:475-495 [DOI] [PubMed] [Google Scholar]
- 26.Shimano M, Tsuji Y, Inden Y, Kitamura K, Uchikawa T, Harata S, Nattel S, Murohara T. Pioglitazone, a peroxisome proliferator‐activated receptor‐gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm. 2008; 5:451-459 [DOI] [PubMed] [Google Scholar]
- 27.Cheng XW, Obata K, Kuzuya M, Izawa H, Nakamura K, Asai E, Nagasaka T, Saka M, Kimata T, Noda A, Nagata K, Jin H, Shi GP, Iguchi A, Murohara T, Yokota M. Elastolytic cathepsin induction/activation system exists in myocardium and is upregulated in hypertensive heart failure. Hypertension. 2006; 48:979-987 [DOI] [PubMed] [Google Scholar]
- 28.Saygili E, Rana OR, Reuter H, Frank K, Schwinger RH, Muller‐Ehmsen J, Zobel C. Losartan prevents stretch‐induced electrical remodeling in cultured atrial neonatal myocytes. Am J Physiol Heart Circ Physiol. 2007; 292:H2898-H2905 [DOI] [PubMed] [Google Scholar]
- 29.Cheng XW, Kuzuya M, Nakamura K, Di Q, Liu Z, Sasaki T, Kanda S, Jin H, Shi GP, Murohara T, Yokota M, Iguchi A. Localization of cysteine protease, cathepsin S, to the surface of vascular smooth muscle cells by association with integrin alphanubeta3. Am J Pathol. 2006; 168:685-694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kallergis EM, Manios EG, Kanoupakis EM, Mavrakis HE, Arfanakis DA, Maliaraki NE, Lathourakis CE, Chlouverakis GI, Vardas PE. Extracellular matrix alterations in patients with paroxysmal and persistent atrial fibrillation: biochemical assessment of collagen type‐I turnover. J Am Coll Cardiol. 2008; 52:211-215 [DOI] [PubMed] [Google Scholar]
- 31.Okumura Y, Watanabe I, Nakai T, Ohkubo K, Kofune T, Kofune M, Nagashima K, Mano H, Sonoda K, Kasamaki Y, Hirayama A. Impact of biomarkers of inflammation and extracellular matrix turnover on the outcome of atrial fibrillation ablation: importance of matrix metalloproteinase‐2 as a predictor of atrial fibrillation recurrence. J Cardiovasc Electrophysiol. 2010; 22:987-993 [DOI] [PubMed] [Google Scholar]
- 32.Kozawa E, Nishida Y, Cheng XW, Urakawa H, Arai E, Futamura N, Shi GP, Kuzuya M, Hu L, Sasaki T, Ishiguro N. Osteoarthritic change is delayed in a cathepsin K knockout mouse model of osteoarthritis. Arthritis Rheum. 2012; 64:54-64 [DOI] [PubMed] [Google Scholar]
- 33.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Mitogen‐activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension. 2008; 52:679-686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee KW, Everett TH, IV, Rahmutula D, Guerra JM, Wilson E, Ding C, Olgin JE. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006; 114:1703-1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CL, Huang SK, Lin JL, Lai LP, Lai SC, Liu CW, Chen WC, Wen CH, Lin CS. Upregulation of matrix metalloproteinase‐9 and tissue inhibitors of metalloproteinases in rapid atrial pacing‐induced atrial fibrillation. J Mol Cell Cardiol. 2008; 45:742-753 [DOI] [PubMed] [Google Scholar]
- 36.Goette A, Bukowska A, Lendeckel U, Erxleben M, Hammwohner M, Strugala D, Pfeiffenberger J, Rohl FW, Huth C, Ebert MP, Klein HU, Rocken C. Angiotensin II receptor blockade reduces tachycardia‐induced atrial adhesion molecule expression. Circulation. 2008; 117:732-742 [DOI] [PubMed] [Google Scholar]
- 37.Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose‐induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003; 42:206-212 [DOI] [PubMed] [Google Scholar]
- 38.Reilly SN, Jayaram R, Nahar K, Antoniades C, Verheule S, Channon KM, Alp NJ, Schotten U, Casadei B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2012; 124:1107-1117 [DOI] [PubMed] [Google Scholar]