

Abstract
Background
No prospective studies exist on the relationship between change in periodontal clinical and microbiological status and progression of carotid atherosclerosis.
Methods and Results
The Oral Infections and Vascular Disease Epidemiology Study examined 420 participants at baseline (68±8 years old) and follow‐up. Over a 3‐year median follow‐up time, clinical probing depth (PD) measurements were made at 75 766 periodontal sites, and 5008 subgingival samples were collected from dentate participants (average of 7 samples/subject per visit over 2 visits) and quantitatively assessed for 11 known periodontal bacterial species by DNA‐DNA checkerboard hybridization. Common carotid artery intima‐medial thickness (CCA‐IMT) was measured using high‐resolution ultrasound. In 2 separate analyses, change in periodontal status (follow‐up to baseline), defined as (1) longitudinal change in the extent of sites with a ≥3‐mm probing depth (Δ%PD≥3) and (2) longitudinal change in the relative predominance of bacteria causative of periodontal disease over other bacteria in the subgingival plaque (Δetiologic dominance), was regressed on longitudinal CCA‐IMT progression adjusting for age, sex, race/ethnicity, diabetes, smoking status, education, body mass index, systolic blood pressure, and low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol. Mean (SE) CCA‐IMT increased during follow‐up by 0.139±0.008 mm. Longitudinal IMT progression attenuated with improvement in clinical or microbial periodontal status. Mean CCA‐IMT progression varied inversely across quartiles of longitudinal improvement in clinical periodontal status (Δ%PD≥3) by 0.18 (0.02), 0.16 (0.01), 0.14 (0.01), and 0.07 (0.01) mm (P for trend<0.0001). Likewise, mean CCA‐IMT increased by 0.20 (0.02), 0.18 (0.02), 0.15 (0.02), and 0.12 (0.02) mm (P<0.0001) across quartiles of longitudinal improvement in periodontal microbial status (Δetiologic dominance).
Conclusion
Longitudinal improvement in clinical and microbial periodontal status is related to a decreased rate of carotid artery IMT progression at 3‐year average follow‐up.
Keywords: atherosclerosis, infection, inflammation, periodontal, progression
Introduction
Studies have linked periodontal disease (clinical manifestation of chronic periodontal infections and inflammation) to both cardiovascular disease (CVD) and atherosclerosis.1–5 The clinical evidence was extended to serological studies linking elevated periodontal bacteria antibody titers to atherosclerotic vascular disease,6–10 and we have reported cross‐sectional evidence of greater carotid intima‐media thickness with increasing proportion of “etiologic” periodontal bacteria in the subgingival plaque.11 One unanswered question is evaluating the relationship between temporal change in chronic periodontal infections levels and subclinical atherosclerosis progression. No prospective studies exist on the parallel evolution of chronic low‐grade infections, including periodontal infections, and subclinical vascular disease. Prospective studies of this nature are important for establishing or refuting causality, thus, filling a critical gap, as recently summarized in an American Heart Association statement regarding the association between periodontal disease and atherosclerotic vascular disease.12
The Oral Infections and Vascular Disease Epidemiology Study (INVEST) was specifically designed to study the hypothesis that periodontal infections predispose to accelerated progression of carotid atherosclerosis and subsequent CVD. In addition to clinical assessments of periodontal status, the study examined the level of a number of bacterial species in the subgingival environment adjacent to selected teeth, namely, those considered to be etiologically related to periodontitis and a selection of others as controls. In this report, we investigated whether changes in periodontal status, assessed clinically and microbiologically, were associated with progression of carotid atherosclerosis longitudinally. Our a priori hypothesis was that improvement in periodontal status and reduction in the proportion of “etiologic” periodontal bacteria in the subgingival plaque would be related to slower intima‐medial thickness (IMT) progression, whereas worsening periodontal infections would increase IMT progression.
Methods
INVEST is a randomly sampled prospective cohort study investigating the relationship between oral infections, carotid atherosclerosis, and CVD in participants residing between 145th Street and 218th Street in Manhattan.11,13 Hispanics, blacks, and whites live together in this area with similar access to medical care. The selection process was derived from the Northern Manhattan Study.14
INVEST baseline eligibility criteria were: (1) resident >3 months of zip codes 10031, 10032, 10033, 10034, or 10040; (2) contacted by random‐digit dialing; (3) age ≥55 years; (4) no history of stroke, myocardial infarction, or chronic inflammatory conditions; and (5) ambulatory. This protocol was approved by the Columbia University and University of Miami institutional review boards. All participants provided written informed consent.
Of 1056 participants enrolled at baseline, 842 were dentate, and 626 were alive and attended a follow‐up visit (81% of surviving dentate participants). Two hundred six subjects were excluded from the present analysis for antibiotic prophylaxis requirements (n=81), missing baseline cardiovascular data (n=63), logistical reasons (n=47), or no longer being dentate (n=15), a necessary condition for periodontal and microbiological assessments. Thus, we include 420 for analyses of longitudinal change in clinical periodontal status and progression of IMT, representing 79% of dentate subjects not requiring premedication, in accordance with American Heart Association criteria.15 Among these 420, 364 (87%) also had longitudinal bacteriological data.
Oral Examination
Participants were examined by calibrated dental examiners.16 Briefly, full‐mouth periodontal assessment for all teeth present included probing depth (PD) and attachment loss (AL) measurements (both in millimeters) at 6 sites/tooth (mesiobuccal, midbuccal, distobuccal, mesiolingual, midlingual, and distolingual) using a UNC‐15 manual probe (HuFriedy, Chicago, IL). Intraexaminer reliability studies on 14 participants yielded intraclass correlations of 0.97 and 0.94 between repeat measures of mean PD or AL, respectively. Mean absolute differences between repeat examinations for mean PD or AL were 0.09 and 0.17 mm.
Subgingival Plaque Collection and Processing
Up to 8 subgingival bacterial plaque samples were collected per participant (65% contributed 8 biofilms and 91% contributed ≥4 biofilms) from the 2 most posterior teeth (mesiolingual in maxilla and mesiobuccal in mandible) in each quadrant to avoid biased collection of samples from the most diseased sites. Thus, we report on 5008 plaque samples over 2 visits. At baseline and follow‐up, digoxigenin‐labeled whole‐genomic probes were used to assess microbial levels for 11 bacterial strains selected to include species considered to be (1) etiologically linked to periodontal disease or frequently encountered in pathological periodontal conditions17–18 (Aggregatibacter actinomycetemcomitans [ATCC 43718], Porphyromonas gingivalis [ATCC 33277], Tannerella forsythia [ATCC 43037], Treponema denticola [ATCC 35404]); (2) putative periodontal pathogens17 (Prevotella intermedia [ATCC 25611], Fusobacterium nucleatum [ATCC 10953], and Micromonas micros [ATCC 33270], Campylobacter rectus [ATCC 33238], and Eikenella corrodens [ATCC 23834]); and (3) primarily associated with gingivitis or healthy periodontal conditions (Veillonella parvula [ATCC 10790] and Actinomyces naeslundii [ATCC 49340]), using checkerboard DNA–DNA hybridization11,17 in all 5008 subgingival plaque samples.
Gingival Crevicular Fluid Interleukin‐1β
Methylcellulose filter paper strips (IDE, Amityville, New York) were inserted into the mesiobuccal crevice/pocket of each quadrant's most posterior tooth until reaching its base, and kept in place for 30 seconds. Gingival crevicular fluid (GCF) volume determinations were performed using a calibrated Periotron 6000. Interleukin‐1β (IL‐1β) levels (in picograms per milliliter) were determined by ELISA (Multikine Kit; Cistron Biotechnology, Pine Brook, NJ.
Carotid Ultrasound
Bilateral carotid arteries were scanned longitudinally in the common carotid artery (CCA), internal carotid artery and bifurcation, as described.11 The optimal angle of insonation was used to measure the CCA‐IMT in the near and far walls extending from 10 mm distal to the flow divider and stopping at 20 mm below the flow divider. In addition, to minimize measurement error, a rigorous follow‐up protocol involved (1) identification of internal landmarks, (2) utilization of a carotid mask to maximize superimposable visualizations at both visits, and (3) utilization of the same ultrasound machine at both visits.
Carotid IMT measurements were performed off‐line using IMAGE‐Pro V.5.1 (Microsoft) image analysis software, outside of plaque by 1 reader.11 In accordance with the third Manheim consensus,19 we selected digital frames corresponding to the minimum observed arterial diameter during the cardiac cycle, which represents diastolic phase. The reader visualized blood‐intima and media‐adventitia boundaries with a mouse‐controlled tracer (caliper) within the carotid segment. The measurement algorithm takes a minimum of 100 measurements within the 10‐mm segment. Bilateral IMT values were averaged across the near and far walls of either (1) the maximal CCA‐IMT or (2) all 12 sites (C‐IMT), that is, the maximal CCA‐IMT, the maximal bifurcation IMT, and the maximal internal carotid artery IMT.
Interultrasonographer reliability studies yielded a mean absolute difference between repeat measures of 0.04 and 0.05 mm for C‐IMT and CCA‐IMT, respectively; intraclass correlation coefficients were 0.77 and 0.75 and consistent with previous findings from longitudinal studies of IMT change.20 Ultrasound examiners were unaware of periodontal status at IMT scanning and reading. Similarly, periodontal laboratory technicians were blinded to risk factors and IMT results.
Risk Factor Assessment
Subjects were interviewed regarding sociodemographic characteristics, cardiovascular risk factors, and other medical conditions using standardized questions.16 Research assistants measured height and weight with calibrated scales, blood pressure (2 measurements average) using a calibrated sphygmomanometer (Omron).13 Fasting glucose and lipids were measured.21 Diabetes mellitus was defined by a fasting glucose ≥126 mg/dL (7.7 mmol/L) or self‐report of physician‐diagnosed diabetes. Smoking was assessed both categorically (current/former/never) and continuously (pack‐years).16
Statistical Analysis
Analyses were performed using SAS 9.2. Periodontal status was defined on the basis of clinical and bacterial periodontal measures as follows.
Clinical periodontal status was defined by either (1) full‐mouth mean probing depth (MPD) values or (2) the percentage of measured sites with PD≥3 mm (%PD≥3). These variables were selected a priori on the basis of previous research demonstrating their strong correlation with underlying bacterial profiles22 and clinical signs of inflammation.23 We also considered mean attachment loss and the percentage of sites with AL≥5 mm (%AL≥5) to assess the sensitivity of associations to commonly used epidemiological definitions of periodontitis.24
For each microbial species, bacterial counts were natural log–transformed, averaged within the mouth, and standardized by dividing these values by the natural log–transformed population standard deviation. As a result, 1 standard deviation on the natural log scale was equivalent across microbes.11 Thus, for each person and both evaluations, and based on a priori evidence at baseline (ie, the consensus of the 1996 World Workshop in Periodontics25 and Socransky's Red Complex,26) we operationalized periodontal bacterial data in 3 ways according to the cumulative sum of colonization levels as follows: (1) combined levels of P gingivalis, T forsythia, A actinomycetemcomitans, and T denticola were defined as “etiologic bacterial burden” (EB); (2) combined levels of C rectus, E corodens, F nucleatum, M micros, and P intermedia were defined as “putative bacterial burden”; and (3) combined levels of A naeslundii and V parvula were defined as “health‐associated” bacterial burden as previously,11,22 with the last 2 groups being used as controls. Finally, etiologic dominance (ED) was defined as the relative predominance of the etiologic bacterial group in the studied ecological niche by dividing EB by the cumulative sum of all 3 aforementioned bacterial burdens (EB, putative bacterial burden, and health‐associated bacterial burden). Etiologic dominance informs the specific effect of etiologic species on IMT change via division, rather than statistical adjustment.
Both baseline and longitudinal change in the aforementioned periodontal variables were considered independent variables in statistical models. The dependent variable was defined as change in either the CCA‐IMT (ΔCCA‐IMT) or the C‐IMT (ΔC‐IMT). With both measurements providing similar conclusions, CCA‐IMT results are reported.
Change in either independent or dependent variables were calculated as follow‐up−baseline (positive value indicates worsening, negative value indicates improvement). The Δ symbol signifies longitudinal change.
Multivariable linear regression models assessed the association between quartiles of either baseline or change in periodontal status and IMT change, adjusting for age, sex, race/ethnicity, diabetes, smoking status, education, body mass index, systolic blood pressure, low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol. We opted to not include results arising from models adjusted for baseline IMT to avoid bias27–28 in the regression estimates that might exaggerate any observed associations. Analyses adjusting for baseline IMT did not change results. Tests for linear trend were conducted by modeling the 3‐level ordinal categorical exposure variable as a linear term.
Results
The mean±SD age of participants was 68±8 years at baseline; 62% were female, 60% Hispanic, 20% black, 19% white, and 1% other (Table 1). The median follow‐up time was 3.1 years (range, 1.8 to 7.2 years), and 94% were reexamined within 2 to 4 years.
Table 1.
General Characteristics According to Quartiles of Progression in Periodontal Pockets (Δ%PD≥3)
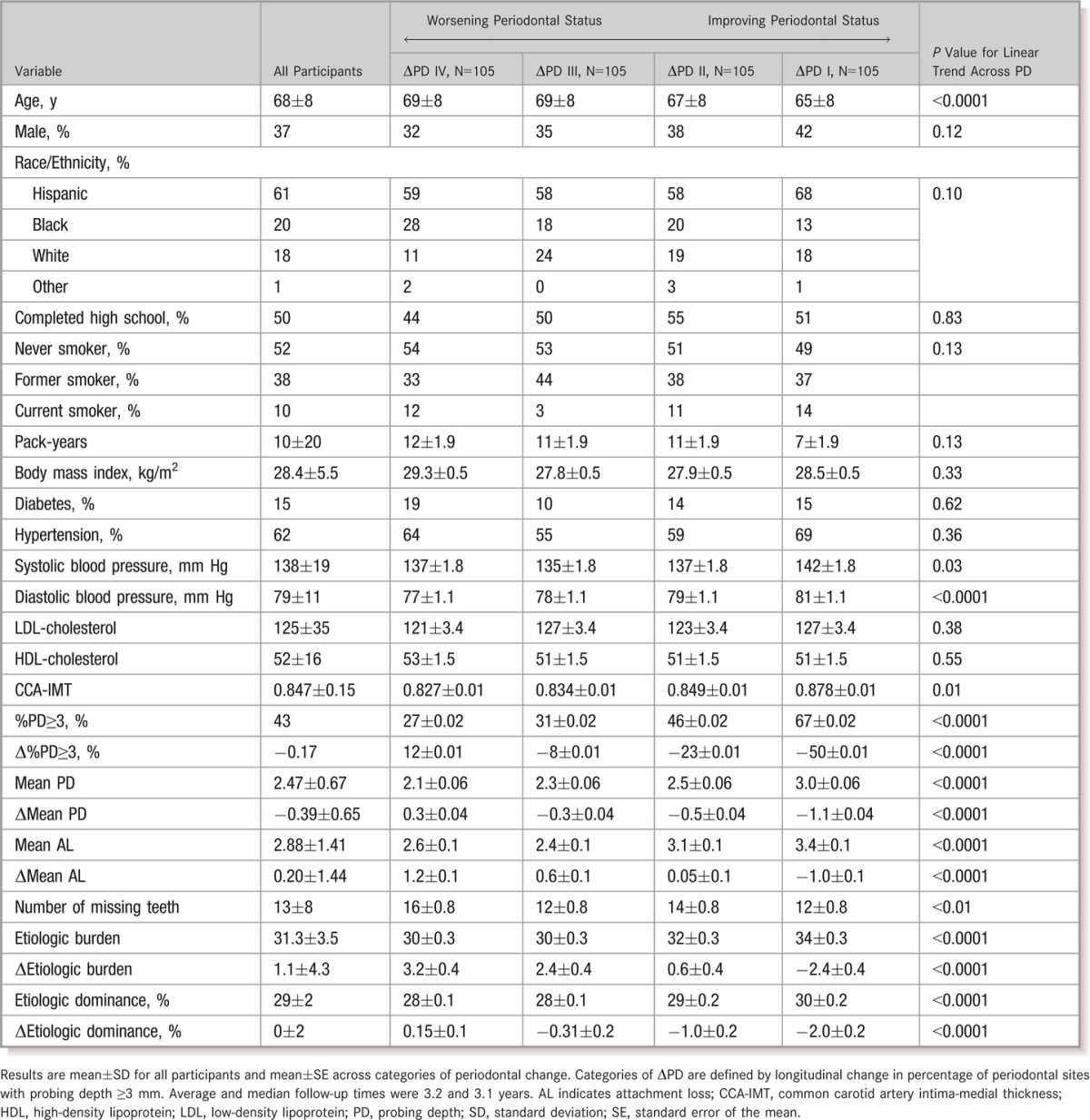
Participants with greater levels of clinical periodontal disease at baseline generally experienced more improvement in periodontal health longitudinally, reflecting a strong inverse correlation between baseline MPD and ΔMPD (r=−0.57, P<0.0001) or between baseline %PD≥3 and Δ%PD≥3 (r=−0.64, P<0.0001). Baseline bacterial EB and ΔEB were also strongly inversely correlated (r=−0.86, P<0.0001). Change in MPD was correlated with change in EB (r=0.37, P<0.0001).
Mean baseline CCA‐IMT was 0.847±0.154 mm (range, 0.52 to 1.45 mm). Mean CCA‐IMT increased by 0.139±0.170 mm (range, −0.533 to 0.516 mm). The annualized rate of CCA‐IMT change was 0.044±0.054 mm/year. Baseline CCA‐IMT was positively correlated with follow‐up CCA‐IMT (0.34, P<0.0001) but inversely correlated with ΔCCA‐IMT (−0.62, P<0.0001).
General characteristics according to categories of periodontal change are in Table 1. Primary correlates of prospective decline in periodontal status were greater age, lower blood pressure, and corresponding changes in other periodontal measures. Correlates of ΔED were similar (data not shown).
Validation of A Priori Concepts Regarding Periodontal Bacteria
Periodontal bacterial levels were related to clinical periodontal status at baseline.11 Further, Figure 1 shows that change in etiologic dominance (ΔED) was directly related to changes in both MPD and IL‐1β in the GCF. Similarly, mean change values (Δ%PD≥3) across quartiles of change of ΔED bacterial values were −33%, −17%, −6%, and −5% (P for trend<0.0001).
Figure 1.
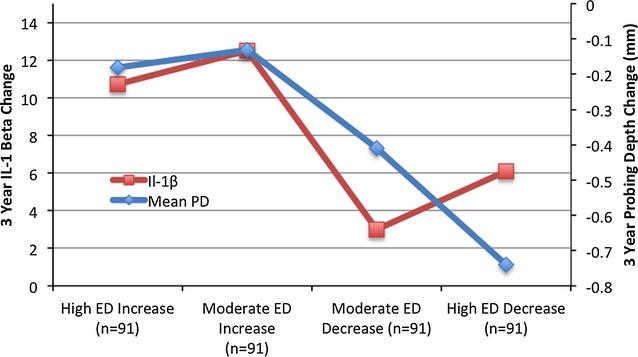
Association between 3‐year bacterial etiologic dominance change and concurrent change in gingival IL‐1β and periodontal probing depth adjusted for age, sex, race/ethnicity, diabetes, smoking status, education, body mass index, systolic blood pressure, LDL‐cholesterol, and HDL‐cholesterol. Mean probing depth P for linear trend<0.0001; IL‐1β P for linear trend=0.12. ED indicates etiologic dominance; HDL, high‐density lipoprotein; IL‐1β, interleukin‐1β; LDL, low‐density lipoprotein; PD, probing depth.
Change in Clinical Periodontal Status and Atherosclerotic Progression
Longitudinal change in either mean PD or AL was positively associated with IMT change; the difference in IMT between participants with the greatest worsening in PD versus those with the greatest improvement was 0.072 mm, whereas this difference was 0.049 mm when comparing worsening versus improving AL (Table 2). Results were stronger and more linear when modeling Δ%PD≥3 as the exposure (Figure 2A). The extent of longitudinal attachment loss was also related, although not statistically, to IMT progression: multivariable‐adjusted mean CCA‐IMT change across quartiles of Δ%AL≥5 was 0.094, 0.146, 0.158, and 0.145 mm (P for linear trend=0.09).
Table 2.
Longitudinal Change in Common Carotid Artery Intima‐Media Thickness Across Quartiles of Longitudinal Change in Clinical Periodontal Probing Depth or Attachment Loss
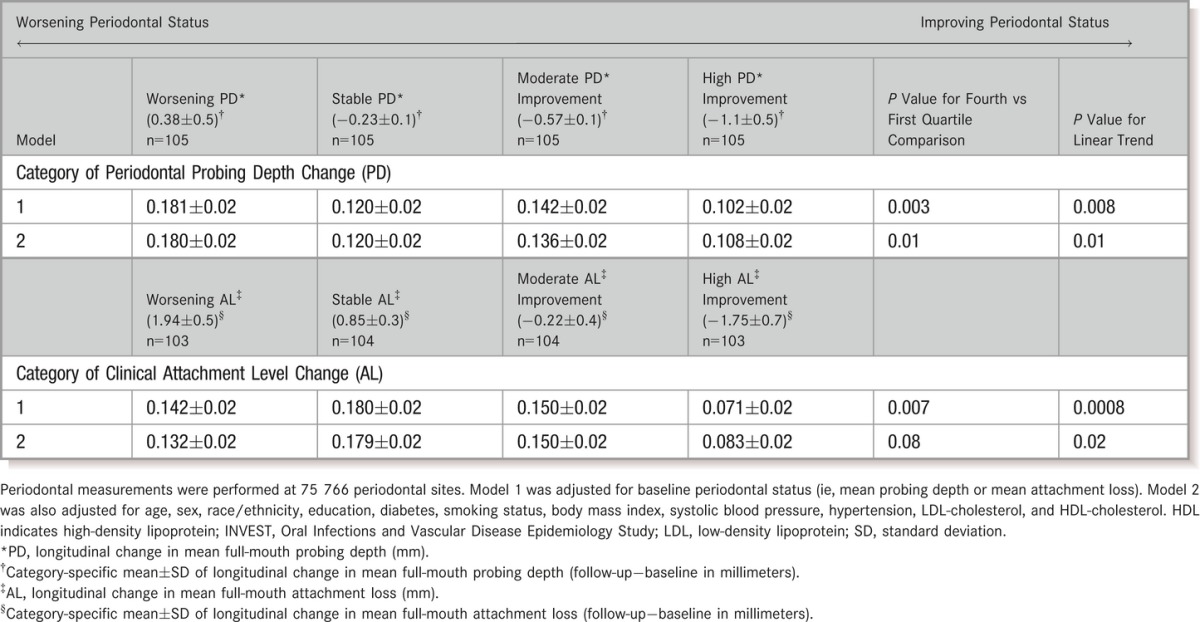
Figure 2.
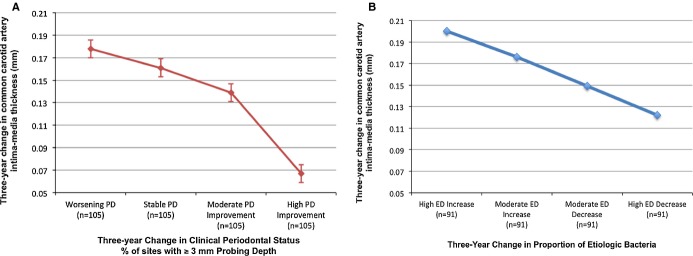
Relationship between longitudinal change in periodontal status and change in common carotid artery intima‐media thickness. Periodontal status (A) defined clinically, according to percentage of sites with ≥3‐mm probing depth (P for linear trend<0.0001); or (B) defined microbiologically—etiologic dominance (ED)—as the proportion of oral bacteria believed to be periodontally etiologic (P for linear trend<0.01). Both were adjusted for age, sex, race/ethnicity, diabetes, smoking status, education, body mass index, systolic blood pressure, LDL‐cholesterol, and HDL‐cholesterol. HDL indicates high‐density lipoprotein; LDL, low‐density lipoprotein; PD, probing depth.
Adjusting for change in periodontal status, baseline levels of clinical periodontal disease were unrelated to IMT change. Mean±SE ΔCCA‐IMT values across quartiles of increasing baseline MPD were 0.124±0.02, 0.169±0.02, 0.124±0.02, and 0.128±0.02 (P for linear trend=0.64). Mean ΔCCA‐IMT changes across quartiles of increasing baseline mean AL were 0.147, 0.122, 0.117, and 0.158 (P for linear trend=0.61). ΔCCA‐IMT values across baseline quartiles of %AL≥5 were: 0.142, 0.101, 0.144, and 0.169 mm (P for linear trend=0.07).
Change in Periodontal Bacteria and Atherosclerotic Progression
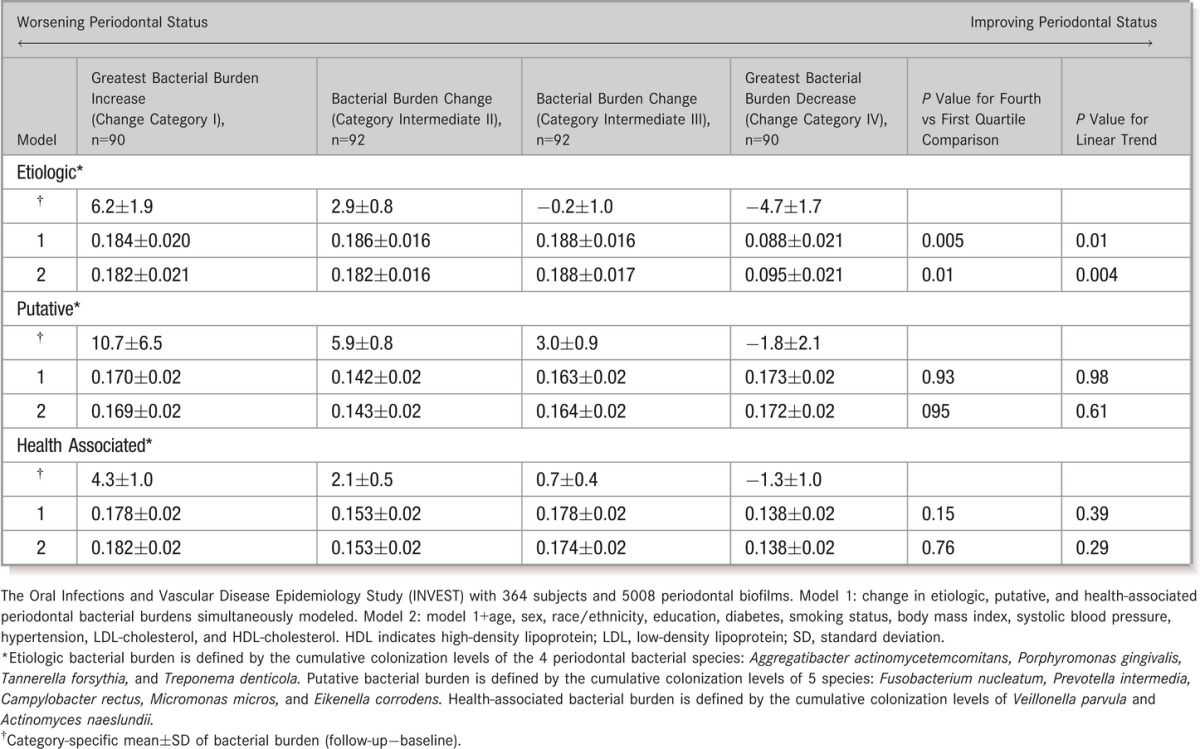
Compared with participants who experienced decreases in etiologic bacterial levels, those who experienced little change or increased burden of etiologic bacteria during follow‐up also experienced greater progression of CCA‐IMT (Table 3). This association was not observed for changes in levels of either putative or health‐associated burden, adjusting for changes in etiologic burden (Table 3). In analyses assessing the association between change in etiologic dominance and IMT progression, mean carotid IMT progressed in a direct and dose‐responsive manner, with increasing dominance of the etiologic bacterial group during follow‐up (Figure 2B). The results for ΔED were unchanged when further adjusting for GCF IL‐1β or MPD. Interestingly, in the model mutually adjusting for microbiological (ΔED) and clinical (ΔMPD) status, the ΔCCA‐IMT trends across quartiles of ΔMPD were attenuated: 0.190, 0.159, 0.174, and 0.152 mm (P for trend=0.47), with only the bacterial exposure (ΔED) remaining statistically significant. Adjustment for follow‐up time did not change results in either the clinical or bacteriological periodontal models related to atherosclerotic progression.
Table 3.
Longitudinal Change in Common Carotid Artery Intima‐Media Thickness Across Quartiles of Longitudinal Change in Etiologic, Putative, and Health‐Associated Periodontal Bacterial Colonization Patterns
Baseline hypertension was the only traditional CVD risk factor that predicted longitudinal IMT change but in the direction opposite that expected; ΔCCA‐IMT was 0.040 mm greater among normotensive participants compared with hypertensive participants (P=0.03). However, adjustment for longitudinal change in systolic blood pressure removed this association, consistent with the observation that systolic blood pressure decreased by an average of 11 mm Hg among baseline hypertensives but increased by 4 mm Hg among normotensives (P<0.0001). Longitudinal change in systolic blood pressure was positively, though not statistically, associated with ΔCCA‐IMT.
Discussion
We had previously reported that higher levels of etiologic bacteria were cross sectionally associated with thicker carotid IMT at baseline.11 We now report longitudinal change in periodontal status to be concurrent with longitudinal carotid artery IMT progression over an average period of 3 years: relative to participants who experienced deterioration in their periodontal status, those who experienced improvements in periodontal status (clinical and bacteriological) also realized slower CCA‐IMT progression during follow‐up. These findings held after multivariable CVD risk factor adjustments and across multiple definitions of periodontal status.
The observed associations were strongest for periodontal exposures defined using either (1) etiologic periodontal bacterial levels or (2) clinical periodontal measures previously demonstrated to have strong associations with etiologic bacterial colonization in this population.22 We found the relative predominance of bacteria traditionally considered causally related to periodontal disease to be most closely linked to atherosclerotic progression. These bacterial species, strongly associated with baseline clinical periodontal disease,23 are now associated with change in both clinical periodontal status and subgingival inflammation (GCF interleukin‐1β).
In addition to measuring established etiologic bacterial species, we also measured 7 species serving as internal controls to minimize the potential for confounding by healthy behaviors. Because participants who brush and floss more regularly have lower absolute colonization levels of all species11 (etiologic or otherwise) and these participants are also more likely to engage in CVD health‐promoting behaviors,11 confounding would be possible. The specificity of the relationship to the periodontal etiologic fraction of bacteria significantly alleviates that concern, adding credence to our findings.
The clinical periodontal definitions that demonstrated the strongest associations with atherosclerotic changes were consistent with our a priori expectations and methodological investigations showing low‐threshold periodontal measures to correlate more strongly with etiologic periodontal bacteria22 and inflammation.23 Of particular note is the dose‐responsive relationship between IMT progression and change in the extent of ≥3‐mm periodontal pockets, a probing depth threshold generally regarded as compatible with periodontal health. This suggests that future intervention studies might consider defining periodontal eligibility criteria using low‐threshold (“preclinical”) characteristics. Our results are consistent with Pussinen et al's29 report that serum antibody levels to periodontal bacteria (and thus systemic translation of local infection) are more related to bacterial levels than overt clinical disease. Hence, as an exposure of significance for systemic disease, “preclinical” periodontal disease cannot be ignored.30
In analyses jointly modeling bacterial exposures and either clinical exposures or GCF IL‐1β levels, only bacterial exposures remained statistically significantly associated with IMT progression, identifying bacterial exposure constructs as more systemically relevant.
The potential for oral microbes to contribute to atherogenesis is biologically plausible (for reviews, see references 31–32). Oral bacterial species can induce immune system activation characterized by chronic elevations in systemic inflammatory markers,11,33–34 possibly resulting from bacteremias of oral origin,35 which may in turn initiate or exacerbate the inflammatory aspect of atherogenesis,36 as in animal models.37–39 In humans, anti‐infective periodontal therapy improves endothelial function in as little as 2 months.40
We have found an ≈0.1 mm difference in IMT change among participants with deteriorating versus improving periodontal health during a relatively short time (ie, 3 years). A baseline difference of this magnitude has been found relevant by Salonen and Salonen,41 O'Leary et al,42 and Polak and colleagues43 in predicting CVD. In progression studies, Hodis et al44 reported that a 0.03 mm/year increase in carotid IMT (equivalent to a ≈0.1 mm progression over 3 years, as here) is associated with a 2.3‐fold increased risk for coronary events. Interventional studies with statins reported as clinically significant a difference of 0.0082 mm/year in carotid IMT between annualized progression rates with placebo (0.067 mm) and pravastatin (0.059).45 Thus, our findings of ≈0.1‐mm difference in progression during an average follow‐up time of 3 years appear to meet the threshold of clinical significance.
The lack of any strong associations between baseline periodontal status or CVD risk factors and IMT change may reflect that periodontal status changed substantially for many (Table 1). Similar trends were observed for blood pressure (see Results). It is likely that as a consequence of receiving examination results during baseline enrollment, participants sought advice from health professionals (dentists for periodontal findings and physicians for hypertension findings) who could have then intervened on risk factors differentially according to baseline indication (ie, levels of periodontal disease or blood pressure).
We measured only 11 bacterial species, whereas hundreds of species are known to colonize the subgingival space.46 The 4 species presently defined as “etiologic” were selected a priori at the time of baseline enrollment on the basis of contemporary scientific evidence.25–26 We believe that from an epidemiological standpoint, the combined sum of these species remains a robust etiologic meter of periodontal pathology, either as directly causal or correlate of yet‐unmeasured pathogenic bacterial species.22–23 Nevertheless, additional pathogens could conceivably modify the overall relationship.
Our results should be interpreted within the context and methodology of our randomly selected cohort and the timing of our exposure and outcome assessments. Our IMT progression rates are slightly higher than what has been previously reported.47 This is likely explained by the older age (≈10 years older) and higher prevalence of diabetes (≈10% to 15% higher prevalence) in our population relative to studies included in a previous meta‐analysis.47 Although we did not use explicit cardiac gating, we identified the end‐diastole frames for measurement based on lumen diameter in accordance with the Mannheim consensus,19 and we only present results from the CCA as previously recommended.47 Our high follow‐up rate (80%) for an observational cohort with a mean age of 69 years at baseline is a strength, although the possibility for bias induced by loss to follow‐up cannot be completely dismissed. However, it is most likely that, if anything, loss to follow‐up would bias our findings toward the null because it is usually the least healthy individuals (ie, greater baseline periodontal disease levels and higher IMT progression rates) who do not return for in‐person exams. Finally, as longitudinal change in exposure and outcome were assessed concurrently, the potential issue of lag time remains unresolved, and we must await further follow‐up of INVEST for clarification and translation to clinical events.
In summary, we report the first evidence that improvement in periodontal status—defined both clinically and microbiologically—is associated with less progression in carotid atherosclerosis in a randomly selected population‐based sample of men and women. These findings were observed during a relatively short period, strengthening the hypothesis that accelerated atherosclerotic progression is a mechanistic explanation for previous reports linking periodontal disease and clinical CVD. Because they were observed in a population setting, they also emphasize the importance of primary periodontal care as a possible preventive health measure. Future randomized clinical trials are required to definitively determine whether anti‐infective periodontal interventions can reduce atherosclerotic progression and prevent subsequent clinical cardiovascular events, and we provide a potential target and timeline for so doing. In INVEST, carotid atherosclerosis progression paralleled progression in both bacterial periodontal profiles and clinical periodontal measures, in the dual directions of worsening and improvement, providing change‐on‐change population‐based results equivalent to a quasi‐experimental design.48
Sources of Funding
This research is supported by NIH grants R01 DE‐13094 (to Dr Desvarieux) and NS‐29993 (to Dr Sacco). Dr Demmer is also supported by R00 DE‐018739, and Dr Rundek is also supported by NINDS R01 NS‐047655, all from the NIH. This research was also partially supported by an INSERM Chair of Excellence from the Institut National de la Santé et de la Recherche Médicale (INSERM) and a Chair in Chronic Disease, École des Hautes Études en Santé Publique, France (both to Dr Desvarieux) and a Mayo Chair Endowment, School of Public Health, University of Minnesota (to Dr Jacobs).
Disclosures
None.
Acknowledgments
We thank INVEST staff George Loo, Mariana Cukier, Yira Flores, Publio Silfa, Kenia Martinez, Shantanu Lal, Bernadette Boden‐Albala, and Janet DeRosa for their devoted patient care; Miriam Herrera‐Abreu, Romi Celenti and Jun Yang for the laboratory analysis of the dental plaque samples; and most importantly the participants. Participants were seen at the Columbia University General Clinical Research Center, NIH grants 1UL1RR024156 and UL1 TR000040.
References
- 1.Wu T, Trevisan M, Genco RJ, Dorn JP, Falkner KL, Sempos CT. Periodontal disease and risk of cerebrovascular disease: the first national health and nutrition examination survey and its follow‐up study. Arch Intern Med. 2000; 160:2749-2755 [DOI] [PubMed] [Google Scholar]
- 2.DeStefano F, Anda RF, Kahn HS, Williamson DF, Russell CM. Dental disease and risk of coronary heart disease and mortality. BMJ. 1993; 306:688-691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck JD, Elter JR, Heiss G, Couper D, Mauriello SM, Offenbacher S. Relationship of periodontal disease to carotid artery intima‐media wall thickness: the atherosclerosis risk in communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2001; 21:1816-1822 [DOI] [PubMed] [Google Scholar]
- 4.Jimenez M, Krall EA, Garcia RI, Vokonas PS, Dietrich T. Periodontitis and incidence of cerebrovascular disease in men. Ann Neurol. 2009; 66:505-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietrich T, Jimenez M, Krall Kaye EA, Vokonas PS, Garcia RI. Age‐dependent associations between chronic periodontitis/edentulism and risk of coronary heart disease. Circulation. 2008; 117:1668-1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pussinen PJ, Nyyssonen K, Alfthan G, Salonen R, Laukkanen JA, Salonen JT. Serum antibody levels to actinobacillus actinomycetemcomitans predict the risk for coronary heart disease. Arterioscler Thromb Vasc Biol. 2005; 25:833-838 [DOI] [PubMed] [Google Scholar]
- 7.Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2007; 193:222-228 [DOI] [PubMed] [Google Scholar]
- 8.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007; 27:1433-1439 [DOI] [PubMed] [Google Scholar]
- 9.Beck JD, Eke P, Lin D, Madianos P, Couper D, Moss K, Elter J, Heiss G, Offenbacher S. Associations between IgG antibody to oral organisms and carotid intima‐medial thickness in community‐dwelling adults. Atherosclerosis. 2005; 183:342-348 [DOI] [PubMed] [Google Scholar]
- 10.Beck JD, Eke P, Heiss G, Madianos P, Couper D, Lin D, Moss K, Elter J, Offenbacher S. Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation. 2005; 112:19-24 [DOI] [PubMed] [Google Scholar]
- 11.Desvarieux M, Demmer RT, Rundek T, Boden‐Albala B, Jacobs DR, Jr, Sacco RL, Papapanou PN. Periodontal microbiota and carotid intima‐media thickness: the oral infections and vascular disease epidemiology study (INVEST). Circulation. 2005; 111:576-582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, Taubert KA, Newburger JW, Gornik HL, Gewitz MH, Wilson WR, Smith SC, Jr, Baddour LM. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: a scientific statement from the American Heart Association. Circulation. 2012; 125:2520-2544 [DOI] [PubMed] [Google Scholar]
- 13.Desvarieux M, Demmer RT, Jacobs DR, Jr, Rundek T, Boden‐Albala B, Sacco RL, Papapanou PN. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J Hypertens. 2010; 28:1413-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sacco RL, Boden‐Albala B, Gan R, Chen X, Kargman DE, Shea S, Paik MC, Hauser WA. Stroke incidence among white, black, and Hispanic residents of an urban community: the Northern Manhattan Stroke Study. Am J Epidemiol. 1998; 147:259-268 [DOI] [PubMed] [Google Scholar]
- 15.Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007; 116:1736-1754 [DOI] [PubMed] [Google Scholar]
- 16.Desvarieux M, Demmer RT, Rundek T, Boden‐Albala B, Jacobs DR, Jr, Papapanou PN, Sacco RL. Relationship between periodontal disease, tooth loss, and carotid artery plaque: the oral infections and vascular disease epidemiology study (INVEST). Stroke. 2003; 34:2120-2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Socransky SS, Smith C, Martin L, Paster BJ, Dewhirst FE, Levin AE. “Checkerboard” DNA‐DNA hybridization. Biotechniques. 1994; 17:788-792 [PubMed] [Google Scholar]
- 18.Gunaratnam M, Smith GL, Socransky SS, Smith CM, Haffajee AD. Enumeration of subgingival species on primary isolation plates using colony lifts. Oral Microbiol Immunol. 1992; 7:14-18 [DOI] [PubMed] [Google Scholar]
- 19.Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, Csiba L, Desvarieux M, Ebrahim S, Hernandez Hernandez R, Jaff M, Kownator S, Naqvi T, Prati P, Rundek T, Sitzer M, Schminke U, Tardif JC, Taylor A, Vicaut E, Woo KS. Mannheim carotid intima‐media thickness and plaque consensus (2004–2006–2011). An update on behalf of the advisory board of the 3rd, 4th and 5th watching the risk symposia, at the 13th, 15th and 20th European Stroke Conferences, Mannheim, Germany, 2004, Brussels, Belgium, 2006, and Hamburg, Germany, 2011. Cerebrovasc Dis. 2012; 34:290-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambless LE, Folsom AR, Davis V, Sharrett R, Heiss G, Sorlie P, Szklo M, Howard G, Evans GW. Risk factors for progression of common carotid atherosclerosis: the Atherosclerosis Risk in Communities Study, 1987–1998. Am J Epidemiol. 2002; 155:38-47 [DOI] [PubMed] [Google Scholar]
- 21.Sacco RL, Elkind M, Boden‐Albala B, Lin IF, Kargman DE, Hauser WA, Shea S, Paik MC. The protective effect of moderate alcohol consumption on ischemic stroke. JAMA. 1999; 281:53-60 [DOI] [PubMed] [Google Scholar]
- 22.Demmer RT, Papapanou PN, Jacobs DR, Jr, Desvarieux M. Evaluating clinical periodontal measures as surrogates for bacterial exposure: the oral infections and vascular disease epidemiology study (INVEST). BMC Med Res Methodol. 2010; 10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demmer RT, Papapanou PN, Jacobs DR, Jr, Desvarieux M. Bleeding on probing differentially relates to bacterial profiles: the Oral Infections and Vascular Disease Epidemiology Study. J Clin Periodontol. 2008; 35:479-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Page RC, Eke PI. Case definitions for use in population‐based surveillance of periodontitis. J Periodontol. 2007; 78:1387-1399 [DOI] [PubMed] [Google Scholar]
- 25. Consensus report. Periodontal diseases: pathogenesis and microbial factors. Ann Periodontol. 1996; 1:926-932 [DOI] [PubMed] [Google Scholar]
- 26.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RLJ. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998; 25:134-144 [DOI] [PubMed] [Google Scholar]
- 27.Glymour MM, Weuve J, Berkman LF, Kawachi I, Robins JM. When is baseline adjustment useful in analyses of change? An example with education and cognitive change. Am J Epidemiol. 2005; 162:267-278 [DOI] [PubMed] [Google Scholar]
- 28.Yanez ND, III, Kronmal RA, Shemanski LR. The effects of measurement error in response variables and tests of association of explanatory variables in change models. Stat Med. 1998; 17:2597-2606 [DOI] [PubMed] [Google Scholar]
- 29.Pussinen PJ, Kononen E, Paju S, Hyvarinen K, Gursoy UK, Huumonen S, Knuuttila M, Suominen AL. Periodontal pathogen carriage, rather than periodontitis, determines the serum antibody levels. J Clin Periodontol. 2011; 38:405-411 [DOI] [PubMed] [Google Scholar]
- 30.Demmer RT, Kocher T, Schwahn C, Volzke H, Jacobs DR, Jr, Desvarieux M. Refining exposure definitions for studies of periodontal disease and systemic disease associations. Community Dent Oral Epidemiol. 2008; 36:493-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease: the heart of the matter. J Am Dent Assoc. 2006; 137suppl:14S-20S [DOI] [PubMed] [Google Scholar]
- 32.Kebschull M, Demmer RT, Papapanou PN. “Gum bug, leave my heart alone!”—epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010; 89:879-902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slade GD, Offenbacher S, Beck JD, Heiss G, Pankow JS. Acute‐phase inflammatory response to periodontal disease in the US population. J Dent Res. 2000; 79:49-57 [DOI] [PubMed] [Google Scholar]
- 34.Ebersole JL, Machen RL, Steffen MJ, Willmann DE. Systemic acute‐phase reactants, C‐reactive protein and haptoglobin, in adult periodontitis. Clin Exp Immunol. 1997; 107:347-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinane DF, Riggio MP, Walker KF, MacKenzie D, Shearer B. Bacteraemia following periodontal procedures. J Clin Periodontol. 2005; 32:708-713 [DOI] [PubMed] [Google Scholar]
- 36.Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999; 340:115-126 [DOI] [PubMed] [Google Scholar]
- 37.Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, Lu Y, Papapanou PN, Schmidt AM. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E‐null mice. Arterioscler Thromb Vasc Biol. 2003; 23:1405-1411 [DOI] [PubMed] [Google Scholar]
- 38.Li L, Messas E, Batista ELJ, Levine RA, Amar S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E‐deficient murine model. Circulation. 2002; 105:861-867 [DOI] [PubMed] [Google Scholar]
- 39.Gitlin JM, Loftin CD. Cyclooxygenase‐2 inhibition increases lipopolysaccharide‐induced atherosclerosis in mice. Cardiovasc Res. 2009; 81:400-407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, Suvan J, Hingorani AD, Vallance P, Deanfield J. Treatment of periodontitis and endothelial function. N Engl J Med. 2007; 356:911-920 [DOI] [PubMed] [Google Scholar]
- 41.Salonen JT, Salonen R. Ultrasound B‐mode imaging in observational studies of atherosclerotic progression. Circulation. 1993; 87:II56-II65 [PubMed] [Google Scholar]
- 42.O'Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK., Jr Carotid‐artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999; 340:14-22 [DOI] [PubMed] [Google Scholar]
- 43.Polak JF, Pencina MJ, Pencina KM, O'Donnell CJ, Wolf PA, D'Agostino RB., Sr Carotid‐wall intima‐media thickness and cardiovascular events. N Engl J Med. 2011; 365:213-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hodis HN, Mack WJ, LaBree L, Selzer RH, Liu CR, Liu CH, Azen SP. The role of carotid arterial intima‐media thickness in predicting clinical coronary events. Ann Intern Med. 1998; 128:262-269 [DOI] [PubMed] [Google Scholar]
- 45.Crouse JR, III, Byington R, Bond MG, Espeland MA, Craven T, Sprinkle JW, McGovern ME, Furberg CD. Pravastatin, lipids, and atherosclerosis in the carotid arteries (PLAC‐II). Am J Cardiol. 1995; 75:455-459 [DOI] [PubMed] [Google Scholar]
- 46.Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006; 42:80-87 [DOI] [PubMed] [Google Scholar]
- 47.Bots ML, Evans GW, Riley WA, Grobbee DE. Carotid intima‐media thickness measurements in intervention studies: design options, progression rates, and sample size considerations: a point of view. Stroke. 2003; 34:2985-2994 [DOI] [PubMed] [Google Scholar]
- 48.Campbell DT, Stanley JC, Gage NL. Experimental and Quasi‐Experimental Designs for Research. 1966Chicago: R. McNally [Google Scholar]