

Abstract
Copper-mediated trifluoromethylation of unsaturated organotrifluoroborates with the Langlois reagent (NaSO2CF3) and TBHP allows the introduction of trifluoromethyl groups into a variety of organic substructures. The reactions are easy to set up, the conditions are mild and general, and the process provides access to trifluoromethylated alkynes, alkenes, arenes and heteroarenes in fair to good yields.
The unique properties of the trifluoromethyl group have been appreciated in diverse fields of chemistry.1 The CF3 group is notably popular with medicinal chemists as a compact, lipophilic group that slows the metabolic breakdown of aromatics.2 This has led to the development of many new reagents and methods for its introduction.3,4 Among the substrates used for these trifluoromethylations, organoboron compounds have emerged as precursors of choice because of their increasing and pivotal role in synthetic organic chemistry.5 Indeed, the past years have witnessed the emergence of new methods involving different classes of borylated starting materials reacting with nucleophilic, electrophilic and radical CF3 species, all of which can be mediated or catalyzed by copper sources (Scheme 1).
Scheme 1.
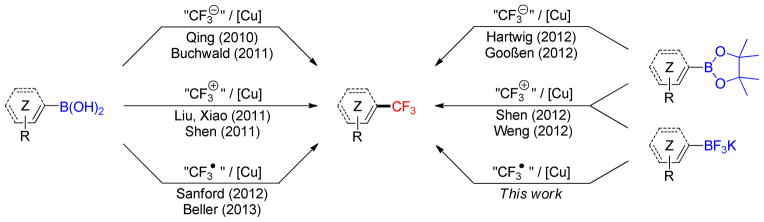
Trifluoromethylations of organoboron compounds
Boronic acids were the first class of compounds studied, and in a pioneering study Qing described the oxidative trifluoromethylation of aryl-, heteroaryl-, and alkenylboronic acids by the nucleophilic Ruppert- Prakash reagent (TMSCF3)6 in a process similar to Chan-Lam-Evans coupling.7,8,9 Shortly thereafter, Buchwald developed a related, but more practical, catalytic version of this reaction,10 also achieved by Qing one year later.11 Importantly, Grushin developed a protocol using simple fluoroform12 together with alkylboronic acids that could be used in a strategy similar to that reported by Fu.13
The complementary use of electrophilic trifluoromethylation reagents has also been intensively studied in the case of boronic acids. Whereas Liu14 and Xiao15 described the use of trifluoromethylsulfonium salts, Shen expanded this reactivity to Togni’s reagent.16 As protodeborylation of the boronic acid starting material was observed as the main side reaction in all of these examples, the trifluoromethylation of protected boron species was also investigated. Hartwig and Goosen reported the oxidative nucleophilic trifluoromethylation of aryl pinacol boronates, but these required the use of either [(phen)CuCF3] or K[CF3B(OMe)3].17,18 The use of the electrophilic Togni reagent [3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole] was found to be more general as it could be used for arylboronates,19 aryltrifluoroborates20 or alkenyltrifluoroborates.21
Sanford recently made a very interesting contribution to the field.22 Her group reported the conversion of aryl- and heteroarylboronic acids by trifluoromethyl radicals generated in situ from the inexpensive Langlois reagent (NaSO2CF3) and tert-butyl hydroperoxide (THBP).23 This reagent was already used for the C–H trifluoromethylation of heterocycles 24 and similar copper-mediated conditions were subsequently reported by Beller with an extension to alkenylboronic acids.25 Importantly, Sanford’s protocol is very practical, as the reaction proceeds under ambient conditions, and isolation of the products is straightforward. Owing to their added value compared to their boronic acid and -ester counterparts,26,27,28 an extension of this method to organotrifluoroborates was envisioned because only two examples had been reported in a study published during the course of our investigation.25 Herein, efforts toward the development of such a general trifluoromethylation method are revealed.
Potassium organotrifluoroborates can be efficiently used in radical reactions,29 even under aqueous conditions.30 Therefore, in a quest for a general protocol, investigations were initiated under the previously reported conditions for boronic acids: NaSO2CF3 (3.0 equiv), TBHP (5.0 equiv), CuCl (1.0 equiv) in CH2Cl2/MeOH/H2O (1:1:0.8) at rt for electron-rich substrates and NaSO2CF3 (3.0 equiv), TBHP (4.0 equiv), (CH3CN)CuPF6 (1.0 equiv), NaHCO3 (1.0 equiv) in MeOH at rt for electron-poor substrates. Although an excess of the Langlois reagent was required, the overall process remains one of the more cost-effective owing to the lower cost of this reagent compared to other trifluoromethylation reagents. The results obtained with aryl-and heteroaryltrifluoroborates are summarized in Table 1. In all of these examples and similar to the case of boronic acids, isolation of the desired products is very easy, as these materials can be obtained with good purity by a simple aqueous work-up. As expected, electron-rich substrates such as 1a and 1c were efficiently trifluoromethylated, but a decreased yield was observed in the case of ortho-substituted products such as 2c, which may be attributed to steric hindrance. Additionally, the reaction performed with 1b led to a poor yield (21%). The case of unactivated substrates was less straightforward. The simple trifluoromethylbenzene 2d was generated from the corresponding trifluoroborate in 28% yield under the same conditions, and electron-poor arenes such as 1e and 1f required the use of modified conditions B. Under these conditions, 2e and 2f were obtained in yields of 61% and 27%, respectively. We next turned our attention to the case of heteroaryl substrates. Five- and six-membered heteroaryltrifluoroborates afforded the corresponding trifluoromethylated products in 6–67% yields, and the observed trend was the same as that seen for simple arenes. A broad variety of electron-rich substrates such as indole 1g, pyrazole 1h, thiazole 1i and benzofuran 1j afforded the desired products in good yields under generic conditions A. However, the case of electron-poor quinoline was once again problematic, and 2k was obtained in a poor 6% yield. Even though the yields obtained with electron-poor potassium organotrifluoroborates are in general lower than the ones observed with the corresponding boronic acids,22 the increased stability of the trifluoroborate salts as compared to boronic acids makes the use of the former reagents very attractive.
Table 1.
Scope of the Trifluoromethylation of Aryl- and Heteroaryltrifluoroboratesa
| ||||
|---|---|---|---|---|
| Entry | Substrate | Method a | Product | Yield b |
| 1 |
1a |
A |
2a |
99% (93%) |
| 2 |
 1b |
A |
 2b |
21% (14%) |
| 3 |
 1c |
A |
 2c |
66% (50%) |
| 4 |
 1d |
A |
 2d |
28% (−) |
| 5 |
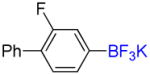 1e |
B |
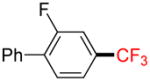 2e |
61% (34%) |
| 6 |
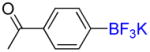 1f |
B |
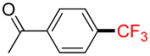 2f |
27% (−) |
| 7 |
1g |
A |
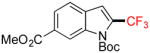 2g |
67% (64%) |
| 8 |
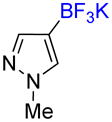 1h |
A |
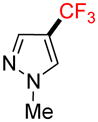 2h |
48% (−) |
| 9 |
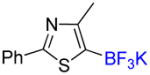 1i |
A |
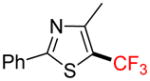 2i |
43% (24%) |
| 10 |
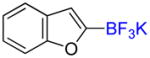 1j |
A |
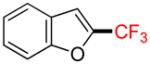 2j |
53% (−) |
| 11 |
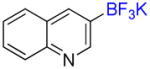 1k |
B |
 2k |
6% (−) |
Conditions A: NaSO2CF3 (3.0 equiv), TBHP (5.0 equiv), CuCl (1.0 equiv), CH2Cl2/MeOH/H2O, 1:1:0.8 ([1] = 0.1 M), open flask, rt, 12 h; conditions B: NaSO2CF3 (3.0 equiv), TBHP (4.0 equiv), (CH3CN)CuPF6 (1.0 equiv), NaHCO3 (1.0 equiv), MeOH ([1] = 0.1 M), open flask, rt, 12 h.
Yields determined by 19F analysis; isolated yields are reported in brackets.
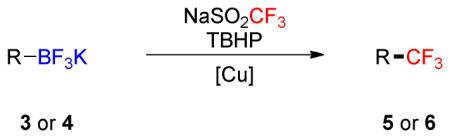
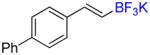
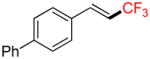
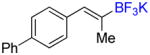
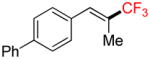
To investigate the scope of the reaction further, we also evaluated the case of alkynyl- and alkenyltrifluoroborates (Table 2). Owing to the electron-rich nature of these substrates, only conditions A mentioned above were applied. Alkynyltrifluoroborates 3a and 3d afforded the desired products in fair yields, opening a new route to the preparation of trifluoromethyl-substituted alkynes. The case of alkenyltrifluoroborates 4a–d afforded valuable information. Under exactly the same conditions, simple styrene derivatives 6a and 6b were obtained in fair to good yields similar of those reported by other methods,21,25 and only a small amount (~5%) of isomerization was detected. Moreover, the substitution pattern of the olefin was found to be crucial. When the trifluoroborate group was placed at the β-position of the aryl ring, the trisubstituted olefin 6c was obtained in a similarly good yield of 70%, but in the case where the CF3 resides in the α-position, the yield in 6d decreased to 12%. Ultimately, no product was observed in the case of cyclic olefins such as 4e, even using specifically designed conditions for alkenyltrifluoroborates.21
Table 2.
Scope of the Trifluoromethylation of Alkynyl- and Alkenyltrifluoroborates
| ||||
|---|---|---|---|---|
| Entry | Substrate | Method a | Product | Yield b |
| 1 |
3a |
A |
5a |
50% (−) |
| 2 |
3b |
A |
5b |
51% (45%) |
| 3 |
 4a |
A |
 6a |
54% (−)c |
| 4 |
 4b |
A |
 6b |
77% (77%)c |
| 5 |
 4c |
A |
 6c |
70% (59%) |
| 6 |
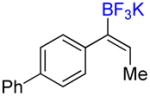 4d |
A |
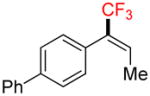 6d |
12% (10%) |
| 7 |
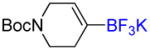 4e |
A | - | - |
Conditions A: NaSO2CF3 (3.0 equiv), TBHP (5.0 equiv), CuCl (1.0 equiv), CH2Cl2/MeOH/H2O, 1:1:0.8 ([3 or 4] = 0.1 M), open flask, rt, 12 h.
Yields determined by 19F analysis; isolated yields are reported in brackets.
With ~5% of the (Z) product.
In conclusion, the copper-mediated radical trifluoromethylation of unsaturated potassium organotrifluoroborates is described. The conditions previously reported were successfully extended to various trifluoroborato-containing arenes and heteroarenes. Additionally, the trifluoromethylation of alkynyltrifluoroborates as well as mono- and di-substituted alkenyltrifluoroborates was achieved. These results, in conjunction with the simplicity of this protocol, demonstrate the usefulness of the radical trifluoromethylation of boronated substrates toward a generic trifluoromethylation protocol.
Experimental Section
Preparation of starting materials 1, 3 and 4
General procedure C from commercially available boronated derivatives
In air, the boronated starting material (1.0 mmol, 1.0 equiv) was weighed in a round-bottomed flask and solubilized in MeOH (5 mL, [SM] = 0.2 M). Sat. aq. KHF2 (0.9 mL, 4.5 M, 4.0 equiv) was added. The flask was closed with a septum, and the resulting mixture stirred at rt for 1 h. The reaction mixture was evaporated to dryness, and the resulting salt was extracted with hot acetone using a Soxhlet apparatus overnight. The filtrate was concentrated to ca. 5 mL, and precipitation was achieved by dropwise addition of the filtrate to Et2O (100 mL) at 0 °C. The product was collected by gravity filtration on a fritted funnel and dried to afford the corresponding potassium organotrifluoroborate.
Potassium 3-Benzyloxyphenyltrifluoroborate 1b
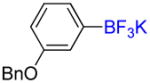
Following general procedure C, the reaction performed with 3-benzyloxyphenylboronic acid (2.0 g, 8.77 mmol) afforded 1b (2.24 g, 88%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C13H11BF3O (M – K)− 251.0861, found 251.0854. 1H NMR (400 MHz / DMSO-d6): δ 7.46 – 7.41 (m, 2H), 7.41 – 7.34 (m, 2H), 7.34 – 7.27 (m, 1H), 7.03 – 6.89 (m, 3H), 6.65 (ddd, J = 8, 3, 1 Hz, 1H), 5.02 (s, 2H). 13C NMR (100 MHz / DMSO-d6): δ 157.2 (C), 138.0 (C), 128.3 (2 CH), 127.5 (CH), 127.4 (2 CH), 127.1 (CH), 124.1 (CH), 117.3 (CH), 111.5 (CH), 68.64 (CH2). 11B NMR (128 MHz / DMSO-d6): δ 3.08 (br). 19F NMR (376 MHz / DMSO-d6): δ −139.2 (br).
Potassium 2-Benzyloxyphenyltrifluoroborate 1c
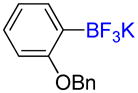
Following general procedure C, the reaction performed with 2-benzyloxyphenylboronic acid (0.5 g, 2.19 mmol) afforded 1c (597 mg, 94%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C13H11BF3O (M – K)− 251.0861, found 251.0856. 1H NMR (400 MHz / DMSO-d6): δ 7.54 (d, J = 7 Hz, 2H), 7.40 – 7.29 (m, 3H), 7.29 – 7.22 (m, 1H), 6.98 (td, J = 8, 2 Hz, 1H), 6.75 – 6.65 (m, 2H), 4.99 (s, 2H). 13C NMR (100 MHz / DMSO-d6): δ 161.5 (C), 138.9 (C), 133.4 (CH), 133.4 (CH), 127.9 (2 CH), 126.8 (2 CH), 126.3 (CH), 119.4 (CH), 111.7 (CH), 68.8 (CH2). 11B NMR (128 MHz / DMSOd6): δ 3.09 (br). 19F NMR (376 MHz / DMSO-d6): δ −136.8 (br).
Potassium 1-Boc-6-(Methoxycarbonyl)indolyl-2-trifluoroborate 1g
Following general procedure C, the reaction performed with 1-Boc-6-(methoxycarbonyl)indole-2-boronic acid (1.0 g, 3.13 mmol) afforded 1g (903 mg, 76%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C15H16BF3NO4 (M – K)− 342.1130, found 342.1130. 1H NMR (400 MHz / DMSO-d6): δ 8.71 (s, 1H), 7.69 (dd, J = 8, 1 Hz, 1H), 7.49 (d, J = 8 Hz, 1H), 6.51 (d, J = 1 Hz, 1H), 3.85 (s, 3H), 1.58 (s, 9H). 13C NMR (100 MHz / DMSO-d6): δ 167.2 (C), 151.1 (C), 136.9 (C), 134.6 (C), 122.6 (C), 122.3 (CH), 119.0 (CH), 115.9 (CH), 111.4 (CH), 82.0 (C), 51.7 (CH3), 27.5 (3 CH3). 11B NMR (128 MHz / DMSO-d6): δ 1.29 (br). 19F NMR (376 MHz / DMSO-d6): δ −136.7 (br).
Potassium 4-Methyl-2-phenyl-5-(trifluoroborato)-1,3-thiazole 1i
Following general procedure C, the reaction performed with 4-methyl-2-phenyl-5-boronic-1,3-thiazole acid pinacol ester (0.5 g, 1.66 mmol) afforded 1i (386 mg, 83%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C10H8BF3NS (M – K)− 242.0428, found 242.0421. 1H NMR (400 MHz / DMSO-d6): δ 7.86 – 7.79 (m, 2H), 7.45 – 7.37 (m, 2H), 7.37 – 7.31 (m, 1H), 2.35 (s, 3H). 13C NMR (100 MHz / DMSO-d6): δ 163.4 (C), 152.5 (C), 134.5 (C), 128.8 (2 CH), 128.5 (CH), 125.5 (2 CH), 16.8 (CH3). 11B NMR (128 MHz / DMSO-d6): δ 2.25 (br). 19F NMR (376 MHz / DMSO-d6): δ −132.3 (br).
Potassium (E)-4-Phenylstyryltrifluoroborate 4b
Following general procedure C, the reaction performed with (E)-4-phenylstyrylboronic acid (535 mg, 2.39 mmol) afforded 4b (292 mg, 43%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C14H11BF3 (M – K)− 247.0911, found 247.0904. 1H NMR (400 MHz / DMSO-d6): δ 7.67 – 7.62 (m, 2H), 7.56 (d, J = 8 Hz, 2H), 7.48 – 7.37 (m, 4H), 7.36 – 7.29 (m, 1H), 6.52 (d, J = 18 Hz, 1H), 6.30 – 6.20 (m, 1H). 13C NMR (100 MHz / DMSO-d6): δ 140.1 (C), 139.5 (C), 137.5 (C), 132.5 (CH), 128.9 (2 CH), 127.0 (CH), 126.5 (2 CH), 126.3 (2 CH), 125.9 (2 CH). 11B NMR (128 MHz / DMSO-d6): δ 2.67 (br). 19F NMR (376 MHz / DMSO-d6): δ −137.8 (br).
Potassium N-Boc-1,2,5,6-Tetrahydropyridinyl-4-trifluoroborate 4e
Following general procedure C, the reaction performed with N-Boc-1,2,5,6-tetrahydropyridine-4-boronic acid pinacol ester (0.5 g, 1.62 mmol) afforded 4e (396 mg, 85%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C10H16BF3NO2 (M – K)− 250.1232, found 250.1237. 1H NMR (400 MHz / acetone-d6): δ 5.56 (br, 1H), 3.72 (br, 2H), 3.33 (t, J = 6 Hz, 2H), 2.07 (br, 2H), 1.42 (s, 9H). 13C NMR (100 MHz / acetone-d6): δ 155.4 (C), 121.3 (CH), 78.7 (C), 44.7 (CH2), 42.3 (CH2), 28.8 (3 CH3), 27.6 (CH2). 11B NMR (128 MHz / acetone-d6): δ 2.91 (br). 19F NMR (376 MHz / acetone-d6): δ −146.7 (br).
Preparation of 3b
31 In air, 4-ethynylbiphenyl (1.0 g, 5.61 mmol, 1.0 equiv) was weighed in a 50 mL round bottom flask equipped with a stir bar. The flask was closed with a septum, evacuated and backfilled with N2. THF (10 mL, [SM] = 0.5 M) was added and the mixture was cooled to −78 °C. n-BuLi (2.2 mL, 2.5 M in hexanes, 5.61 mmol, 1.0 equiv) was added slowly, and the reaction was stirred 1 h at −78 °C. Triisopropyl borate (1.9 mL, 8.42 mmol, 1.5 equiv) was added, and the reaction was stirred 1 h at −78 °C and then warmed to −20 °C in 1 h. Sat. aq. KHF2 (7.5 mL, 4.5 M, 6.0 equiv) was added, and the reaction was stirred at rt for 2 h. The reaction mixture was evaporated to dryness, and the resulting salt was extracted with hot acetone using a Soxhlet apparatus overnight. The filtrate was concentrated to ca. 5 mL, and precipitation was achieved by dropwise addition of the filtrate to Et2O (100 mL) at 0 °C. The resulting product was collected by gravity filtration on a fritted funnel and dried to afford 3b (280 mg, Purity = 95%, 17%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C14H9BF3 (M – K)− 245.0755, found 245.0750. 1H NMR (400 MHz / DMSO-d6): δ 7.66 (d, J = 7 Hz, 2H), 7.59 (d, J = 8 Hz, 2H), 7.46 (dd, J = 8, 8 Hz, 2H), 7.40 – 7.33 (m, 3H). 13C NMR (100 MHz / DMSO-d6): δ 139.5 (C), 138.2 (C), 131.5 (2 CH), 128.9 (2 CH), 127.4 (CH), 126.4 (2 CH), 126.4 (2 CH), 124.7 (C), 89.0 (C). 11B NMR (128 MHz / DMSO-d6): δ −1.56 (br). 19F NMR (376 MHz / DMSO-d6): δ −131.7 (br).
Preparation of 4c and 4d
4-(Propyn-1-yl)-biphenyl
In air, 4-ethynylbiphenyl (1.0 g, 5.61 mmol, 1.0 equiv) was weighed in a round bottom flask equipped with a stir bar. The flask was closed with a septum, evacuated, and backfilled with N2. THF (20 mL, [SM] = 0.25 M) was added, and the mixture was cooled to −78 °C. n-BuLi (2.5 mL, 2.5 M in hexanes, 6.17 mmol, 1.1 equiv) was slowly added (by syringe) and the reaction was stirred 15 min at −78 °C. MeI (0.4 mL, 6.17 mmol, 1.1 equiv) was added, and the reaction was allowed to warm to rt over 2 h and then stirred at rt for 4 h. Subsequently, the reaction mixture was poured into sat. aq. NH4Cl (20 mL), and the resulting solution was extracted twice with EtOAc (20 mL). The combined organic layers were washed with brine (50 mL), dried (MgSO4) and evaporated to afford the crude product. Purification by flash column chromatography (80 g SiO2, heptane to EtOAc/heptane, 1:9) afforded the title compound (654 mg, 60%) as a white solid, mp = 69–70 ºC. 1H NMR (400 MHz / CDCl3): δ 7.63 – 7.58 (m, 2H), 7.57 – 7.53 (m, 2H), 7.51 – 7.43 (m, 4H), 7.40 - 7.340 (m, 1H), 2.10 (s, 3H). Spectral data were consistent with that previously reported.32
Compound 4c
33 In air, CuCl (5 mg, 0.05 mmol, 5 mol %), Ph3P (27 mg, 0.10 mmol, 10 mol %) and NaOt-Bu (20 mg, 0.21 mmol, 20 mol %) were weighed in a 2–5 mL MW vial equipped with a stir bar. The vial was sealed, evacuated, and backfilled with N2 (x3). THF (0.5 mL) was added and the resulting mixture stirred at rt for 30 min. A solution of bis(pinacolato)diboron (290 mg, 1.14 mmol, 1.1 equiv) in THF (1 mL) was added, and the mixture stirred at rt for 5 min. A solution of 4-(propyn-1-yl)-biphenyl (200 mg, 1.04 mmol, 1.0 equiv) in THF (0.5 mL) and MeOH (84 μL, 2.08 mmol, 2.0 equiv) were successively added, and the reaction stirred at rt for 14 h. Then, the reaction mixture was diluted with Et2O (10 mL) and filtered through a pad of Celite. The filtrate was evaporated and purified by flash column chromatography (24 g SiO2, heptane to EtOAc/heptane, 1/4). In air, this boronate was charged in a round-bottomed flask and solubilized in THF (10 mL). Sat. aq. KHF2 (0.9 mL, 4.5 M, 4.2 mmol, 4.0 equiv) was added. The flask was closed with a septum, and the resulting mixture stirred at rt for 2 h. The reaction mixture was evaporated to dryness, and the resulting salt was extracted with hot acetone using a Soxhlet overnight. The filtrate was concentrated to ca. 5 mL, and precipitation was achieved by dropwise addition of the filtrate to Et2O (100 mL) at 0 °C. The product was collected by gravity filtration on a fritted funnel and dried to afford 4c (208 mg, 67%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C15H13BF3 (M – K)− 261.1068, found 261.1065. 1H NMR (400 MHz / DMSO-d6): δ 7.65 (d, J = 8 Hz, 2H), 7.57 (d, J = 8 Hz, 2H), 7.44 (dd, J = 8, 8 Hz, 2H), 7.36 – 7.30 (m, 1H), 7.27 (d, J = 8 Hz, 2H), 6.41 (s, 1H), 1.76 (s, 3H). 13C NMR (100 MHz / DMSO-d6): δ 140.2 (C), 139.9 (C), 136.1 (C), 129.0 (2 CH), 128.8 (2 CH), 126.9 (CH), 126.3 (2 CH), 126.0 (2 CH), 125.6 (CH), 16.7 (CH3). 11B NMR (128 MHz / DMSO-d6): δ 3.08 (br). 19F NMR (376 MHz / DMSO-d6): δ −143.6 (br).
Compound 4d
34 In air, CuCl (4 mg, 0.04 mmol, 5 mol %), xantphos (51 mg, 0.09 mmol, 10 mol %) and NaOt- Bu (17 mg, 0.18 mmol, 20 mol %) were weighed in a 2–5 mL MW vial equipped with a stir bar. The vessel was sealed, evacuated, and backfilled with N2 (x3). Toluene (1 mL) was added and the mixture stirred at rt for 15 min. Pinacolborane (0.2 mL, 1.33 mol, 1.5 equiv) was added, and the mixture stirred at rt for 5 min. A solution of 4-(propyn-1-yl)-biphenyl (171 mg, 0.89 mmol, 1.0 equiv) in toluene (1 mL) was added and the reaction stirred at rt for 3 d. The reaction mixture was diluted with EtOAc (10 mL) and filtered through a pad of Celite. The filtrate was evaporated to afford the crude boronate intermediate. In air, this boronate was charged in a round-bottomed flask and solubilized in THF (10 mL). Sat. aq. KHF2 (0.8 mL, 4.5 M, 3.6 mmol, 4.0 equiv) was added. The flask was closed with a septum, and the resulting mixture was stirred at rt for 2 h. The reaction mixture was evaporated to dryness, and the resulting salt was extracted with hot acetone using a Soxhlet apparatus overnight. The filtrate was concentrated to ca. 5 mL, and precipitation was achieved by dropwise addition of the filtrate to Et2O (100 mL) at 0 °C. The product was collected by gravity filtration on a fritted funnel and dried to afford 4d (69 mg, 26%) as a white solid, mp > 200 °C (dec). HRMS (ESI): m/z calcd. for C15H13BF3 (M – K)− 261.1068, found 261.1058. 1H NMR (400 MHz / DMSO-d6): δ 7.63 (dd, J = 8, 1 Hz, 2H), 7.50 – 7.40 (m, 4H), 7.33 – 7.27 (m, 1H), 7.12 (d, J = 8 Hz, 2H), 5.69 (q, J = 7 Hz, 1H), 1.48 (t, J = 7 Hz, 3H). 13C NMR (100 MHz / DMSO-d6): δ 145.2 (C), 140.8 (C), 135.4 (C), 128.9 (2 CH), 128.7 (2 CH), 126.6 (CH), 126.2 (2 CH), 125.2 (2 CH), 122.6 (CH), 15.0 (CH3). 11B NMR (128 MHz / DMSO-d6): δ 2.73 (br). 19F NMR (376 MHz / DMSO-d6): δ −139.4 (br).
Radical Trifluoromethylation of Unsaturated Potassium Organotrifluoroborates
General procedure A for the preparation of trifluoromethylated compounds 2, 5 and 6
In air, potassium organotrifluoroborates 1 or 3 or 4 (0.5 mmol, 1.0 equiv), NaSO2CF3 (234 mg, 1.5 mmol, 3.0 equiv) and CuCl (50 mg, 0.5 mmol, 1.0 equiv) were weighed in a 2–5 mL MW vial equipped with a stir bar. MeOH (1 mL), CH2Cl2 (1 mL) and distilled H2O (0.8 mL) were successively added, and the tube was sealed with a tap open to air by a needle. This solution was cooled to 0 °C and TBHP (0.35 mL, 70% in H2O, 2.5 mmol, 5.0 equiv) was slowly added. The reaction was allowed to warm to rt and stirred at rt for 12 h. The reaction mixture was diluted with Et2O (10 mL) and this solution was washed successively with sat. aq. NaHCO3 (5 mL) and 5% aq. Na2S2O3 (5 mL). The organic layer was dried (MgSO4) and 1,3,5-trifluorobenzene (51.7 μL, 0.5 mmol, 1.0 equiv) was added as an internal standard. The solution was analyzed by 19F NMR and GCMS. Additionally, this solution can be evaporated and purified by flash column chromatography (SiO2, mixtures of EtOAc and heptane) to afford the pure compound 2 or 5 or 6.
General procedure B for the preparation of trifluoromethylated compounds 2
In air, potassium organotrifluoroborates 1 (0.5 mmol, 1.0 equiv), NaSO2CF3 (234 mg, 1.5 mmol, 3.0 equiv) and (MeCN)4CuPF6 (157 mg, 0.5 mmol, 1.0 equiv) were weighed in a 2–5 mL MW vial equipped with a stir bar. MeOH (3 mL) was added, and the tube was sealed with a tap open to air by a needle. This solution was cooled to 0 °C and TBHP (0.28 mL, 70% in H2O, 2.0 mmol, 4.0 equiv) was slowly added. The reaction was allowed to warm to rt and stirred at rt for 12 h. The reaction mixture was diluted with Et2O (10 mL) and this solution was washed successively with sat. aq. NaHCO3 (5 mL) and 5% aq. Na2S2O3 (5 mL). The organic layer was dried (MgSO4) and 1,3,5-trifluorobenzene (51.7 μL, 0.5 mmol, 1.0 equiv) was added as an internal standard. The solution was analyzed by 19F NMR and GCMS. Additionally, this solution can be evaporated and purified by flash column chromatography (SiO2, mixtures of EtOAc and heptane) to afford the pure compound 2.
1-(Benzyloxy)-4-(trifluoromethyl)benzene 2a
Following general procedure A, the reaction performed with 1a (145 mg, 0.5 mmol) afforded 2a in 19F yield = 99% and, after purification, 117 mg (93%) as a white solid, mp = 79–80 ºC. GC-MS: 5.438 min (m/z 252 (M)+). 19F NMR (376 MHz / CDCl3): δ −61.4 (s). Spectral data were consistent with that previously reported.16
1-(Benzyloxy)-3-(trifluoromethyl)benzene 2b
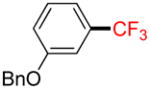
Following general procedure A, the reaction performed with 1b (145 mg, 0.5 mmol) afforded 2b in 19F yield = 21% and, after purification, 18 mg (14%) as a colorless oil. GC-MS: 5.241 min (m/z 252 (M)+). 19F NMR (376 MHz / CDCl3): δ −62.9 (s). Spectral data were consistent with that previously reported.35
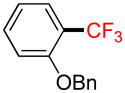
1-(Benzyloxy)-2-(trifluoromethyl)benzene 2c
Following general procedure A, the reaction performed with 1c (145 mg, 0.5 mmol) afforded 2c in 19F yield = 66% and, after purification, 63 mg (50%) as a colorless oil. GC-MS: 5.462 min (m/z 252 (M)+). HRMS (ESI): m/z calcd. for C14H11F3O (M)+ 252.0762, found 252.0748. 1H NMR (400 MHz / CDCl3): δ 7.64 (dd, J = 8, 1 Hz, 1H), 7.52 – 7.46 (m, 3H), 7.46 – 7.40 (m, 2H), 7.39 - 7.33 (m, 1H), 7.11 – 7.00 (m, 2H), 5.22 (s, 2H). 13C NMR (100 MHz / CDCl3): δ 156.4 (C), 136.3 (C), 133.2 (CH), 128.6 (2 CH), 127.9 (CH), 127.1 (q, J = 5 Hz, CH), 126.8 (2 CH), 123.8 (q, J = 272 Hz, C), 120.2 (CH), 119.2 (q, J = 31 Hz, C), 113.2 (CH), 70.2 (CH2). 19F NMR (376 MHz / CDCl3): δ −62.3 (s).
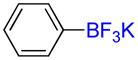
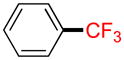
Trifluoromethylbenzene 2d
Following general procedure A, the reaction performed with 1d (92 mg, 0.5 mmol) afforded 2d in 19F yield = 28%. GC-MS: 0.624 min (m/z 146 (M)+). 19F NMR (376 MHz / CDCl3): δ − 62.9 (s). Spectral data were consistent with that previously reported.22
1-Fluoro-2-phenyl-5-trifluoromethylbenzene 2e
Following general procedure B, the reaction performed with 1e (139 mg, 0.5 mmol) afforded 2e in 19F yield = 61% and, after purification, 41 mg (34%) as a colorless oil. GC-MS: 4.151 min (m/z 240 (M)+). 19F NMR (376 MHz / CDCl3): δ −62.8 (s, 3F), 115.7 (s, 1F). Spectral data were consistent with that previously reported.16
4-Acetyl-trifluoromethylbenzene 2f
Following general procedure B, the reaction performed with 1f (113 mg, 0.5 mmol) afforded 2f in 19F yield = 27%. GC-MS: 2.437 min (m/z 188 (M)+). 19F NMR (376 MHz / CDCl3): δ−63.2 (s). Spectral data were consistent with that previously reported.22
1-Boc-2-Trifluoromethyl-6-(methoxycarbonyl)indole 2g
Following general procedure A, the reaction performed with 1g (191 mg, 0.5 mmol) afforded 2g in 19F yield = 67% and, after purification, 110 mg (64%) as a white solid, mp = 83–84 °C. GC-MS: 7.321 min (m/z 343 (M)+). HRMS (ESI): m/z calcd. for C11H8F3NO2 (M-Boc+H)+ 243.0507, found 243.0533. 1H NMR (400 MHz / CDCl3): δ 8.99 (s, 1H), 7.99 (d, J = 8 Hz, 1H), 7.66 (d, J = 8 Hz, 1H), 7.16 (s, 1H), 3.97 (s, 3H), 1.70 (s, 9H). 13C NMR (100 MHz / CDCl3): δ 167.1 (C), 148.0 (C), 137.1 (C), 129.9 (C), 129.5 (q, J = 40 Hz, C), 128.6 (C), 124.5 (CH), 121.8 (CH), 120.4 (q, J = 268 Hz, C), 118.1 (CH), 112.8 (q, J = 5 Hz, CH), 86.1 (C), 52.2 (CH3), 27.8 (3 CH3). 19F NMR (376 MHz / CDCl3): δ −58.4 (s).
1-Methyl-4-trifluoromethyl-1H-pyrazole 2h
Following general procedure A, the reaction performed with 1h (47 mg, 0.25 mmol) afforded 2f in 19F yield = 48% (in this case, 2.0 equiv of internal standard were used). GCMS: 0.882 min (m/z 150 (M)+). 19F NMR (376 MHz / CDCl3): δ −56.4 (s). Spectral data were consistent with that previously reported.36
4-Methyl-2-phenyl-5-trifluoromethyl-1,3-thiazole 2i
Following general procedure A, the reaction performed with 1i (140 mg, 0.5 mmol) afforded 2i in 19F yield = 43% and, after purification, 37 mg (24%) as a colorless oil (contaminated by 5% of the bis-trifluoromethylated product). GC-MS: 4.642 min (m/z 243 (M)+). HRMS (ESI): m/z calcd. for C11H8F3NS (M)+ 243.0330, found 243.0331. 1H NMR (400 MHz / CDCl3): δ 7.95 – 7.89 (m, 2H), 7.50 – 7.43 (m, 3H), 2.62 (q, J = 2 Hz, 3H). 13C NMR (100 MHz / CDCl3): δ 168.4 (C), 155.2 (C), 132.5 (C), 131.0 (CH), 129.1 (2 CH), 126.7 (2 CH), 122.7 (q, J = 269 Hz, C), 119.7 (q, J = 37 Hz, C), 16.1 (CH3). 19F NMR (376 MHz / CDCl3): δ −53.0 (s).
2-Trifluoromethylbenzofuran 2j
Following general procedure A, the reaction performed with 1j (124 mg, 0.5 mmol) afforded 2j in 19F yield = 53%. GC-MS: 1.902 min (m/z 186 (M)+). 19F NMR (376 MHz / CDCl3): δ − 65.0 (s). Spectral data were consistent with that previously reported.22
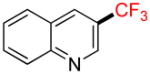
3-Trifluoromethylquinoline 2k
Following general procedure B, the reaction performed with 1k (117 mg, 0.5 mmol) afforded 2k in 19F yield = 6%. GC-MS: 3.277 min (m/z 197 (M)+). 19F NMR (376 MHz / CDCl3): δ −62.0 (s). Spectral data were consistent with that previously reported.16
1′-Trifluoromethylphenylacetylene 5a
Following general procedure A, the reaction performed with 1j (104 mg, 0.5 mmol) afforded 5a in 19F yield = 50%. GC-MS: 1.344 min (m/z 170 (M)+). 19F NMR (376 MHz / CDCl3): δ −49.9 (s). Spectral data were consistent with that previously reported.37
1′-Trifluoromethyl-1-biphenylacetylene 5b
Following general procedure A, the reaction performed with 3b (149 mg, 0.5 mmol) afforded 5b in 19F yield = 51% and, after purification, 56 mg (45%) as a white solid, mp = 82–83 ºC. GC-MS: 5.471 min [m/z 246 (M)+]. 19F NMR (376 MHz / CDCl3): δ −49.9 (s). Spectral data were consistent with that previously reported.37
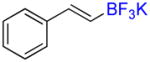
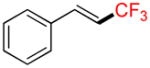
Compound 6a
Following general procedure A, the reaction performed with 4a (105 mg, 0.5 mmol) afforded 6a in 19F yield = 54% (contaminated by 5% of the isomerized (Z)-product). GC-MS: 1.806 min (m/z 172 (M)+). 19F NMR (376 MHz / CDCl3): δ −63.4 (s). Spectral data were consistent with that previously reported.38
Compound 6b
Following general procedure A, the reaction performed with 4b (143 mg, 0.5 mmol) afforded 6b in 19F yield = 77% and, after purification, 101 mg (77%) as a white solid (contaminated by 5% of the isomerized (Z)-product). GC-MS: 5.890 min (m/z 248 (M)+). 19F NMR (376 MHz / CDCl3): δ −63.4 (s). Spectral data were consistent with that previously reported.16
Compound 6c
Following general procedure A, the reaction performed with 4c (75 mg, 0.25 mmol) afforded 6c in 19F yield = 70% (in this case, 2.0 equiv of internal standard were used) and, after purification, 39 mg (59%) as a white solid, mp = 82–83 °C. GC-MS: 6.183 min (m/z 262 (M)+). HRMS (ESI): m/z calcd. for C16H13F3 (M)+ 262.0969, found 262.0970. 1H NMR (400 MHz / CDCl3): δ 7.73 – 7.60 (m, 4H), 7.54 – 7.35 (m, 5H), 7.12 (s, 1H), 2.10 (s, 3H). 13C NMR (100 MHz / CDCl3): δ 141.0 (C), 140.3 (C), 133.5 (C), 130.9 (q, J = 6 Hz, CH), 129.7 (2 CH), 128.9 (2 CH), 127.6 (CH), 127.1 (2 CH), 127.0 (2 CH), 126.2 (q, J = 29 Hz, C), 124.6 (q, J = 272 Hz, C), 12.3 (CH3). 19F NMR (376 MHz / CDCl3): δ −69.5 (s).
Compound 6d
Following general procedure A, the reaction performed with 4d (62 mg, 0.21 mmol) afforded 6d in 19F yield = 12% (in this case, 2.0 equiv of internal standard were used) and, after purification, 6 mg (10%) as a white solid, mp = 64–65 °C. GC-MS: 5.862 min (m/z 262 (M)+). HRMS (ESI): m/z calcd. for C16H13F3 (M)+ 262.0969, found 262.0963. 1H NMR (400 MHz / CDCl3): δ 7.68 – 7.57 (m, 5H), 7.54 – 7.42 (m, 2H), 7.42 – 7.30 (m, 2H) 6.58 (m, 1H), 1.74 (m, 3H). 13C NMR (100 MHz / CDCl3): δ 141.2 (C), 140.5 (C), 131.9 (C), 131.6 (q, J = 5 Hz, CH), 131.3 (q, J = 45 Hz, C), 130.1 (2 CH), 128.8 (2 CH), 128.6 (q, J = 275 Hz, C), 127.5 (CH), 127.1 (4 CH), 14.3 (CH3). 19F NMR (376 MHz / CDCl3): δ −65.7 (s).
Supplementary Material
Acknowledgments
This research was supported by NIH (NIGMS R01 GM081376) and the Neuroscience Medicinal Chemistry Department of Janssen. Michel Carpentier and Guy Winderickx (Janssen) are acknowledged for obtaining HRMS data.
Footnotes
General experimental considerations and copies of GCMS monitoring and NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Frederik Rombouts, Email: frombout@its.jnj.com.
Gary A. Molander, Email: gmolandr@sas.upenn.edu.
References
- 1.Schlosser M. Angew Chem Int Ed. 2006;45:5432. doi: 10.1002/anie.200600449. [DOI] [PubMed] [Google Scholar]
- 2.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 3.Liu T, Shen Q. Eur J Org Chem. 2012:6679. [Google Scholar]
- 4.Liang T, Neumann CN, Ritter T. Angew Chem Int Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- 5.Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- 6.Chu L, Qing FL. Org Lett. 2010;12:5060. doi: 10.1021/ol1023135. [DOI] [PubMed] [Google Scholar]
- 7.Chan DMT, Monaco KL, Wang RP, Winters MP. Tetrahedron Lett. 1998;39:2933. [Google Scholar]
- 8.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937. [Google Scholar]
- 9.Lam PYS, Clark CG, Saubern S, Adams J, Winters MP, Chan DMT, Combs A. Tetrahedron Lett. 1998;39:2941. [Google Scholar]
- 10.Senecal TD, Parsons AT, Buchwald SL. J Org Chem. 2011;76:1174. doi: 10.1021/jo1023377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang X, Chu L, Qing FL. J Org Chem. 2012;77:1251. doi: 10.1021/jo202566h. [DOI] [PubMed] [Google Scholar]
- 12.Novak P, Lishchynskyi A, Grushin VV. Angew Chem Int Ed. 2012;51:7767. doi: 10.1002/anie.201201613. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Xiao B, Xie CQ, Luo DF, Liu L, Fu Y. Angew Chem Int Ed. 2012;51:12551. doi: 10.1002/anie.201206681. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, Luo DF, Liu ZJ, Gong TJ, Fu Y, Liu L. Chem Commun. 2011;47:4300. doi: 10.1039/c1cc10359h. [DOI] [PubMed] [Google Scholar]
- 15.Zhang CP, Cai J, Zhou CB, Wang XP, Zheng X, Gu YC, Xiao JC. Chem Commun. 2011;47:9516. doi: 10.1039/c1cc13460d. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, Shen Q. Org Lett. 2011;13:2342. doi: 10.1021/ol2005903. [DOI] [PubMed] [Google Scholar]
- 17.Litvinas ND, Fier PS, Hartwig JF. Angew Chem Int Ed. 2012;51:536. doi: 10.1002/anie.201106668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan BA, Buba AE, Goosen LJ. Chem Eur J. 2012;18:1577. doi: 10.1002/chem.201102652. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Shao X, Wu Y, Shen Q. Angew Chem Int Ed. 2012;51:540. doi: 10.1002/anie.201106673. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Fang X, Lin X, Li H, He W, Huang KW, Yuan Y, Weng Z. Tetrahedron. 2012;68:9949. [Google Scholar]
- 21.Parsons AT, Senecal TD, Buchwald SL. Angew Chem Int Ed. 2012;51:2947. doi: 10.1002/anie.201108267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye Y, Kunzi SA, Sanford MS. Org Lett. 2012;14:4979. doi: 10.1021/ol3022726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langlois BR, Laurent E, Roidot N. Tetrahedron Lett. 1991;32:7525. [Google Scholar]
- 24.Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci USA. 2011;108:14411. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Wu L, Neumann H, Beller M. Chem Commun. 2013;49:2628. doi: 10.1039/c2cc36554e. [DOI] [PubMed] [Google Scholar]
- 26.Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49. [Google Scholar]
- 27.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 28.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 29.Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Presset M, Fleury-Bregeot N, Oehlrich D, Rombouts F, Molander GA. J Org Chem. 2013:4615. doi: 10.1021/jo4005519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molander GA, Katona BW, Machrouhi F. J Org Chem. 2002;67:8416. doi: 10.1021/jo0262356. [DOI] [PubMed] [Google Scholar]
- 32.Chen M, Zheng X, Li W, He J, Lei A. J Am Chem Soc. 2010;132:4101. doi: 10.1021/ja100630p. [DOI] [PubMed] [Google Scholar]
- 33.Kim HR, Yun J. Chem Commun. 2011;47:2943. doi: 10.1039/c0cc04496b. [DOI] [PubMed] [Google Scholar]
- 34.Semba K, Fujihara T, Terao J, Tsuji Y. Chem Eur J. 2012;18:4179. doi: 10.1002/chem.201103612. [DOI] [PubMed] [Google Scholar]
- 35.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlik JW, Ayudhaya TIN, Pandit CR, Tantayanon S. J Heterocycl Chem. 2004;41:61. [Google Scholar]
- 37.Kawatsura M, Yamamoto M, Namioka J, Kajita K, Hirakawa T, Itoh T. Org Lett. 2011;13:1001. doi: 10.1021/ol1030734. [DOI] [PubMed] [Google Scholar]
- 38.van Alem K, Belder G, Lodder G, Zuilhof H. J Org Chem. 2005;70:179. doi: 10.1021/jo0487956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.