

Abstract
The expression of pathogen recognition receptors in human FOXP3+ T regulatory cells is established, yet the function of these receptors is currently obscure. In the process of studying the function of both peripheral and lamina propria FOXP3+ lymphocytes in patients with the human inflammatory bowel disease Crohn’s disease, we observed a clear deficiency in the quantity of FOXP3+ lymphocytes in patients with disease-associated polymorphisms in the pathogen recognition receptor gene NOD2. Subsequently, we determined that the NOD2 ligand, muramyl dipeptide (MDP), activates NF-κB in primary human FOXP3+ T cells. This activation is functionally relevant, as MDP-stimulated human FOXP3+ T cells are protected from death receptor Fas-mediated apoptosis. Importantly, apoptosis protection was not evident in MDP-stimulated FOXP3+ T cells isolated from a patient with the disease-associated polymorphism. Thus, we propose that one function of pathogen recognition receptors in human T regulatory cells is the protection against death receptor-mediated apoptosis in a Fas ligand-rich environment, such as that of the inflamed intestinal subepithelial space.
Crohn’s disease (CD) is a common, incurable, relapsing disorder characterized by chronic inflammation of the small and large intestine that causes intense suffering to patients, that often requires disfiguring surgery, and for which current therapies are inadequate. A preponderance of data from murine models of colitis (1), ex vivo studies of human T cells, and genome-wide association studies indicate an important role for unregulated T cell activation in the instigation and perpetuation of chronic intestinal inflammation in CD (2, 3). Thus, insight into T cell regulation is critical to the understanding of CD pathophysiology.
The existence of a T cell subset with a regulatory phenotype in humans is now well established. Although the field of T regulatory cells (Treg cells) is rapidly expanding and frequently contradictory, two points of consensus relevant to this report have been reached. First, naturally occurring, thymus-derived CD4+CD25+FOXP3+ Treg cells exist in the peripheral circulation in humans, and the biology of these human Treg cells has far-reaching implications for the field of autoimmunity and cancer (4). Second, the transcription factor forkhead box P3 (FOXP3) appears to be critically important for Treg cell development and function. Mutation of FOXP3 in humans results in the immune proliferative syndrome known as IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome), and transduction of non-Treg cells with FOXP3 imparts suppressive function in both in vitro suppression assays and murine models of autoimmunity (5, 6). Thus, human Treg cells are critical to immune homeostasis and are dependent upon the transcription factor FOXP3 for development and function.
Advances in understanding CD pathogenesis have occurred through the discovery of disease susceptibility genes (7–10). Although >20 inflammatory bowel disease susceptibility loci have been identified, the statistical effects of most identified susceptibility loci are modest. One notable exception is the risk conferred by polymorphic variants of the gene NOD2 (2). In North American populations, carriage of two variant alleles for NOD2 occurs in 15–20% of CD populations, and the odds ratio (for carriers of two variant alleles) for CD development is estimated between 20 and 40 (11). NOD2 is an intracellular protein that controls the activation of transcription factor NF-κB in response to muramyl dipeptide (MDP), a peptide present in the bacterial cell wall (12, 13). Variant NOD2 alleles have been linked to CD, but the mechanism responsible for variant NOD2-mediated immune activation is not known.
Previous work from our laboratory has focused on the relevance of pathogen recognition receptors to Treg cell biology (14). In this study, we report for the first time the functional relevance of NOD2 signaling in human Treg cells. We demonstrate that 1) NOD2 variants are associated with a deficiency of FOXP3+ Treg cells in the colonic lamina propria (LP), 2) NOD2 variants are associated with impaired NF-κB activation in lymphocytes, 3) NF-κB activation promotes Treg cell survival and protection from Fas-mediated apoptosis, and 4) NOD2 signaling results in the upregulation of antiapoptotic genes in human Treg cells. These studies provide new insight into the obscure pathogenic mechanism of NOD2 variant alleles in CD.
Materials and Methods
Human studies
All studies were reviewed and approved by the Institutional Review Board of the Mayo Clinic, and informed consent was obtained from all patients included in the study.
Subjects
Quantification of LP Treg cells
Surgical resection specimens or endoscopic biopsy specimens from 30 patients with CD (mean age, 45.9 ± 3.5; 17 women, 13 men) and endoscopic biopsy specimens from 10 otherwise healthy individuals (mean age, 56.2 ± 6.8; 7 women, 3 men) undergoing colonoscopy for a noninflammatory condition (8 with a diagnosis of constipation, 2 with a diagnosis of hemorrhoids) were obtained from archived tissue for immunohistochemistry. Histological disease severity was graded by a blinded single pathologist (T.S.). Medical records were reviewed to abstract the medication profile of each patient.
Cell isolation and flow cytometry
Cell isolation for peripheral blood assays
Primary human Treg cells were isolated from patients with CD or healthy blood donors by Ficoll separation and magnetic bead sorting (CD4+CD25+ T Cell Isolation kit, 130-091-301; Miltenyi Biotec, Auburn, CA). Cells with the highest CD25 expression (CD25++) were selected through incubation with a limiting quantity of anti-CD25 beads, as described (2 μl anti-CD25 beads/107 cells) (15). FOXP3 enrichment was found to be >75% by flow cytometry. CD4 positivity was greater than 98% by flow cytometry.
Cell isolation for LP assays
Pooled tissue from 8 to 16 endoscopic mucosal biopsies was the source of LP lymphocytes. Tissue initially underwent mechanical disruption in the presence of collagenase (0.1 mg/ml), DNase (0.1 mg/ml), and trypsin inhibitor (0.1 mg/ml) for 1 h at 37°C. After passage through a 70-μm cell strainer, CD4+ LP cells were isolated using magnetic bead sorting (CD4+ T Cell Isolation kit, 130-091-155; Miltenyi Biotec). CD4+ LP lymphocytes were then stained for the brightest population of CD25-expressing cells (typically 3% of the CD4 population). CD25++ LP lymphocytes were enriched in FOXP3 >66%.
Immunohistochemistry
The 4-μm–thick slides were prepared by deparaffinizing the mucosal specimens via EDTA pretreatment and heat-mediated Ag retrieval. Slides were then placed in the Dako Autostainer automated stainer (Dako, Glostrup, Denmark), using anti-CD4 mouse mAb (clone 4B12; Novocastra/Leica, Bannockburn, IL) and anti-FOXP3 mouse mAb (clone 236A/E7; Abcam, Cambridge, MA/Novus Biologicals, Littleton, CO) as the primary Abs. Diaminobenzidine followed by hemotoxylin counterstaining was then performed. Slides were reviewed by a pathologist who was blinded to the subject’s genotype (T.S.).
Suppression assays
For LP suppression assays, CD4+CD25++ cells sorted from LP cell isolates were expanded over 10–14 d, using anti-CD3/anti-CD28 conjugated Dynabeads (Miltenyi Biotec), as previously described (16). MLRs were performed using 5 ×103 CD4+CD25−T responder cells and 5 ×104 irradiated (3300 rads) T cell-depleted APCs isolated from healthy blood donors. Aliquots of the same donor were stored at −80°C for reproducibility of assays. LP Treg cells were added to cell culture at titrations of 1:32–1:1. The culture medium was cRPMI supplemented with 10% human serum and 2.5 μg/ml anti-CD3 (UCHT1) and anti-CD28 (17). Proliferation was read at 7 d upon addition of 1 μCi tridiated thymidine for the last 18 h of culture.
Transfection and luciferase assays
CD4+CD25++ cells were transfected using previously described methodology (18). Isolated cells were briefly electroporated using a BTX model 820 square wave electroporator (BTX Harvard Apparatus, Holliston, MA) with 332 V at 1-pulse, 10-pulse length/ms and transfected with the P106 NF-κB 3 luciferase reporter construct (19). Cells were treated for 4 h with 1 μg/ml MDP. The luciferase reading was made following overnight incubation of transfected cells on a Berthold Lumat LB 9507 luminometer (Berthold Technologies, Oak Ridge, TN) and controlled for cell death and transfection efficiency with internal standard renilla, per protocols previously described (Promega Dual-Luciferase Reporter Assay System technical manual [Promega, Madison, WI]).
ELISA
Freshly isolated CD4+CD25++ Treg cells were treated over a time course with 1 μg/ml MDP (MDP, BACHEM; 0565698). Nuclear extracts were prepared according to the manufacturer’s protocol (Nuclear Extract kit, 40010; Active Motif, Carlsbad, CA) for use in ELISA assay (TransAM NFκB Activation Assay, 40096; Active Motif). NF-κB activity was quantified using the VMax Kinetic Microplate Reader instrument (Molecular Devices, Sunnyvale, CA). The detection limit for the assay was <0.2 μg nuclear extract/well.
Western blotting
CD4+ T cells and CD4− flow-through cells were isolated by MACS columns as above. Protein was isolated from 5 × 105–1 × 106 cells (primary cells, THP-1 cells, or 293T transfected cells) for Western blotting. Protein was run for 6 h on 7.5% gradient gels and transferred to PVDF membrane for 4 h. Membranes were probed overnight with primary Ab (anti-NOD2, polyclonal rabbit, Imgenex 5831 [Imgenex, San Diego, CA]; anti-NALP3, monoclonal mouse, Abcam 17267; and anti–β-Actin, polyclonal rabbit, BioLegend 622101 [BioLegend, San Diego, CA]). For NOD2, the concentration of primary Ab was 1:500 and that of secondary Ab was 1:2500.
Small interfering RNA studies
CD4− T cells isolated by MACS column (as above) were used for nucleofection program U-014 per the manufacterer’s protocol (Lonza, Basel, Switzerland). A total of 1 × 106 cells per 100 μl were nucleofected with 2.5 μg small interfering RNA (siRNA). Human NOD2 siRNA (siGE-NOME SMARTpool siRNA; Thermo Scientific, Dharmacon, Lafayette, CO) or a nontargeting pool control siRNA (siGENOME; Dharmacon) was used for nucleofection.
Microscopy
Freshly isolated primary human Treg cells were treated for 4 h with and without 1 μg/ml MDP, followed by an overnight incubation with 100 ng/ml Fas ligand (FasL) (Bender MedSystems, Vienna, Austria; BMS309). The cells were fixed after 18 h and treated with 1 μg/ml Hoechst stain (Sigma-Aldrich, St. Louis, MO; B1155). The percentage of apoptotic nuclei was measured in blinded fashion, using a Zeiss Axioplan2 light microscope (Zeiss, Oberkochen, Germany).
Caspase assay
Apoptosis was measured through CaspaTag assay. Treg and non-Treg cells were incubated for 4 h with 1 μg/ml MDP, and subsequently treated for a 16-h time course with 100 ng/ml FasL. Cell samples were stained using a CaspaTag apoptosis detection kit (Millipore, Billerica, MA) and analyzed via flow cytometry.
Real Time PCR
RNA was isolated from CD4+CD25++ Treg cells (~5 × 105 per experiment), CD4− PBMCs, 293T cells, and murine EGFP+FOXP3+ splenocytes for real-time PCR by the Qiagen RNeasy kit per the manufacturer’s protocol (Qiagen, Valencia, CA; 74104). The oligonucleotide primers used were as follows: HUMAN NOD2, 5′-CTGAAGAATGCCCGCAAGGT-3′ FORWARD and 5′-GTCTCTTGGAGCAGGCGGATG-3′ REVERSE; HUMAN CD11b, 5′-AGGCAGACAGAGAGGGAGTCATTCGCTACGTCA-3′ FORWARD and 5′-ATCACGAGGCGGCTTGGATGCGATGGTATTAAG-3′ REVERSE; MOUSE NOD2, 5′-TGTCAGCGCTCCTCAGGAAGTTCGTTCGCACAG-3′ FORWARD and 5′-AGGTTCCCGGTGGTGCTTTCTCAGGTACAGTTG-3′ REVERSE.
Treg and non-Treg cells were treated with 1 μg/ml MDP for 4 h, and RNA for PCR was isolated using the RNeasy kit per the manufacturer’s protocol. The oligonucleotide primers used were as fo0llows: BCL-2 V BETA, 5′-CAGCCAGCTGCACCTGACGCCCTTC-3′ FORWARD and 5′-CAGACTCACATCACCAAGTGCACC-3′ REVERSE; BCL-2 V ALPHA, 5′-GAGGAGCTCTTCAGGGACGGGGTG-3′ FORWARD and 5′-CTTCAGAGACAGCCAGGAGAAATC-3′ REVERSE; FLIP, 5′-CTGGAGCCTGTGTACTGCGGACAT-3′ FORWARD and 5′-AGAGTGTGCTGCAGCCAGACATAA-3′ REVERSE; BCL XL, 5′-GCACTGTGCGTGGAAAGCGTAGAC-3′ FORWARD and 5′-CGGCTGCTGCATTGTTCCCATAGA-3′ REVERSE; CIAP 1, 5′-ATTCCAACAGAAGATGTTTCAGGTCT-3′ FORWARD and 5′-GAGAAATGTACGAACAGTACCCTTGA-3′ REVERSE; CIAP 2, 5′-TGGCACTTTTTCAACATTTGACTTGT-3′ FORWARD and 5′-CCGCAATTGTTCTTCCACTGGTAGAT-3′-REVERSE; XIAP, 5′-TGGGGTTCAGTTTCAAGGACATTAAG-3′ FORWARD and 5′-GACTGTGTAGCACATGGGACACTTGT-3′ REVERSE; ACTIN, 5′-GAGAAAATCTGGCACCACACCTTCTACAATG-3′ FORWARD and 5′-CAGGGATAGCACAGCCTGGATAGCAACGTAC-3′ REVERSE.
Quantitative PCR was performed using the 7900HT real-time PCR instrument (Applied Biosystems, Foster City, CA). Detection was Power SYBR Green from Applied Biosystems.
Genotyping of NOD2 polymorphisms
PCR was performed on peripheral blood and mucosal specimens, using a biotinylated primer targeting the three major functional mutations observed in patients with CD: Arg702Trp (single nucleotide polymorphism [SNP] 8), Gly908Arg (SNP 12), and Leu1007fsinsC (SNP 13) (20). Processing was performed using a Pyrosequencer (PSQ 96), and results were analyzed using PSQ evaluation software (Biotage AB, Uppsala, Sweden).
Statistical considerations
All experiments were performed a minimum of three times. In experiments with results that can be quantified (quantification of Tregs in LP, suppression assays, luciferase assays, survival and apoptosis assays), experiments were performed in triplicate with mean and SD reported. Tests of significance in paired samples included the two-tailed paired t test with confidence intervals set for 95%, p < 0.05 (GraphPad Prism statistical software).
Results
Functional characterization of FOXP3+ T cells isolated from the LP of normal control subjects and patients with CD
Immunohistochemistry staining for FOXP3 and CD4 was performed on 30 surgical or endoscopic biopsy samples from the inflamed colonic mucosa of patients with CD and compared with biopsy tissue from 10 samples of normal colonic mucosa. The numbers of FOXP3+ T cells and CD4+ T cells were counted per high-power field (0.307 mm2). Results were reported as the number of FOXP3+ T cells per 100 CD4+ T cells to adjust for an overall lymphocyte response to inflammation. In patients with CD, there was a significant increase in FOXP3+ cells present in the LP compared with normal colonic mucosa (Fig. 1A). To perform functional assays of isolated FOXP3+ cells, we optimized flow cytometric identification of LP FOXP3+ T cells, using cell surface staining Abs within LP tissue isolated from mucosal endoscopic biopsies.
FIGURE 1.

Increased FOXP3+ Treg cells in the LP of patients with CD maintain normal suppressive capacity. A, Mean number of FOXP3+ T cells per 100 CD4+ T cells is increased in the LP of patients with CD compared with normal colonic mucosa (45.7 ± 20.2 versus 20.7 ± 4.4; p = 0.005). B, FACS analysis reveals CD4+CD25++ LP T cells to be enriched for the expression of FOXP3 in both patients with CD (n = 22) and healthy control subjects (n = 6). The dot plot on the left is a representative example of FOXP3 and CD25 staining among CD4+ cells isolated from the LP of a CD patient. The inset box represents the gate upon which cells are sorted. Aliquots of sorted cells are then stained for FOXP3, demonstrating consistent FOXP3 enrichment (right panel, CD mean/SE, 72.8% ± 5.0, and control, 66.67% ± 5.9). C, In health and in the setting of CD, LP Treg cells suppress a common T responder population. Presented are the mean and SD of thymidine counts collected from three consecutive healthy subjects or patients with CD. Individual experiments were performed in triplicate, and the combined means/SEs for all three patients are demonstrated. D, T responder cells isolated from the LP of patients with active CD can be suppressed by control Treg cells. Presented are the mean and SD of thymidine counts collected from four consecutive patients with CD. Individual experiments were performed in triplicate, and the combined means/SEs for all three patients are demonstrated in D.
Flow cytometric cell sorting for the population of CD4+ LP lymphocytes exhibiting the highest expression of CD25 (Fig. 1B, left panel) allowed mean FOXP3 enrichment of >66% (Fig. 1B, right panel) among patients with CD (n = 22) and control subjects (n = 6). Flow sorting for the CD4+CD25++ cells returned significantly higher numbers in patients with active disease (n = 6) than in those with inactive disease (n = 16 patients; mean 10,554 cells versus 3413; p < 0.05). Postflow cytometry cell sorting cell numbers greater than 7500 allowed in vitro expansion over 10 d and subsequent evaluation for suppressive function. Expanded FOXP3+ T cells were titrated into cell culture preparations with CD4+CD25− (FOXP3−) T responder cells and irradiated APCs. To adequately compare Treg cell suppressive function in individual healthy control subjects with that in patients with CD, we used a single control T responder cell population and APC population frozen in aliquots. Thus in this set of experiments, FOXP3+ cells from patients and controls were compared for suppressive function against the same T responder cell population on the same APCs. Fig. 1C demonstrates the suppressive capacity of LP Treg cells in both CD patients (n = 3) and normal subjects (n = 3). In addition, we evaluated the sensitivity of LP T responder cells isolated from CD patients to the suppressive function of peripheral Treg cells isolated from healthy control subjects. Fig. 1D demonstrates normal suppression of LP T responder cells from CD patients (n = 4).
Patients with the variant NOD2 genotype are deficient in LP Treg cells
These observations were evaluated by NOD2 genotype. Genotyping for the three disease-associated SNPs of the NOD2 gene (Arg702Trp, Gly908Arg, and Leu1007fsinsC) was performed on the 30 tissue samples presented in Fig. 1A. Among CD samples, there were 17 wild-type (wt), 9 heterozygous variants, and 4 homozygous variants of the NOD2 gene. The patients with homozygous variant genotypes had LP FOXP3+ T cell counts significantly lower than those found in the other CD patients (Fig. 2A). To exclude an effect on FOXP3+ lymphocyte number by disease severity and medication usage, we stratified the 30 disease samples by both variables. Fig. 2B and 2C show no association of FOXP3+ cell number with either disease severity or medication. Representative histological sections demonstrate the paucity of FOXP3+ LP lymphocytes in a patient homozygous for the Leu1007fsincC genotype (Fig. 3A) when compared with a patient with the homozygous wt genotype (Fig. 3B).
FIGURE 2.

Homozygosity for variant NOD2 genotype is associated with a deficiency of LP Treg cells. A, Thirty samples isolated from colonic biopsy specimens or surgical specimens of patients with active CD were studied by immunohistochemistry for CD4+ and FOXP3+ lymphocytes. DNA obtained from paraffin blocks was examined for polymorphic variation in NOD2, and the quantification of FOXP3+ cells was stratified by genotype. Patients with homozygosity for NOD2 polymorphisms demonstrated significantly fewer FOXP3+ lymphocytes when compared with heterozyogous or wt genotypes. B, The observed deficiency in NOD2 variant genotypes was seen in mild, moderate, or severe disease. Pathological interpretation of clinically indicated biopsies was used to define mild, moderate, and severe categories listed along the x-axis. C, Quantification of FOXP3+ lymphocytes was stratified by medication. No significant relationship between FOXP3+ lymphocytes and medication usage was found. Anti-TNF, infliximab or adalimumab; immunomod, azathiaprine, 6-mercaptopurine, or methotrexate; A + I, anti-TNF + immunomodulator; S + A or I, corticosteroids plus anti-TNF or immunomodulator.
FIGURE 3.

Representative section of immunohistochemistry for FOXP3+ lymphocytes demonstrates a paucity of FOXP3+ T cells (brown stain) in a patient homozygous for NOD2 variation (Leu1007fsincC) (A) when compared with a patient with wt genotype (B) (original magnification ×10, brown stain represents anti-FOXP3).
NOD2 is expressed in human CD4+ T cells
We began our investigation into the potential functional relevance of NOD2 signaling in T cells by confirming the expression of NOD2 in human CD4+ T cells. Freshly isolated human PBMCs were separated into CD4+CD25++FOXP3−-enriched Treg cells and CD4−FOXP3-poor cells by magnetic bead separation. Protein concentration from highly enriched Treg cells isolated from individual blood donors was ultimately insufficient to perform reliable Western blotting. Thus, mRNA was isolated from Treg cells and CD4− PBMCs from individual healthy blood donors (n = 6), and the expression of Nod2 mRNA by real-time PCR was measured. Fig. 4A demonstrates an approximate 10-fold increased expression of Nod2 mRNA in CD25++ human CD4 T cells when compared with the human embryonic kidney cell line, 293T, a cell line known to not express NOD2 (mean, 9.34 ± 1.30; p = 0.0077) (12, 13). CD4− cells (flow through from the magnetic column separation containing APCs) provide the positive control (mean, 29.73 ± 3.55). To evaluate for significant contamination of the CD25++CD4+ Treg cells with CD4− PBMCs, we performed flow cytometry for CD4 and demonstrate >98% purity for CD4+ T cells (Fig. 4B, FACS plot and bar graph). This cell fraction was also enriched in the expression of FOXP3 (Fig. 4B, mean, 78.9% ±2.0). Restricted expression of the integrin CD11b (integrin α M, present on the cell surface of innate immune leukocytes) to the CD4− cell subset supports adequate stringency of cell separation (inset DNA gel, Fig. 4A). As further evidence that the measured increased Nod2 mRNA exists in Treg cells and not a contaminating population of CD4− innate immune cells, we isolated by flow cytometry FOXP3-expressing cells from the FOXP3EGFP mouse (B6.Cg-Foxp3tm2Tch/J; The Jackson Laboratory, Bar Harbor, ME), a C57/BL6 strain in which Foxp3 mRNA expression strictly segregates with EGFP+ T cells (21). In Fig. 4C, we demonstrate significant mRNA expression in EGFP+ FOXP3+ Treg cells compared with mRNA isolated from murine muscle (mean/SE, 21.35 ±0.48 versus 1.0; p < 0.0005). Of interest, in murine splenocytes, Nod2 mRNA expression was consistently higher in Treg cells than in CD4− splenocytes (mean/SE, 21.35 ± 0.48 versus 15.58 ± 0.58; p = 0.016). These experiments were repeated using CD4+CD25++ splenocytes isolated from three wt C57/BL6 mice with identical results (data not shown). Thus, using quantitative PCR we demonstrate expression of Nod2 mRNA in human and murine Treg cells.
FIGURE 4.

Nod2 mRNA is expressed in human Treg cells. A, RNA extracted from freshly isolated CD4+CD25++ Treg cells, CD4− PBMCs, and 293T cells is probed for Nod2, CD11b, and actin. Expression of Nod2 mRNA within Treg cells and CD4− PBMCs (CD4−) is significantly increased over 293T cells (mean, 9.34 ± 1.30, 29.73 ± 3.55; p = 0.0077). The inset DNA gel demonstrates an absence of the CD11b mRNA transcript in Treg cells, evidence against significant cell subset contamination. Six unique healthy blood donors were used for these experiments, and results represent the mean/SE of each independent experiment. B, The CD4+CD25++ human T cell population is highly purified. The upper flow cytometry dot plots demonstrate >98% purity of isolated cells for CD4 and 78% purity for FOXP3. The bar graph demonstrates the mean (SE) of CD4/FOXP3 enrichment for the six unique blood donors. C, RNA extracted from EGFP+FOXP3+ murine splenocytes or CD4− murine splenocytes demonstrates significant upregulation of murine Nod2 mRNA transcripts compared with murine muscle negative control (21.35 ± 0.48, p < 0.0005; 15.58 ± 0.58, p = 0.016, respectively).
As it is well accepted that the NOD2 signaling pathway results in NF-κB activation (22), we sought to confirm functional relevance of NOD2 in human CD4+ T cells through assessment of NF-κB activation in response to MDP stimulation. Given the possibility that MDP may alternatively signal through the adaptor cryopyrin, NALP3 (23), we performed Western blotting for NALP3 in human T cells. Fig. 5 demonstrates expression of NALP3 in both human CD4− PBMCs and adherent human PBMCs (Ad, Fig. 5), but not CD4+ T cells.
FIGURE 5.
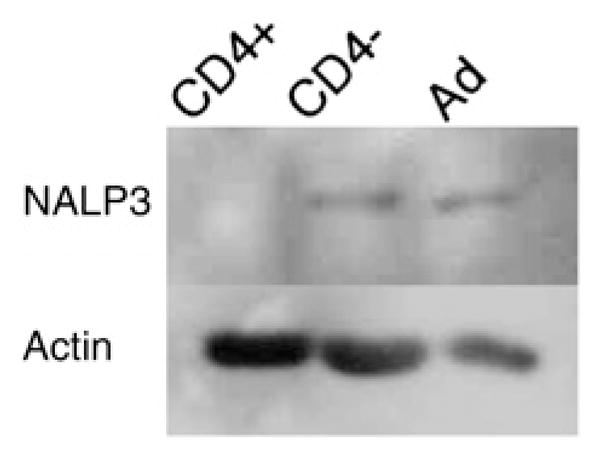
CD4+ T cells do not express NALP3. Protein lysates from freshly isolated human CD4+ T cells, CD4− PBMCs, or adherent PBMCs were probed from NALP3 and actin. NALP3 is not present in human CD4+ T cells. Ad, adherent.
We performed three independent assays of NF-κB activation in primary human T cells. First, Treg cells were transfected with an NF-κB luciferase reporter construct by electroporation. Upon incubation with the NOD2 ligand, MDP, CD25++ Treg cells demonstrated robust luciferase activity, evidence for functionally relevant NOD2 expression (Fig. 6A).
FIGURE 6.

NOD2 is functionally active in human Treg cells. A, Luciferase assay using Treg cells transfected with the P106 NF-κB 3 luciferase reporter construct demonstrates significant increase in luciferase activity upon MDP treatment (mean relative light unit ± SE, unstimulated Treg 350 ± 15.0 versus stimulated Treg, 2767.0 ± 145.3; p < 0.05). B, At 30 min, both p50 and p65 active NF-κB subunits are present in the nucleus in samples treated with MDP (black column, 0.659 ± 0.086 and 0.243 ± 0.082, respectively) when compared with untreated cells (open column, 0.377 ± 0.092 and 0.112 ± 0.046, respectively). C, FACS analysis performed on freshly isolated Treg cells demonstrates robust activation of the p65 NF-κB subunit upon treatment with 1 μg MDP (70.46% positive cells) compared with untreated cells (28.36% positive cells). Short-term (15 min) stimulation with PMA serves as a relevant positive control (upper right panel).
We subsequently performed two additional independent assays of NF-κB activation to confirm the luciferase results. Upon treatment with the NOD2 ligand, MDP, nuclear extract was isolated from fresh human Treg cells at time points of 30, 60, and 120 min. Active p50, p65, and c-Rel subunits were quantified by incubation and capture in consensus-binding site impregnated wells per the manufacturer’s recommendations (TransAM ELISA, Active Motif). Fig. 6B demonstrates translocation of the active NF-κB subunits p50 and p65 at 30 min in response to MDP. The MDP effect abated over 2 h (data not shown). No significant differences were seen in c-Rel (data not shown).
Finally, we studied activation of the p65 NF-κB subunit by flow cytometry. Using a fluorophore-labeled mAb specific for the nuclear localization sequence of p65 (Chemicon/Millipore, MAB3026), we subjected MDP-stimulated and unstimulated freshly isolated human Treg cells to flow cytometry at 30, 60, and 120 min post-MDP stimulation. At 60 min, a peak fluorescent signal was evident in MDP-treated cells (lower right panel, Fig. 6C) when compared with untreated cells. The fluorescent intensity of PMA-treated cells served as a positive control (Fig. 6C). Thus, human Treg cells express Nod2 mRNA, and NOD2 appears to be functionally relevant as measured by NF-κB activation in response to MDP. Understanding that NF-κB is relevant to T cell apoptotic programs, we next investigated an association between NOD2 ligation and apoptosis in primary human T cells.
MDP ligation protects Treg cells from Fas-mediated apoptosis
The well-characterized Fas-dependent apoptotic program is critically important to lymphocyte homeostasis in humans. Indeed, our examination of FasR expression in LP CD4+ lymphocytes demonstrated marked upregulation of FasR in FOXP3+ T cells (Fig. 7A), an observation that may contribute to the established unique susceptibility of Treg cells to Fas ligation in the resting state (24, 25). As functional assays of freshly isolated Treg cells could not be performed with the small number of LP isolates, the following studies of apoptosis are performed in peripherally isolated Treg cells. Peripheral FOXP3+ T cells also overexpress the FasR when compared with FOXP3−CD4+ T cells (Fig. 7B).
FIGURE 7.
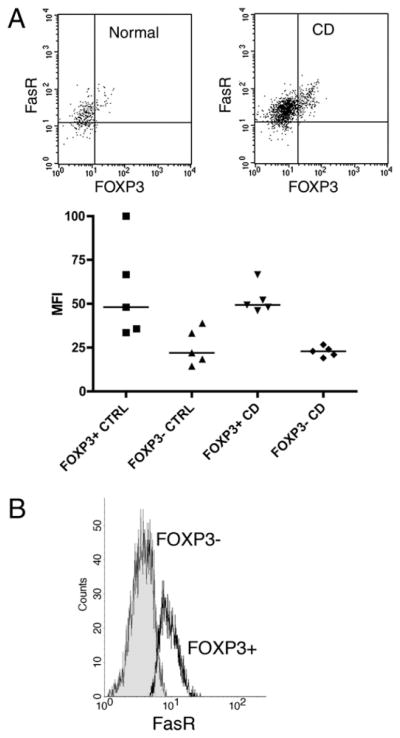
FOXP3+ LP and peripheral Treg cells demonstrate high-level expression of the Fas receptor. A, FACS performed on LP CD4+ T cells stained for Fas receptor and FOXP3 reveals significantly greater Fas receptor expression in FOXP3+ cells as compared with FOXP3− cells. A representative dot plot demonstrates high-level expression of the Fas receptor in FOXP3+ cells in both normal patients (left dot plot) and patients with CD (right plot). Below, the mean fluorescent intensity demonstrates consistency of this observation across five consecutive normal subjects and patients with CD. B, FACS performed on peripheral CD4+ T cells stained for Fas receptor and FOXP3 reveals significantly greater Fas receptor expression in FOXP3+ cells (open histogram) as compared with FOXP3− cells (closed histogram). CTRL, control.
To begin, we performed an assay of cell survival by incubating freshly isolated peripheral Treg cells with and without MDP over 6 d. We measured survival by Trypan blue exclusion. Fig. 8A demonstrates significant percent survival in MDP-stimulated Treg cells over 6 d when compared with unstimulated cells (65% ± 4.8 versus 26.7% ± 7.3; p < 0.05). To evaluate the Fas signaling pathway in response to NOD2 ligation, we performed the following study. Freshly isolated peripheral Treg cells were exposed to FasL with and without preincubation with MDP. Two independent measurement tools were applied to assay apoptotic cells. Initially, we performed a simple measurement of apoptotic cells by fixation and nuclear staining with Hoechst reagent. Apoptotic cells were identified by morphology and counted by a single blinded study participant (P.S.). Fig. 8B demonstrates protection against Fas-induced apoptosis in Treg cells treated with MDP (percent apoptosis, 26.8% ± 3.5% versus 61.6% ± 7.5%; p < 0.05). A requirement for NF-κB activation to achieve apoptosis protection is demonstrated by the abolition of MDP protection by the NF-κB pharmacological inhibitor Bay 11-7082 (Fig. 8B, mean 66% ± 7.8% apoptosis). Incubation with Bay 11-7082 alone did not induce apoptosis over the time course (data not shown). Subsequently, we corroborated the above results with a more mechanistic assay of apoptotic cell death, flow cytometric determination of effector caspases in response to Fas ligation.
FIGURE 8.

MDP stimulation of Treg cells protects cells from Fas-mediated apoptosis. A, Peripheral Treg cells were cultured in RPMI 1640 fortified media (17) for 6 d, and cell survival was measured by Trypan blue exclusion. The addition of 100 ng/ml of MDP resulted in significantly enhanced survival over 6 d (mean/SE, 63.3% ± 4.8 versus 26.7% ± 7.2; p < 0.05). B, Hoechst staining performed on FasL-treated (closed histogram) and untreated (open histogram) T cells shows a significant decrease in Fas-induced (100 ng/ml FasL) apoptotic Treg cells upon MDP incubation (percent apoptosis 26.8% ± 3.5% versus 61.6% ± 7.5%; p < 0.05). Pharmacological inhibition of NF-κB (Treg + MDP + Bay11) abrogated the suppressive effect of MDP (mean 66% ± 7.8% apoptosis). C, Flow cytometry for active caspase 3/7 demonstrates significant protection against Fas-mediated apoptosis at 10 h in Treg cells treated with MDP (1 μg/ml, closed histogram) compared with untreated cells (open histogram, 36.1 ±5.3 versus 21.0 ±1.7; p <0.05). D, Inhibition of NOD2 signaling through siRNA knockdown, NOD2 polymorphic variation, or genetic deletion abolishes the MDP-preventive effect. Each column represents the absolute protection against apoptosis provided by incubation with MDP (1 μg/ml) for 4 h prior to treatment with FasL (100 ng/ml). The open histograms demonstrate the significant apoptosis prevention of MDP treatment in wt human Treg cells (mean ± SE, 37.25 ± 7.2% protection) and mouse spleen Treg cells (mean ± SE, 28.75 ±6.0% protection). This protective effect is lost in human Treg cells isolated from a patient with variant NOD2 genotype (human mt NOD2, 4.0 ±3.2% protection), human Treg cells transfected with NOD2 siRNA (human wt NOD2 + RNAi, 1.7 ± 3.8% protection), and in mouse Treg cells isolated from the NOD2 knockout mouse strain (Mouse KO NOD2, 0.75 ± 3.6% protection). E, Freshly isolated CD4− human PBMCs were nucleofected with pooled siRNA specific for NOD2 (NOD2 siRNA) or pooled nonspecific siRNA (Scramble siRNA), and protein was isolated 96 h post nucleofection. The absence of a band in the NOD2 siRNA lane confirms adequate knockdown in primary human cells. 293T cells transfected with NOD2 and THP-1 cells serve as positive controls.
Fas-dependent activation of effector caspases 3/7 was measured through the use of a fluorochrome-labeled caspase substrate (Caspa Tag, FAM-DEVD-FMK) and flow cytometry. Indicative of Fas-dependent caspase activation, time course experiments demonstrated a progressively increasing percentage of fluorophore-labeled cells after Fas ligation in control Treg cells (open column, Fig. 8C). Preincubation of cells with MDP significantly inhibited caspase activation over the first 10 h of Fas treatment (closed column, Fig. 8C; 36.1 ± 5.3 versus 21.0 ±1.7; p < 0.05).
To directly demonstrate the protective function of NOD2 in Treg apoptosis, we interfered with NOD2 signaling in three ways. First, we transiently interfered with NOD2 expression through transfection of NOD2-specific siRNA (siGENOME SMARTpool siRNA; Thermo Scientific, Dharmacon) into human CD25++ Treg cells and exposed transfected and mock-transfected cells to FasL. Second, we exposed CD25++ Treg cells isolated from the spleen of wt or NOD2-deficient C57/BL6 mice (The Jackson Laboratory) to FasL and quantified apoptosis. Third, we prospectively identified a patient with homozygous NOD2 variant genotype (Leu1007f-sinsC) on no anti-Crohn’s therapy that might confound the above apoptosis-inducing experiments. As demonstrated in Fig. 8D, MDP preincubation results in a significant reduction in FasL-induced apoptosis in both human and mouse CD25++ Treg cells (mean 37.25% reduction in human Treg cells and 28.75% reduction in murine Treg cells, open histograms). Conversely, in Treg cells bearing transient knockdown of NOD2 (wt NOD2+ RNAi, Fig. 8D), genetic deletion of NOD2 (mouse knockout NOD2, Fig. 8D), or variant NOD2 genotype (mutant NOD2, Fig. 8D), preincubation with MDP does not protect against apoptosis as measured by Hoescht staining (closed histograms, Fig. 8D). Transient knockdown of NOD2 in freshly isolated primary CD4− PBMCs is demonstrated in Fig. 8E. 293T cells transfected with NOD2 and THP-1 cells serve as positive controls. Of interest, we observed a high background apoptotic rate over 16 h (mean, 40.2% ± 5.4) in human Treg cells with variant NOD2 genotype, in human Treg cells with transient knockdown of NOD2, and in murine T cells isolated from the NOD2-deficient mouse. Thus, to compare the MDP-induced protective function across these independent model systems, we represent the data as absolute MDP-induced apoptosis prevention (Fig. 8D). In our final set of experiments, we looked for NF-κB–responsive apoptotic genes to explain the above observations.
NOD2 ligation results in X-IAP gene activation
Peripheral Treg cells were isolated from 10 prospectively recruited healthy subjects and incubated with MDP. After 4 h of incubation, mRNA was purified and analyzed for expression of seven established NF-κB–responsive genes relevant to apoptosis by quantitative real-time PCR. For each experiment, cells from two patients collected on the same day were pooled to generate enough mRNA to run the desired assay; thus the experiment was repeated five times with reproducible results. Fig. 9 demonstrates the upregulation of the antiapoptotic gene X-IAP in response to MDP. The expression of Bcl-XL, Bcl-2, C-IAP-1/2, FLIP, and Bfl-1 was not consistently affected by MDP (data not shown).
FIGURE 9.

Quantitative PCR demonstrates upregulation of the NF-κB–responsive proapoptotic gene X-IAP upon treatment with MDP (1 μg/ml).
Discussion
We report two advances in the understanding of human FOXP3+ Treg cell biology. First, the experiments of Treg suppressor function are unique, as they use cells isolated from mucosal biopsies and evaluated for FOXP3 expression. The allogeneic assay derived to suppress healthy T responder cells demonstrates unequivocal in vitro suppressive function of LP Treg cells of patients with CD. In addition, we evaluated the sensitivity to suppression of T responder cells isolated from patients with CD. Second, we describe a deficiency of FOXP3+ Treg cells in the LP of patients homozygous for NOD2 gene variants. We propose a potential mechanism for this deficiency to be the failure of the NOD2 ligand, MDP, to protect Treg cells from Fas-mediated apoptosis. These results are significant, as the association between genetic polymorphisms in the pathogen receptor NOD2 and CD has yet to lead to a consensus on the mechanism of chronic intestinal inflammation in these patients.
The unequivocal suppressive function of FOXP3-expressing Treg cells isolated from the LP will be addressed first and is novel for three reasons. We have assayed T cells enriched for FOXP3 expression rather than CD25 expression, as has been performed in previous studies (26–28). In suppression assays, we used T cells isolated from inflamed LP rather than cultured from biopsy specimens (29) or isolated from mesenteric lymph nodes of surgical resection tissue (30). Using allogeneic T responder cells and Treg cells derived from the peripheral blood of healthy control subjects, we have dissected the suppressive function of Treg cells from the susceptibility to be suppressed by T responder cells. The finding that Treg cells suppress in vitro and T responder cells are “suppressible” when isolated from the inflamed tissue of patients with inflammatory bowel disease is hypothesis generating. Why, if Treg cells are present and functional, is the LP still inflamed? One possibility is that the inflamed milieu enriched in cytokines, such as IL-6 (a cytokine known to inhibit Treg function in vitro) (31), inhibits Treg function in vivo. Our data do not address this possibility, as the demonstrated full suppressive function is ex vivo. A second possibility is that FOXP3 is an unreliable marker of the Treg cell in the human LP, and perhaps FOXP3 expression and regulatory function is not a static phenotype. Indeed, plasticity in cells expressing FOXP3 to effector T cells capable of expressing IL-17 or follicular B Th cells has been recently described in mouse intestine (32, 33). Our data indicating suppressive in vitro function of these FOXP3+ cells argue against this possibility. Recent corroborating data also demonstrate the suppressive function of FOXP3+CD4+ T cells isolated from tumor micro-environments and the LP of several ulcerative colitis patients (34). This group differentiated primarily isolated FOXP3+ T cells from in vitro activated FOXP3+ T cells through effector cytokine expression in the latter (34). Similarly, in our experience, FOXP3+ LP lymphocytes isolated from 22 consecutive patients with CD did not demonstrate intracellular staining for the key effector cytokine IL-17 (data not shown). Our data thus support the idea that, in vivo, FOXP3 continues to be a reliable marker of Treg cells in the LP.
The novel finding that NOD2 gene variation is associated with a deficiency of FOXP3+ Treg cells in the LP will be considered next. Described immune phenomena associated with NOD2 polymorphisms include the impairment of intestinal cryptdin response to bacterial challenge and the augmentation of proinflammatory responses to Toll receptor and NOD2 signaling pathways in mouse models (35–37). Conversely, human studies of peripheral mono-nuclear cells in patients with variant NOD2 have demonstrated disrupted synergy between Toll receptor pathways and NOD2 in the generation of proinflammatory cytokines (38, 39). Thus, there is no consensus regarding the physiological relevance of variant NOD2 genotype and the progression to CD.
This report is the first of functionally relevant expression of NOD proteins in T cells; however, the finding is not wholly surprising, given the frequent report of other pathogen recognition receptors in mouse and human T cells. TLR2 and -4 signaling has been shown to affect proliferation and survival in murine CD4+CD25+ lymphocytes (40, 41). In human CD4+CD25+ T cells, TLR expression and FOXP3 expression are mechanistically linked. Human TLR5 has been shown to upregulate FOXP3 (42), and FOXP3 likewise can drive TLR gene transcription (14). There is also precedent for pathogen recognition receptors enhancing T cell survival. Murine CD4+ T cells expressing TLR3 and -9 demonstrate enhanced survival that is dependent upon activation of NF-κB and perhaps secondary to upregulation of the antiapoptotic Bcl family member, Bcl-xL (43).
Independent evidence supports the overall hypothesis that defective NF-κB activation is linked to CD. A murine model of NF-κB deficiency (p50−/− p65+/−) develops spontaneous colitis (44). The inbred mouse strain C3H/HeJ/Bir, which lacks functional TLR4, demonstrates impaired NF-κB signaling and is highly susceptible to colitis (45). Depending upon the cell type and milieu, NF-κB activation results in both proinflammatory cytokine production and resistance to apoptosis, through the upregulated expression of specific target genes.
Four pieces of evidence support a role for NOD2 in Treg apoptosis: 1) morphological confirmation of protection against Fas-mediated apoptosis in Treg cells preincubated with MDP, 2) decreased caspase activation in cells exposed to FasL and preincubated with MDP, 3) inhibition of NF-κB abrogates the suppressive effects of MDP, and 4) MDP stimulates NF-κB–dependent antiapoptotic gene transcription in human T cells. The NOD2 stimulatory pathway through the serine threonine kinase RICK (RIP2) to NF-κB–dependent signaling pathways is well defined, yet our data do not rule out potentially significant MAPK signaling events in the apoptotic pathway. In particular, the importance of MAPK activation in apoptotic events in leukemic cells has been recently described (46). Further investigation into the precise NOD2-dependent signaling events in human T cells is required.
The importance of Fas death receptor signaling in colitis is established from recent data in a hapten model of murine colitis. Depletion of FOXP3+ Treg cells was demonstrated in the inflamed colon, and this observation was established to be Fas dependent, as the transfer of Fas-insensitive FasR−/− Treg cells was resistant to depletion (25). Our data demonstrating high-level expression of FasR and exquisite sensitivity to Fas-mediated apoptosis are in accordance with this published data.
Our findings support a role for NOD2 signaling in Treg cell survival; however, the possibility exists that APC recruitment of Treg cells from the memory T cell or naive T cell compartment depends upon NOD2 signaling. In support of this possibility, recent data demonstrate the potential for the polymorphic variant NOD2 (Leu1007fsinsC) to inhibit IL-10 production in human monocytes (47). Further study of all three polymorphic variants and ex vivo methodology to convert T cells to Treg cells is required to fully address this possibility.
In conclusion, this report adds to the current published literature in the following ways. Primary CD4+FOXP3+ T cells isolated from the LP of diseased and unaffected tissue in patients with IBD have been shown unequivocally to function as suppressor cells in vitro, supporting the concept that, in vivo, FOXP3 continues to be a useful marker of human Treg cells. Like TLR family members, the pathogen recognition receptor NOD2 is functionally relevant in human Treg cells and, upon stimulation, enhances Treg cell resistance to Fas-mediated apoptosis. Our finding of depleted FOXP3+ T cells in the LP of CD patients with NOD2 variant genotype suggests a contributing role of Treg apoptosis to the underlying pathogenicity of NOD2 genotype variance in CD.
Acknowledgments
This work was supported by the Dana Human Immunology Award and the International Organization of IBD for the Study of Inflammatory Bowel Disease.
Abbreviations used in this paper
- A + I
anti-TNF + immunomodulator
- Ad
adherent
- anti-TNF
infliximab or adalimumab
- CD
Crohn’s disease
- CTRL
control
- FasL
Fas ligand
- immunomod
azathiaprine, 6-mercaptopurine, or methotrexate
- FOXP3
fork-head box P3
- LP
lamina propria
- MDP
muramyl dipeptide
- S + A or I
cortico-steroids plus anti-TNF or immunomodulator
- siRNA
small interfering RNA
- SNP
single nucleotide polymorphism
- Treg cell
T regulatory cell
- wt
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Strober W, I, Fuss J, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 2.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 3.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang Q, Bluestone JA. Regulatory T-cell physiology and application to treat autoimmunity. Immunol Rev. 2006;212:217–237. doi: 10.1111/j.0105-2896.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 6.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 7.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 8.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. [see comment] Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 9.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. [see comment] Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 10.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Wellcome Trust Case Control Consortium. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Girardin SE, Hugot JP, Sansonetti PJ. Lessons from Nod2 studies: towards a link between Crohn’s disease and bacterial sensing. Trends Immunol. 2003;24:652–658. doi: 10.1016/j.it.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 13.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 14.Bell MP, Svingen PA, Rahman MK, Xiong Y, Faubion WA., Jr FOXP3 regulates TLR10 expression in human T regulatory cells. J Immunol. 2007;179:1893–1900. doi: 10.4049/jimmunol.179.3.1893. [DOI] [PubMed] [Google Scholar]
- 15.Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, Knolle PA, Thomas RK, von Bergwelt-Baildon M, Debey S, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–2025. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 16.Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, Blazar BR, Porter SB. In vitro-expanded human CD4(+)CD25(+) T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104:453–461. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 17.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 18.Bell MP, Huntoon CJ, Graham D, McKean DJ. The analysis of costimulatory receptor signaling cascades in normal T lymphocytes using in vitro gene transfer and reporter gene analysis. Nat Med. 2001;7:1155–1158. doi: 10.1038/nm1001-1155. [DOI] [PubMed] [Google Scholar]
- 19.Ten RM, Paya CV, Israël N, Le Bail O, Mattei MG, Virelizier JL, Kourilsky P, Israël A. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappa B indicates that it participates in its own regulation. EMBO J. 1992;11:195–203. doi: 10.1002/j.1460-2075.1992.tb05042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugot JP, Cho JH. Update on genetics of inflammatory bowel disease. Curr Opin Gastroenterol. 2002;18:410–415. doi: 10.1097/00001574-200207000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 22.Girardin SE, I, Boneca G, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 23.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 24.Fritzsching B, Oberle N, Eberhardt N, Quick S, Haas J, Wildemann B, Krammer PH, Suri-Payer E. In contrast to effector T cells, CD4+ CD25+FoxP3+ regulatory T cells are highly susceptible to CD95 ligand- but not to TCR-mediated cell death. J Immunol. 2005;175:32–36. doi: 10.4049/jimmunol.175.1.32. [DOI] [PubMed] [Google Scholar]
- 25.Reardon C, Wang A, McKay DM. Transient local depletion of Foxp3+ regulatory T cells during recovery from colitis via Fas/Fas ligand-induced death. J Immunol. 2008;180:8316–8326. doi: 10.4049/jimmunol.180.12.8316. [DOI] [PubMed] [Google Scholar]
- 26.Makita S, Kanai T, Nemoto Y, Totsuka T, Okamoto R, Tsuchiya K, Yamamoto M, Kiyono H, Watanabe M. Intestinal lamina propria retaining CD4+CD25+ regulatory T cells is a suppressive site of intestinal inflammation. J Immunol. 2007;178:4937–4946. doi: 10.4049/jimmunol.178.8.4937. [DOI] [PubMed] [Google Scholar]
- 27.Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, Nakamura T, Koganei K, Fukushima T, Watanabe M. CD4+ CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol. 2004;173:3119–3130. doi: 10.4049/jimmunol.173.5.3119. [DOI] [PubMed] [Google Scholar]
- 28.Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, Zeitz M, Duchmann R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology. 2005;128:1868–1878. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 29.Kelsen J, Agnholt J, Hoffmann HJ, Rømer JL, Hvas CL, Dahlerup JF. FoxP3(+)CD4(+)CD25(+) T cells with regulatory properties can be cultured from colonic mucosa of patients with Crohn’s disease. Clin Exp Immunol. 2005;141:549–557. doi: 10.1111/j.1365-2249.2005.02876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saruta M, Yu QT, Fleshner PR, Mantel PY, Schmidt-Weber CB, Banham AH, Papadakis KA. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin Immunol. 2007;125:281–290. doi: 10.1016/j.clim.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 32.Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kryczek I, Liu R, Wang G, Wu K, Shu X, Szeliga W, Vatan L, Finlayson E, Huang E, Simeone D, et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res. 2009;69:3995–4000. doi: 10.1158/0008-5472.CAN-08-3804. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 36.Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 38.van Heel DA, Ghosh S, Butler M, Hunt K, Foxwell BM, Mengin-Lecreulx D, Playford RJ. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur J Immunol. 2005;35:2471–2476. doi: 10.1002/eji.200526296. [DOI] [PubMed] [Google Scholar]
- 39.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 40.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. [see comment] J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 43.Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erdman S, Fox JG, Dangler CA, Feldman D, Horwitz BH. Typhlocolitis in NF-kappa B-deficient mice. J Immunol. 2001;166:1443–1447. doi: 10.4049/jimmunol.166.3.1443. [DOI] [PubMed] [Google Scholar]
- 45.Elson CO, Cong Y, Sundberg J. The C3H/HeJBir mouse model: a high susceptibility phenotype for colitis. Int Rev Immunol. 2000;19:63–75. doi: 10.3109/08830180009048390. [DOI] [PubMed] [Google Scholar]
- 46.Nishioka C, Ikezoe T, Yang J, Yokoyama A. Inhibition of MEK/ERK signaling induces apoptosis of acute myelogenous leukemia cells via inhibition of eukaryotic initiation factor 4E-binding protein 1 and down-regulation of Mcl-1. Apoptosis. 2010 Mar 11; doi: 10.1007/s10495-010-0483-y. [DOI] [PubMed] [Google Scholar]
- 47.Noguchi E, Homma Y, Kang X, Netea MG, Ma X. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol. 2009;10:471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]