

Abstract
Lentiviruses contain accessory genes that have evolved to counteract the effects of host cellular defence proteins that inhibit productive infection. One such restriction factor, SAMHD1, inhibits HIV-1 infection of myeloid-lineage cells 1,2 as well as resting CD4+ T cells 3,4 by reducing the cellular dNTP concentration to a level where the viral reverse transcriptase cannot function 5,6. In other lentiviruses, including HIV-2 and related SIVs, SAMHD1 restriction is overcome by the action of viral accessory protein x (Vpx) or the related viral protein r (Vpr) that target and recruit SAMHD1 for proteasomal degradation 7,8. The molecular mechanism by which these viral proteins are able to usurp the host cell’s ubiquitination machinery to destroy the cell’s protection against these viruses has not been defined. We present here the crystal structure of a ternary complex of Vpx with the host cell’s E3 ligase substrate adaptor DCAF1 and the C-terminal region of SAMHD1. Vpx is made up of a three-helical bundle, stabilised by a zinc finger motif and wraps tightly around the disc-shaped DCAF1 molecule to present a new molecular surface. This adapted surface is then able to recruit SAMHD1 via its C-terminus making it a competent substrate for the E3 ligase to mark for proteasomal degradation. The structure provides the first description of how a lentiviral accessory protein is able to subvert the cell’s normal protein degradation pathway to inactivate the cellular viral defence system.
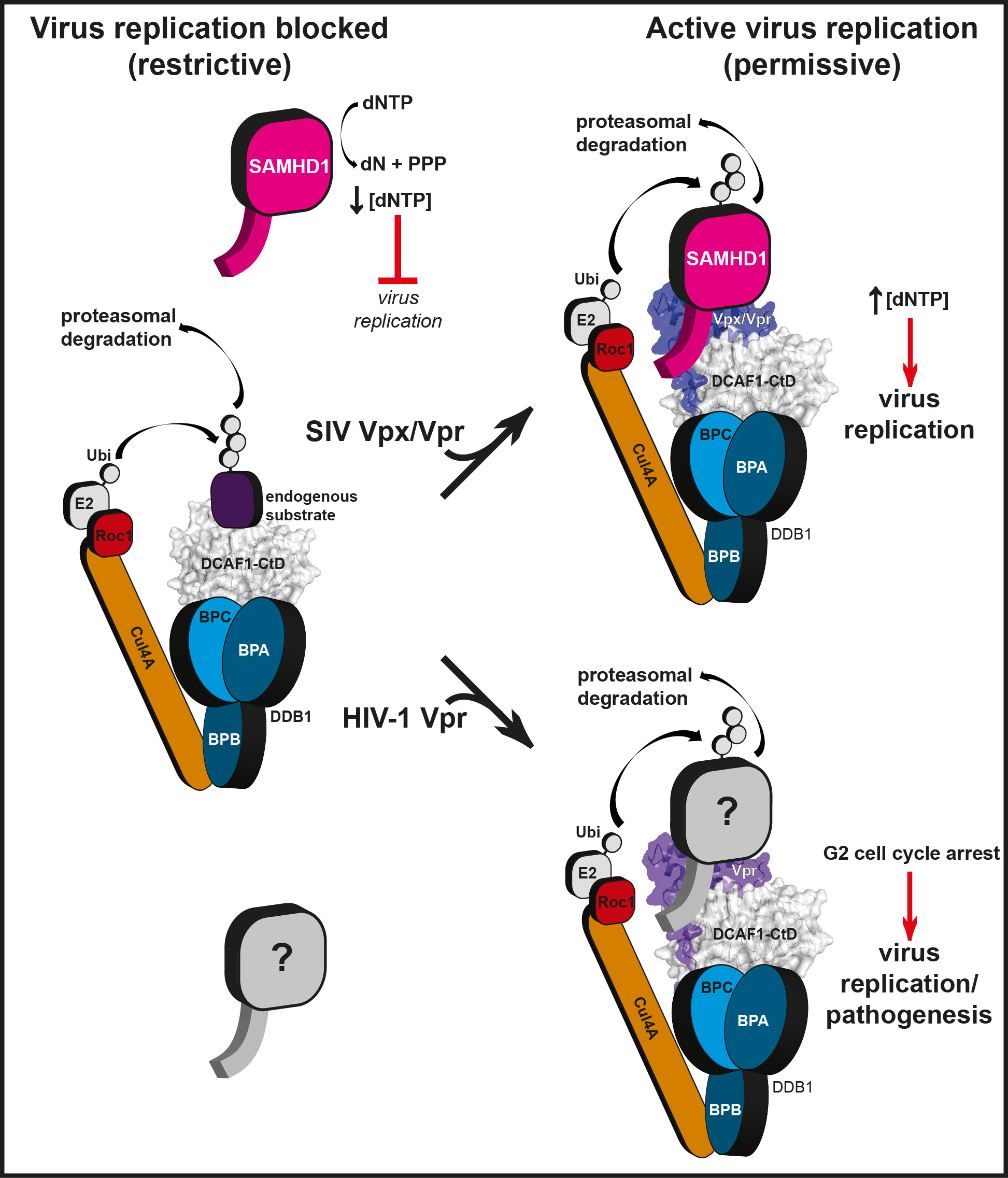
HIV-1 infection of myeloid and CD4+ T cells is inhibited by the post-entry restriction factor SAMHD1. In other primate lentiviruses, including HIV-2 and SIV, this block is overcome by the expression of the Vpx accessory protein. Vpx recruits SAMHD1 to the DDB1/CUL4A/ROC1 E3 ubiquitin ligase complex through interaction with the substrate-adaptor protein DCAF1 and facilitates its degradation through the proteasomal pathway 1,2,7,8. To understand the mechanism of Vpx-mediated recruitment of SAMHD1 we assessed which regions of each molecule (Figure 1a) are required for the interaction. These data reveal that only SAMHD1 molecules containing a C-terminal region (residues 582-626) are able to support ternary complex formation; compare central and left panels (Figure 1b) and that this region alone is sufficient for the interaction, right panel, Figure 1b. We therefore determined the crystal structure of the ternary complex of the C-terminal WD40 domain of DCAF1 (DCAF1-CtD) together with the Vpx of SIV from Sooty mangabey (Vpxsm) and the C-terminal region of SAMHD1 (SAMHD1-CtD). The crystal structure was solved by SAD (Extended Data Figure 1, Table 1) and is shown in Figure 1c. DCAF1-CtD comprises a seven-bladed β-propeller disc-shaped molecule 45 Å in diameter and 20 Å in depth. Vpxsm comprises an antiparallel V-shaped 3-helical bundle that wraps around one side and the top of DCAF1-CtD. This arrangement of helices is conserved in the HIV-1 Vpr solution structure 9. However, the structures differ significantly at the helical termini and in Vpxsm zinc coordinated by His39Vpxsm, His82Vpxsm, Cys87Vpxsm and Cys89Vpxsm (Figure 1c, 2a) brings together the C-termini of helices-1 and 3 to stabilise the structure. Residues Asn606 to Asp624 of SAMHD1-CtD are also well ordered. They form two short perpendicular α-helices, helix-A (Leu610-Ala613) and helix-B (Arg617-Lys622) connected by a three-residue linker (S614-S616) and pack into a cleft between Vpxsm and DCAF1-CtD (Figure 1c).
Figure 1. The SAMHD1-CtD/Vpxsm/DCAF1-CtD complex.

(a) Schematic of proteins, CD – chromo domain, DD – dimerisation domain, SAM – sterile alpha motif, HD – His/Asp domain. Regions coloured grey (DCAF1, 1058-1396), red (SAMHD1, 582-626) and blue (Vpxsm, 1-112) were used for crystallisation. (b) Size exclusion chromatograms (black) of equimolar mixtures of Vpxsm, DCAF1-CtD and SAMHD1(26-583) (left), SAMHD1(26-626) (middle) and SAMHD1(582-626) (right). Chromatograms from individual components are also shown Vpxsm (blue), DCAF1-CtD (grey) and SAMHD1 (red). SDS-PAGE analyses of peaks are inset. Peak1 (void volume) contains unspecific aggregates. (c) Cartoon representation of the ternary complex. DCAF1-CtD, is shown in grey surface, β-propeller blades are numbered. SAMHD1-CtD is red, Vpxsm is blue and a zinc ion shown as grey sphere.
Figure 2. Intermolecular interfaces.

(a) Zn ion (grey sphere) and surrounding residues. Co-ordinating residues are displayed as sticks, co-ordinate bonds as green dashes. (b) Cartoon representation of SAMHD1-CtD/Vpxsm/DCAF1-CtD. DCAF1-Ctd is shown in grey, cylinders represent α-helices in SAMHD1-CtD (red) and Vpxsm (blue), intermolecular interfaces are highlighted by green boxes (I, II, III, IV). (c-f) Views of the interface between SAMHD1-CtD/Vpxsm/DCAF1-CtD (Box I) and Vpxsm/DCAF1-CtD (Box I-IV). Residues contributing to the interface are shown as sticks, hydrogen-bonding interactions as dashed lines and residues important for Vpx function highlighted with an asterisk.
The complex contains four interfacial regions (Figure 2b), a combined Vpxsm/SAMHD1/DCAF1 ternary interface (Figure 2c) and a more extensive DCAF1/Vpxsm binding surface with three sites of interaction (Figure 2d-f). The Vpxsm/SAMHD1 interaction buries 700 Å2 of molecular surface. At the interface the hydrophobic side chains of Leu610, Val618, Leu620 and Phe621 from SAMHD1 helix-A and -B pack into a hydrophobic pocket between the amino-termini of Vpxsm helix-1 and -3 (Figure 2c). The interface also contains electrostatic interactions between acidic residues Glu15Vpxsm and Glu16Vpxsm at the N-terminus of Vpxsm helix-1 and Arg609 and Arg617 part of a tri-basic Arg609/Arg617/Lys622 motif in SAMHD1. By contrast, the contact area between SAMHD1-CtD and DCAF1-CtD is small, just 210Å2. The only direct interaction is between Lys622 in the SAMHD1 tri-basic motif and Asp1092 of DCAF1 in the acidic Glu1091-Asp-Glu1093 loop connecting blade-7 and -1 of the DCAF1-CtD propeller (Figure 2c). Asp1092 is also hydrogen-bonded to Tyr66Vpxsm in an interaction that bridges SAMHD1-CtD and Vpxsm. Several other key residues in Vpxsm also mediate bridging interactions. These include Trp24Vpxsm that stacks against Arg617 of SAMHD1-CtD as well as hydrogen bonding to DCAF1 - Asn1135 (Figure 2e) and Tyr69Vpxsm that packs with Val618 of SAMHD1-CtD and is also hydrogen-bonded to the side chain of Glu1091 in the DCAF1-CtD Glu-Asp-Glu loop.
The DCAF1/Vpxsm interface is much larger, made up from three sites of interaction burying 2000 Å of surface (Figure 2d-f, Extended Data Table 2). The first involves the N-terminal extended region of Vpxsm (Glu6Vpxsm - Ser13Vpxsm) that packs against the concave surface of DCAF1 blade-1 spanning from the underside to the topside of the disc, making several hydrogen bonds and hydrophobic interactions (Figure 2d). Further interactions involve Trp24Vpxsm, Thr28Vpxsm, Ile32Vpxsm and Gln76Vpxsm in helix-1 and -3 that contact residues on the DCAF1-CtD blade-1 and -2 and the interspersing loop (Figure 2e). Helix-3 of Vpxsm lies in a groove between blade-1 and -7 on the upper face of DCAF1-CtD. Interactions between the charged and hydrophobic side chains of Lys77Vpxsm, Arg70Vpxsm, Phe80Vpxsm and Met81Vpxsm along its length with residues in the intra-strand loops of blade-1 and -7 of DCAF-CtD including the Glu-Asp-Glu loop comprise the third region of the DCAF1/Vpx interface (Figure 2f).
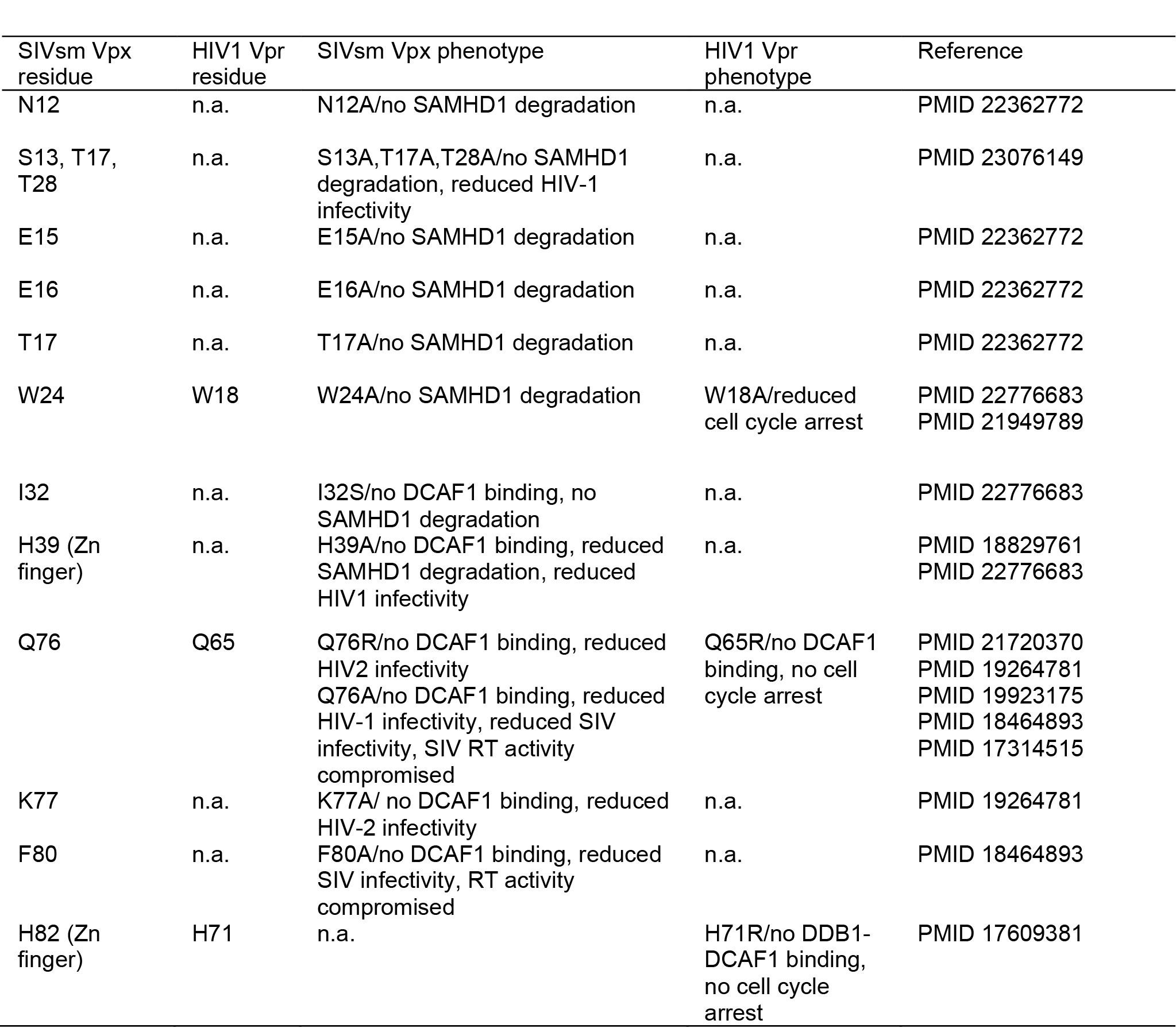
Mutation of many of the residues at these interfaces have been shown previously to reduce viral infectivity, disrupt Vpx binding to DCAF1 and interfere with proteasomal degradation of SAMHD1 (Extended Data Table 3). Examples include: the bridging residue, Trp24vpxsm 10 (Figure 2c, e), Gln76Vpxsm 11 that is hydrogen bonded to Asn1135 and Trp1156 of DCAF1 (Figure 2e) and Lys77Vpxsm 12 that is integral to an extensive salt bridge network that links residues in the DCAF1 Glu-Asp-Glu loop with Arg70Vpxsm, Tyr69Vpxsm and Tyr66Vpxsm in helix-3 of Vpxsm (Figure 2f).
In the structure, SAMHD1-CtD makes salt bridges at both the Vpxsm and DCAF1-CtD interfaces through charged side chains in the tribasic motif. In vitro binding studies show that full length SAMHD1 and SAMHD1-CtD have comparable affinity for the DDB1/Vpx/DCAF1 complex 13. However, to test if SAMHD1-CtD alone is sufficient for recruitment by Vpx and to assess the contribution of the tri-basic motif, an in vivo reporter assay was employed. SAMHD1-CtD was fused to the C-terminus of a tandem NLS-GFP protein that localises to the nucleus 14. 293T Cells transduced with the NLS-GFP-SAMHD1-CtD fusion, (Figure 3a) display GFP fluorescence in their nuclei (Figure 3b). Delivery of Vpx by infection with a SIV-(Vpx+) virus, greatly reduces the number of GFP+ cells, (Figure 3b, c). By contrast, infection with a SIV-(Vpx−) virus or addition of the proteasomal inhibitor MG132 with Vpx+ virus does not reduce the population of GFP+ cells (Figure 3b, c), indicating that loss of GFP fluorescence results from Vpx-mediated proteasomal degradation of NLS-GFP-SAMHD1-CtD. In addition, mutation of residues in the SAMHD1 tribasic motif (Arg609/Arg617/Lys622) to alanine or glutamate also abolishes or severely diminishes the capacity of Vpx to induce degradation of NLS-GFP-SAMHD1-CtD (Figure 3b, c). These data reveal that SAMHD1-CtD in isolation acts as a Vpx-dependent degron to induce the proteasomal turnover of a heterologous protein and that disruption of the protein-protein interactions observed in the crystal structure prevent Vpx-mediated degradation. When the same mutations are introduced into SAMHD1 they also reduce the capacity of Vpx to induce degradation, albeit to varying degrees (Figure 3d). However, all display wildtype restriction of HIV-1 indicating that the CtD is not required for SAMHD1 anti-HIV-1 activity (Figure 3e).
Figure 3. SAMHD1 C-terminal region.

(a) The NLS-GFP-SAMHD1-CtD degron. (b) Microscopic images of uninfected and SIV Vpx+/− infected 293T cells expressing NLS-GFP-SAMHD1-CtD wildtype or mutant R617E. (c) Quantitative analysis of GFPSAMHD1 degron expression following infection with SIV Vpx+/− viruses. Percentages of GFP+ cells are plotted against SIV virus titre. MG132 was added where indicated. Upper left panel is representative of three independent experiments. (d) Immunoblots of 293T cells co-transfected with SAMHD1 and Vpx plasmids (un, untransfected; −, empty vector; +, Vpx) probed with antibodies to SAMHD1 (upper), 14-3-3 (middle) or VpxHIV-2 (lower), representative of two independent experiments. (e) Restriction of HIV-1 infection by WT SAMHD1 and mutants. Restriction is expressed as a ratio of HIV-infected SAMHD1-transduced to infected SAMHD1-negative cells (>3 independent experiments with different viral stocks). Error bars show standard deviation.
To put the DCAF1-CtD/Vpxsm/SAMHD1-CtD structure into the context of the E3 ligase a molecular model of the entire CUL4A/DDB1/ROC1/DCAF/SAMHD1/Vpxsm assembly was constructed by superposition of DCAF1-CtD/Vpxsm/SAMHD1-CtD onto existing structures in the PDB database. First, the β-propellers of DCAF1-CtD and the related substrate adaptor, DDB2 were aligned (Extended Data Figure 2, inset) facilitating substitution of DCAF1-CtD for DDB2 in the existing DDB1-DDB2 structure 15. The DDB1-CUL4A/ROC1 interface is highly flexible 16. Therefore, the DCAF1-CtD/Vpxsm/SAMHD1-CtD/DDB1 model was superposed onto the two most extreme conformations available allowing the range of orientations that CUL4A/ROC can adopt with respect to Vpxsm and SAMHD1 to be visualised (Extended Data Figure 2). The model places Vpxsm and SAMHD1-CtD on the opposite face of the DCAF1-CtD disk from the DCAF1-DDB1 binding site, accessible to the RING domain of ROC1. Moreover, in both conformations SAMHD1 is placed in the proximity of the ROC1 RING domain, ideally located for ubiquitin transfer. Notably, regions of SAMHD1 proximal to the bound SAMHD-CtD are required for catalytic activation/tetramerisation 17 and association with Vpx/DCAF1/DDB1 inhibits SAMHD1 catalysis 13 suggesting that recruitment to the CUL4A/DDB1/ROC1 complex might additionally down-regulate SAMHD1 activity thorough tetramer disassembly.
Reprogramming of the CUL4/DDB1//ROC1 E3 ubiquitin ligase is also employed by paramyxovirus and hepatitis B Virus (HBV) to subvert the cellular antiviral response. These viruses usurp the interaction of DDB1 with DCAF1 by installing the viral substrate recruitment factors V or X in its place 18,19. In lentiviruses, a different strategy has evolved where the substrate recruitment factor, DCAF1, is itself adapted by association with accessory proteins to create a new binding pocket, in the case of Vpxsm and VpxHIV-2 to recruit SAMHD1 through its C-terminal region (Extended Data Figure 3). Notably, the SAMHD1 tribasic motif is conserved amongst primates but is absent in species that are not HIV/SIV hosts (Figure 4a). Similarly, the N-terminal Vpxsm sequence 9PPGNSGEET17 containing Glu15Vpxsm and Glu16Vpxsm that make salt bridges with Arg609 and Arg617 is conserved amongst all Vpx proteins that target human SAMHD1 for degradation (Figure 4b) 7 suggesting that the complementarity between these motifs is a driver of the specificity of the SAMHD1-CtD/Vpx interaction. In Vpx proteins that do not induce degradation of human SAMHD17,20, red-capped Mangabey (Vpxrcm) and Mandrill (Vpxmnd2), 9PPGNSGEET17 is not conserved, (Figure 4b). However, these Vpx proteins still induce degradation of SAMHD1 but do so by targeting it to DCAF1 through sequences located toward the N-terminus of 20.
Figure 4. Species specificity of the SAMHD1-Vpx interaction.

(a) Sequence alignment of the C-termini of SAMHD1. 100% type-conserved residues are boxed red, 60% cyan. Residue numbers refer to hsSAMHD1-CtD. Red stars - side chains involved in Vpxsm-DCAF1-CtD binding, red dots - main chain interactions. Black dots - residues whose mutation impairs DDB1-DCAF1-Vpx binding 8. Species above the red line are HIV-2/SIV hosts, Phos - phosphorylation site. (b) Alignment of Vpx and Vpr proteins, coloured and annotated as in (a) numbering refers to Vpxsm. Red stars - side chains involved in SAMHD1-CtD binding, red dots - main chain interactions and black stars - zinc ligands. Black dots indicate residues whose mutation impairs SAMHD1 degradation. Proteins above the red line induce degradation of human SAMHD1, proteins below cannot. (c) Type-conserved amino acid residues in Vpxsm and VprHIV-1 (orange) mapped onto the Vpxsm (blue ribbon)-DCAF1-CtD (white surface) structure. SAMHD1-CtD is shown as red ribbon.
In some SIVs, the evolutionally related accessory protein Vpr recruits SAMHD1 for degradation 7. By contrast, VprHIV-1 still associates with DCAF1 and the CUL4A/DDB1/ROC1 E3 ligase but results in cell cycle arrest at G2, likely through recruitment of an unidentified cellular factor to the E3 21-23. In the complex, we identified a structural zinc binding-site in Vpxsm (Figure 2a) that is conserved in both Vpx and Vpr proteins (Figure 4b). Mutation of His71 in VprHIV-1, the equivalent of the Vpxsm zinc-coordinating His82, abrogates DDB1-DCAF1 binding and Vpr-induced cell cycle arrest 24. Furthermore, most of the other conserved residues map to the DCAF1 interface (Figure 4c) and mutation of two of these (Gln65Vpr and Trp18Vpr, the equivalents of Gln76Vpx and Trp24Vpx) also results in loss of Vpr function 23,25 (Extended Data Table 3). These observations now suggest a strong structural conservation between these related accessory proteins. Moreover, given that Vpr-induced cell cycle arrest is also mediated through association with DCAF1 and the actions of the CUL4A/DDB1/ROC1 E3 21-23 it is likely that both factors utilise a similar mechanism to target cellular proteins to the CUL4A complex (Extended Data Figure 3). Consequently, although the cellular target(s) of Vpr are unknown the ternary complex presented here provides a structural model for the design of therapeutic agents that target the Vpx− and Vpr-DCAF1 interaction.
METHODS
Cloning, protein expression and purification
The DNA sequence coding for human SAMHD1 residues Q582-M626 (SAMHD1-CtD) was amplified by PCR from cDNA template using the oligonucleotide primer sequences: forward – ggc GGATCC t cag gat ggc gat gtt ata gcc cc, reverse - ggc GCGGCCGC tca tca cat tgg gtc atc ttt aaa aag ctg g. The PCR product was gel-purified and ligated into the pET52b plasmid (Merck Millipore) using standard restriction enzyme cloning to generate an N-terminal Strep-II-tagged fusion protein. N-terminally GST-tagged Sooty mangabey Vpx residues M1-A112 (Vpxsm) in the pET49b plasmid (Merck Millipore) was a generous gift of Dr David Goldstone (University of Auckland). Human DCAF1 residues A1058-E1396 (DCAF1-CtD) were amplified by PCR from cDNA template using the oligonucleotide primer sequences: forward - ggc CCATGG ca tca ttt cca aag tat gga ggg g, reverse - ggc GAGCTC ctc tgc cag acg ctg cct gcc. The PCR product was gel-purified and ligated into the pTriEx-6 plasmid (Merck Millipore) using standard restriction enzyme cloning to generate a C-terminal 10xHis-tagged fusion protein. Insert sequences were verified by DNA sequencing.
SAMHD1-CtD and GST-Vpxsm were expressed in the E. coli strain Rosetta 2 (DE3) (Merck Millipore). Bacterial cultures were grown in an incubator shaker at 37 °C. Protein expression was induced by the addition of 0.1 mM IPTG at A600 = 0.5. Afterwards, cultures were cooled to 18 °C and grown for further 20 h. To produce seleno-methionine (SeMet) -labelled Vpxsm, cells were grown to A600 = 0.5 at 37°C in M9 minimal medium. Then, an amino acid supplement (L-lysine, L-phenylalanine, L-threonine to a final concentration of 100 mg/l, L-isoleucine, L-leucine, L-valine and L-SeMet to a final concentration of 40 mg/l, respectively) was added to inhibit endogenous methionine biosynthesis and start SeMet incorporation 26. Fifteen minutes after addition, the culture was cooled to 18 °C. Protein expression was induced by the addition of 0.5 mM IPTG, and cells were grown for further 20 h.
Cultures were centrifuged for 20 min at 4,500 xg and 4 °C. Cell pellets were resuspended in 30 ml lysis buffer (50 mM Tris-Hcl pH 7.8, 500 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP, 1x EDTA-free mini complete protease inhibitors (Roche), 0.1 U/ml Benzonase (Novagen) per pellet of 1 L bacteria culture. Cells were lysed in an EmulsiFlex-C5 (Avestin). The lysate was cleared by centrifugation for 1 h at 48,000 xg and 4 °C. All further purification steps were performed at 4 °C or on ice. The cleared lysates were applied to 10 ml StrepTactin (IBA, for SAMHD1-CtD) or to 10 ml Glutathione-Sepharose (GE Healthcare, for GST-Vpxsm) columns. Columns were washed with 600 ml wash buffer (50 mM Tris-HCl pH 7.8, 500 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP). Column-immobilized GST-Vpxsm was additionally washed with 250 ml of wash buffer supplemented with 5 mM ATP, followed by 250 ml wash buffer supplemented with 1% CHAPS. SAMHD1-CtD was eluted from the column with 25 mM ammonium bicarbonate pH 7.5, 2.5 mM desthiobiotin. The elution peak was lyophilized, resuspended in 500 μl of 25 mM ammonium bicarbonate pH 7.5 and applied to a Superdex75 16/60 gel filtration column (GE Healthcare) equilibrated with 25 mM ammonium bicarbonate pH 7.5. The peak fractions were pooled, lyophilized and resuspended in 10 mM Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP. Small aliquots at a concentration of approximately 2 mM were flash-frozen in liquid nitrogen and stored at −80 °C. GST-Vpxsm was eluted from the column with wash buffer supplemented with 20 mM glutathione. The elution peak was concentrated to 5 ml and incubated overnight with 1 mg GST-3C protease. Cleaved Vpxsm was further purified on a Superdex200 26/60 gel filtration column (GE Healthcare) equilibrated with 10 mM Tris-HCl pH 7.8, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP. Peak fractions containing Vpxsm were concentrated to approximately 20 mg/ml and flash-frozen in liquid nitrogen in small aliquots for further storage at −80 °C. Seleno-methionine-substituted Vpxsm was purified in the same way.
For the production of DCAF1-CtD, recombinant baculovirus was generated by cotransfecting Sf9 cells with pTriEx-DCAF1-CtD and linearised BAC10:1629KO 27. Sf9 cells were cultured in SF900 II serum free medium (Invitrogen) at 28 °C. In a typical preparation, 2 L of Sf9 cells at 2×106 cells/ml density were infected with 4 ml of high titre DCAF1-CtD virus for 48h. For structure determination, selenium was incorporated into DCAF1-CtD by supplementing 921 Δ series medium (Expression Systems, LLC) with 50 mg/l seleno-methionine.
Sf9 cultures were centrifuged for 20 min at 4500x g and 4°C. Pellets were resuspended in 30 ml lysis buffer (50 mM Tris-HCl pH 7.8, 500 mM NaCl, 4 mM MgCl2, 30 mM imidazole-HCl pH 7.8, 0.5 mM TCEP, 1x EDTA-free mini complete protease inhibitors (Roche), 0.1 U/ml Benzonase (Novagen)) per pellet of 1 L culture. Cells were lysed in an EmulsiFlex-C5 (Avestin). The lysate was cleared by centrifugation for 1 h at 48,000 xg and 4 °C. All further purification steps were performed at 4 °C or on ice. The cleared lysate was applied to a 1 ml Ni-NTA Sepharose column (GE Healthcare). The column was washed with 500 ml wash buffer (50 mM Tris-HCl pH 7.8, 500 mM NaCl, 4 mM MgCl2, 30 mM imidazole-HCl pH 7.8, 0.5 mM TCEP). Protein was eluted with wash buffer supplemented with 300 mM imidazole pH 7.8. Peak fractions were pooled, concentrated to 2 ml and applied on a Superdex200 16/60 gel filtration column (GE Healthcare) equilibrated with 10 mM Tris-HCl pH 7.8, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP. Peak fractions containing DCAF1-CtD were pooled, concentrated to approximately 20 mg/ml and flash-frozen in liquid nitrogen in small aliquots for further storage at −80°C. Seleno-methionine-substituted DCAF1-CtD was purified in the same way.
Assembly of the ternary complex
Initially, the complex was prepared by mixing 100 μg of DCAF1-CtD, Vpxsm and SAMHD1-CtD in 10 mM Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP, followed by incubation overnight on ice. Complex formation was assessed by applying the mixture on an analytical Superdex75 10 300 GL gel filtration column (GE Healthcare), equilibrated with 10 mM Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP and SDS-PAGE analysis (Molecular weight of markers 97, 66.3, 55.4, 36.5, 31.0, 21.5, 14.4, 6.0 and 3.5 kDa). After optimisation, for large scale preparation of the complex, DCAF1-CtD, Vpxsm and SAMHD1-CtD were mixed in a molar ratio of 1:1.5:1.5 in 0.1 M Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP and incubated overnight on ice. The mixture was applied to a Superdex 75 16/60 gel filtration column (GE Healthcare) equilibrated with 10 mM Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP. Peak fractions containing all components were pooled, concentrated to approximately 5 mg/ml and directly used for crystallization experiments, after addition of an equimolar amount of SAMHD1-CtD.
Crystallisation and structure determination
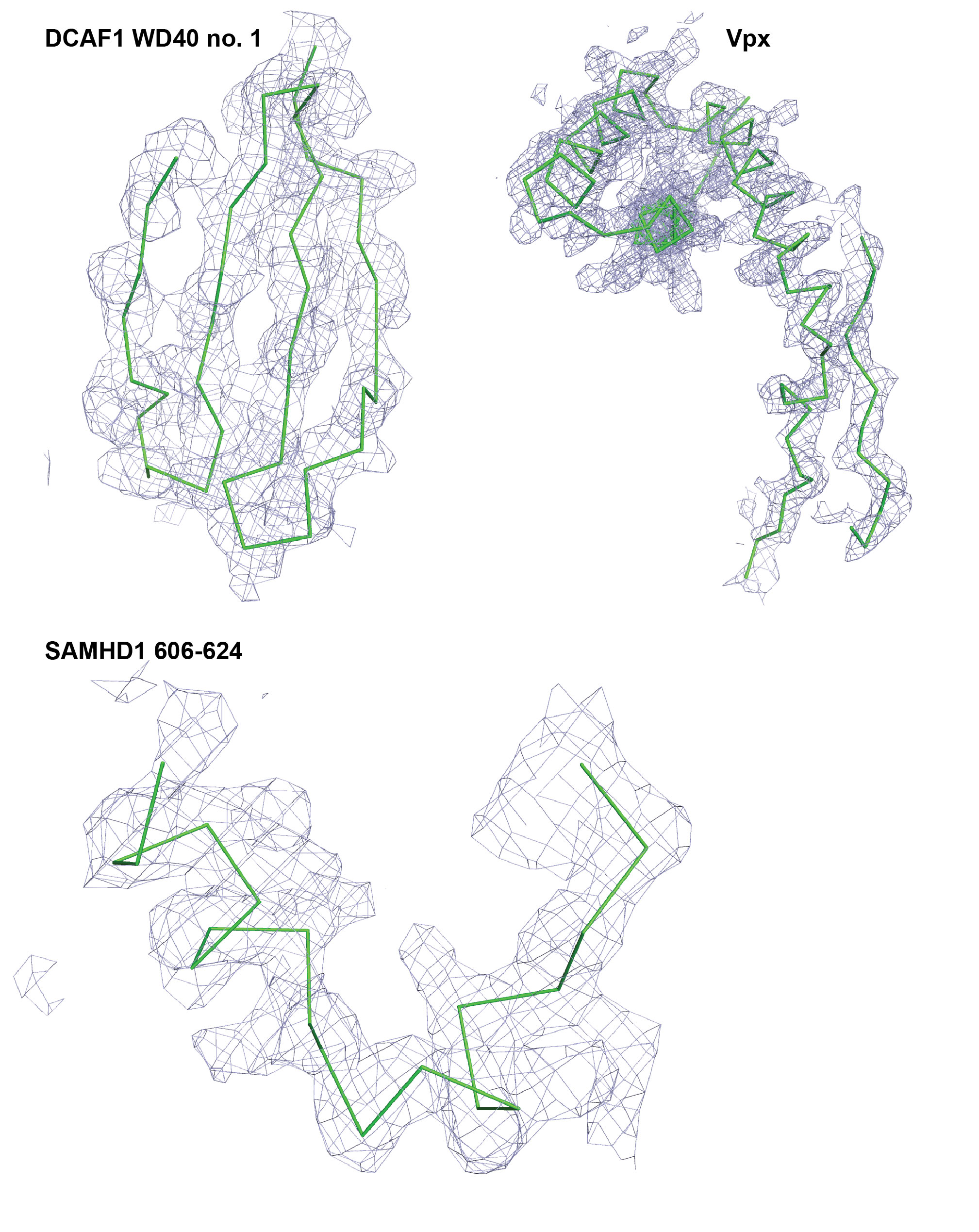
The native DCAF1-CtD-Vpxsm-SAMHD1-CtD complex was crystallised by vapour diffusion using an Oryx crystallisation robot (Douglas Instruments). Crystals were obtained from 0.1 μl droplets containing an equal volume of 2.5 mg/ml protein complex in 10 mM Bis-tris propane pH 8.5, 150 mM NaCl, 4 mM MgCl2, 0.5 mM TCEP mixed with 0.2 M magnesium chloride, 0.1 M Hepes pH 7.5, 15% PEG 400. For data collection, crystals were adjusted to 25 % PEG 400 and cryo-cooled in liquid nitrogen. Native crystals diffracted up to 2.5 Å resolution on beamline I03 at the Diamond Light Source, UK and belong to the space group P212121 with cell dimensions of a=73.79, b=82.03, c=113.29 with a single copy of the complex in the asymmetric unit. Crystals of the complex containing seleno-methionine-substituted DCAF1-CtD and Vpxsm were grown using the vapour diffusion method by mixing 1 μl complex at 5 mg/ml with 1 μl reservoir solution containing 0.4 M magnesium sulphate, 0.1 M MES pH 6.5. Crystals were adjusted to 25 % glycerol and cryo-cooled in liquid nitrogen. A SAD dataset was collected on beamline I04 at the Diamond Light Source, UK at a wavelength of 0.97972 Å, corresponding to the anomalous fʺ peak wavelength for selenium determined from an X-ray fluorescence scan. The crystal diffracted up to 3.5 Å resolution and was nearly isomorphous to the native crystal with the same space group and unit cell dimensions of a=74.25, b=82.88, c=115.56. Data were reduced using the XDS suite 28. An initial set of 5 selenium sites was found using the programs SHELXC and SHELXD 29. These sites were used as input for the program autoSHARP 30, which added 2 more sites and performed density modification, leading to an interpretable electron density map. A nearly complete model for Vpxsm and a partial polyalanine trace of DCAF1-CtD were located in this initial map using the buccaneer chain-tracing program 31. Completion of the polyalanine trace of DCAF1-CtD and placement of the protein backbone of SAMHD1-CtD was then undertaken manually in Coot 32. A round of refinement against native data recorded to 2.5Å produced an improved map that allowed further building of the SAMHD1-CtD and DCAF1-CtD side-chains. Subsequent incorporation of a Zn2+ ion and ligands in Coot combined with positional, real-space, individual B-factor and TLS refinement in phenix.refine 33 produced a final model for residues 1073-1314, 1328-1392 of DCAF1-CtD, 5-90, 100-111 of Vpxsm and 606-624 of SAMHD1-CtD with R-/Rfree-factors of 17.8 %/21.6 %. In the model, 97.1 % of residues have backbone dihedral angles in the favoured region of the Ramachandran plot, 2.66 % fall in the allowed regions and 0.24 % are outliers. Details of data collection and refinement statistics are presented in Extended Data Table 1.
Multiple sequence alignment
Amino acid sequences were aligned using the ClustalW server and adjusted manually. Abbreviations and Uniprot ID, SAMHD1: Hs-Homo sapiens Q9Y3Z3, Pt-Pan troglodytes H6WE97, Ca-Cercocebus atys H6WEA6, Ct-Cercocebus torquatus H6WEA7, Mam-Macaca mulatta F7CA64, Cj-Callithrix jacchus F7IGP7, Cf-Canis familiaris E2QTR2, Mum-Mus musculus Q60710, Rn-Rattus norvegicus D3Z898, Gg- Gallus gallus Q5ZJL9, Xl-Xenopus laevis Q6INN8, Dr-Danio rerio Q502K2, Dd- Dictyostelium discoideum B0G107. Vpx: sm-sooty mangabey (Cercocebus atys) P19508, mac-rhesus macaque (Macaca mulatta) P05917, rcm-red-capped mangabey (Cercocebus torquatus) NCBI GenBank HM803689, mnd-mandrill (Mandrillus sphinx) NCBI Reference Sequence NP_758889, HIV-2A P06939, HIV-2B Q89721. Vpr: HIV-2A P06938, HIV-2B P0C1P6, HIV-1MA P05955, HIV-1MB P35967
Modelling of the cullin/RING ubiquitin ligase assembly
Structural superposition was carried out using SSM 34 implemented in PDBefold (http://www.ebi.ac.uk/msd-srv/ssm/). DCAF1-CtD was first superposed with the DDB2 β-propeller in the DDB1-DDB2 structure (PDB code 3EI3) such that the N-terminal residues and first β-strand of DCAF1-CtD align with equivalent sequence just C-terminal to the helical DDB1-binding element of DDB2, termed the H-box. The second β-propeller domain of DDB1 (BPB), which serves as anchor for the CUL4 scaffold (wheat half-transparent surface), is mobile with respect to the DDB1 BPA and BPC. The two most extreme BPB conformations available in the PDB database were used to model the rotational range of the CUL4 arm with respect to DDB1-BPB (PDB codes 2HYE, to the left and 3I7H, to the right).
Restriction assay
SAMHD1 wildtype sequence was inserted into pLGatewayIeYFP 35 and mutations created by PCR-based site directed mutagenesis. MoMLV-based YFP vectors were made by cotransfecting pVSV-G 36, pKB4 37 and pLgatewaySN_SAMHD1 (wildtype or mutants) into 293T cells, harvesting 48 h post-transfection. HIV-1GFP was made as above by cotransfecting pVSV-G, p8.91 38 and pCSGW 39. U937 cells 40 were maintained in RPMI +[L]Glutamine (GIBCO) with 10 % fetal calf serum (biosera), penicillin and streptomycin. Cells (1 × 106) were transduced by spinoculation at 1,700 rpm for 90 min with 0.5 ml neat virus in the presence of 1 μg/ml polybrene. Cells were differentiated by addition of 100 nM phorbol myristate acetate for 72 hr and infected with HIV-1GFP. Restriction was assessed by 2-colour flow cytometry after 72 hr.
Transfection and immunoblotting
SAMHD1 and HIV-2 Vpx sequences were amplified from pLgatewaySN_SAMHD1 and pIRES2-EGFP-Vpx (gift from Mario Stevenson) respectively and cloned into pCMS28 41. Point mutations were created by PCR-based site directed mutagenesis. 293T cells were cotransfected with 2 μg each pCMS28-SAMHD1 and -Vpx, wild type, mutant proteins or empty vector. Cells were harvested 24 h post-transfection and analysed by SDS PAGE followed by immunoblotting with anti-SAMHD1 3295 (generated in house), anti-HIV-2 Vpx (hybridoma 6D2.6 supernatant, NIH AIDS Reagent Program 2739) and anti-14-3-3 (C-16, Santa Cruz sc-731).
Degron assay
A reporter construct comprising two copies of green fluorescent protein with a N-terminal nuclear localisation signal (NLS) fused to residues 600-626 of SAMHD1 was synthesised (GenArt, NLS-GFP-SAMHD1-CtD, Figure 3a). NLS-GFP-SAMHD1-CtD was inserted into pCMS28 using BglII and EcoRI. All mutants were created by PCR-based site directed mutagenesis. Puromycin virus-like particles were generated by co-transfection of pVSVG, pKB4 and pCMS28-NLS-GFP-SAMHD1-CtD wildtype or mutant as above. The stable cell lines were made by transduction of 293T cells followed by puromycin selection. VLPs SIV Vpx+ and Vpx− were generated by co-transfection of pVSVG and a SIV Gag-Pol expression plasmid pSIV3+ 42 or pSIV3+vpx− (gift from Caroline Goujon and Andreas Cimarelli). Viral titres were quantified using a modified ELISA for reverse transcriptase activity (Cavidi). 293T cells stably expressing SAMHD1-GFP wildtype or mutant were seeded at 5×104 cells per well in a 24-well plate one day prior to infection. Cells were infected with 2-fold serial dilutions of replication defective SIV Vpx+ virus in the presence of 1 μg/ml polybrene. For each experiment, SIV Vpx− VLPs and proteasome inhibitor MG132 (25.2 μM) were included as negative controls. After 48 hours, cells were harvested and the percentage of GFP-positive cells was determined by flow cytometry using a FACSVerse analyser (BD Biosciences). In parallel, GFP expression was analysed by microscopy using an inverted fluorescent microscope (LEICA).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Lesley Haire and Roksana Ogrodowicz for help with crystallisation, Prof. Ian Jones (Reading University) for the provision of the modified A. californica baculovirus bacmid, BAC10: 1629KO, Steve Smerdon and Steve Gamblin for comments on the manuscript. We gratefully acknowledge Diamond Light Source for synchrotron access (Grant No. 7707). This work was supported by the UK Medical Research Council, file references U117565647 (I.A.T) U117592729 (K.N.B) U117512710 (J.P.S), the Wellcome Trust, ref. 084955 (K.N.B) and by an EMBO long-term fellowship co-funded by the European Commission Marie Curie Actions (EMBOCOFUND2010, GA-2010-267146) (D.S).
Footnotes
The coordinates of DCAF1-CtD/Vpxsm/SAMHD1-CtD have been deposited in the Protein Data Bank under accession number 4CC9.
The authors declare no competing financial interests.
References
- 1.Laguette N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. doi: 10.1038/nature10117. pii: nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hrecka K, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. doi:10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldauf HM, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nature medicine. 2012;18:1682–1687. doi: 10.1038/nm.2964. doi:10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Descours B, et al. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology. 2012;9:87. doi: 10.1186/1742-4690-9-87. doi:10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstone DC, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. doi: 10.1038/nature10623. pii: nature10623. [DOI] [PubMed] [Google Scholar]
- 6.Lahouassa H, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nature immunology. 2012;13:223–228. doi: 10.1038/ni.2236. doi:10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim ES, et al. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell host & microbe. 2012;11:194–204. doi: 10.1016/j.chom.2012.01.004. doi: 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn J, et al. HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. The Journal of biological chemistry. 2012;287:12550–12558. doi: 10.1074/jbc.M112.340711. doi:10.1074/jbc.M112.340711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morellet N, Bouaziz S, Petitjean P, Roques BP. NMR structure of the HIV-1 regulatory protein VPR. Journal of molecular biology. 2003;327:215–227. doi: 10.1016/s0022-2836(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 10.Wei W, et al. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cellular microbiology. 2012;14:1745–1756. doi: 10.1111/j.1462-5822.2012.01835.x. doi:10.1111/j.1462-5822.2012.01835.x. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava S, et al. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008;4:e1000059. doi: 10.1371/journal.ppat.1000059. doi:10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergamaschi A, et al. The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J Virol. 2009;83:4854–4860. doi: 10.1128/JVI.00187-09. doi: 10.1128/JVI.00187-09. pii: JVI.00187-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLucia M, Mehrens J, Wu Y, Ahn J. HIV-2 and SIVmac accessory virulence factor Vpx down-regulates SAMHD1 enzyme catalysis prior to proteasome-dependent degradation. The Journal of biological chemistry. 2013;288:19116–19126. doi: 10.1074/jbc.M113.469007. doi:10.1074/jbc.M113.469007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. doi:10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 15.Scrima A, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. doi:10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer ES, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. doi:10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 17.Yan J, et al. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. The Journal of biological chemistry. 2013;288:10406–10417. doi: 10.1074/jbc.M112.443796. doi:10.1074/jbc.M112.443796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angers S, et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. doi:10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nature structural & molecular biology. 2010;17:105–111. doi: 10.1038/nsmb.1719. doi:10.1038/nsmb.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fregoso OI, et al. Evolutionary Toggling of Vpx/Vpr Specificity Results in Divergent Recognition of the Restriction Factor SAMHD1. PLoS pathogens. 2013;9:e1003496. doi: 10.1371/journal.ppat.1003496. doi:10.1371/journal.ppat.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wen X, Duus KM, Friedrich TD, de Noronha CM. The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J Biol Chem. 2007;282:27046–27057. doi: 10.1074/jbc.M703955200. doi: 10.1074/jbc.M703955200. pii: M703955200. [DOI] [PubMed] [Google Scholar]
- 22.Schrofelbauer B, Hakata Y, Landau NR. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4130–4135. doi: 10.1073/pnas.0610167104. doi:10.1073/pnas.0610167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Rouzic E, et al. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6:182–188. doi: 10.4161/cc.6.2.3732. pii: 3732. [DOI] [PubMed] [Google Scholar]
- 24.Hrecka K, et al. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11778–11783. doi: 10.1073/pnas.0702102104. doi:10.1073/pnas.0702102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barnitz RA, Chaigne-Delalande B, Bolton DL, Lenardo MJ. Exposed hydrophobic residues in human immunodeficiency virus type 1 Vpr helix-1 are important for cell cycle arrest and cell death. PLoS ONE. 2011;6:e24924. doi: 10.1371/journal.pone.0024924. doi:10.1371/journal.pone.0024924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods in enzymology. 1997;276:523–530. [PubMed] [Google Scholar]
- 27.Zhao Y, Chapman DA, Jones IM. Improving baculovirus recombination. Nucleic acids research. 2003;31:E6–6. doi: 10.1093/nar/gng006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta crystallographica. Section D, Biological crystallography. 2010;66:133–144. doi: 10.1107/S0907444909047374. doi:10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheldrick GM. A short history of SHELX. Acta crystallographica Section A, Foundations of crystallography. 2008;64:112–122. doi: 10.1107/S0108767307043930. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 30.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods in molecular biology. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. doi:10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 31.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta crystallographica. Section D, Biological crystallography. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. doi:10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta crystallographica. Section D, Biological crystallography. 2012;68:352–367. doi: 10.1107/S0907444912001308. doi:10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 35.Yap MW, Nisole S, Lynch C, Stoye JP. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci U S A. 2004;101:10786–10791. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bock M, Bishop KN, Towers G, Stoye JP. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J Virol. 2000;74:7422–7430. doi: 10.1128/jvi.74.16.7422-7430.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groom HC, et al. Absence of xenotropic murine leukaemia virus-related virus in UK patients with chronic fatigue syndrome. Retrovirology. 2010;7:10. doi: 10.1186/1742-4690-7-10. doi: 10.1186/1742-4690-7-10. pii: 1742-4690-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 39.Bainbridge JW, et al. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene therapy. 2001;8:1665–1668. doi: 10.1038/sj.gt.3301574. doi:10.1038/sj.gt.3301574. [DOI] [PubMed] [Google Scholar]
- 40.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) International journal of cancer. Journal international du cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 41.Gallois-Montbrun S, et al. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. Journal of virology. 2007;81:2165–2178. doi: 10.1128/JVI.02287-06. doi:10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negre D, et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000;7:1613–1623. doi: 10.1038/sj.gt.3301292. doi:10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.