

Abstract
Objectives
Multidrug efflux pumps mediate resistance to antibiotics and other toxic compounds. We studied the role of AcrAB-TolC, the main efflux pump in Escherichia coli, in regulating gene expression.
Methods
Deletion mutants, an acrABp-lacZ fusion and reverse transcription–real-time quantitative PCR experiments were used to study the role of AcrAB-TolC and metabolism in regulating gene expression of the acrAB operon and its transcriptional regulators.
Results
Deletion of the acrB gene increased the expression of the acrAB operon. A similar induction of acrAB was found when acrA or tolC was deleted, and when the pump function was inhibited using phenylalanine-arginine-β-naphthylamide. The induction of acrAB in the ΔacrB strain was totally (AcrR or SoxS) or partially (SoxR or MarA) prevented when the genes for these acrAB regulators were also deleted. The expression of soxS and marA, but not of acrR, was increased in the ΔacrB strain, which also showed altered expression of many other genes related to different cellular processes, including motility. Deletion of the metabolic genes entA and entE (enterobactin biosysnthesis), glpX (gluconeogenesis), cysH (cysteine biosynthesis) and purA (purine biosynthesis) also prevented activation of the acrAB promoter in the ΔacrB strain. Addition of the enterobactin biosynthesis intermediate metabolite 2,3-dihydroxybenzoate induced the expression of acrAB.
Conclusions
These results together suggest a model in which the AcrAB-TolC pump effluxes cellular metabolites that are toxic and/or have a signalling role. If the pump is inactivated or inhibited, these metabolites would accumulate, inactivating AcrR and/or up-regulating soxS and marA expression, ultimately triggering the up-regulation of acrAB expression to restore homeostasis.
Keywords: AcrAB-TolC, multidrug efflux, gene regulation, acrR, soxS, marA
Introduction
AcrAB-TolC is the main multidrug efflux pump in Escherichia coli and other Enterobacteriaceae, and its overexpression is commonly found in multidrug-resistant clinical isolates. The AcrAB-TolC pump effluxes many different classes of antibiotics, including β-lactams, fluoroquinolones and tetracyclines, host factors such as bile salts and antimicrobial peptides, and many other toxic compounds such as acriflavine, triclosan, detergents, dyes and organic solvents.1–3
AcrAB-TolC is a tripartite transporter that captures substrates from the periplasm and effluxes them across the outer membrane and out of the cell. It is composed of the proteins AcrA, AcrB and TolC.1,2 AcrB is an inner membrane resistance–nodulation–cell division efflux protein that also extends into the periplasm, AcrA is a periplasmic adaptor protein and TolC is the outer membrane channel for this pump and at least eight other efflux pumps in E. coli.1,2 However, none of these other pumps seems to significantly contribute to resistance to most drugs in the presence of a functional AcrAB-TolC pump.4,5 The three proteins are encoded in two separate operons, acrAB and tolC-ygiAB, whose expression is activated by the transcriptional regulators MarA, SoxS and Rob.1 The regulator AcrR specifically represses acrAB and its own transcription.1,6
Besides its role in the efflux of exogenous toxic compounds, the AcrAB-TolC pump affects virulence in Salmonella.7 In E. coli, AcrB is also involved in contact-dependent growth inhibition.8 Furthermore, mutants lacking tolC have pleiotropic phenotypes, such as defects in cell division and growth when cultured in minimal glucose medium,9 altered intra- and extracellular concentrations of some metabolites like cAMP, porphyrins, cysteine and enterobactin (see Zgurskaya et al.10 for a review) and increased expression of marA, soxS and MarA/SoxS/Rob-regulated genes and increased Rob activity.11 However, the mechanisms and pump(s) involved in these tolC-associated phenotypes remain unknown because inactivation of acrAB or other known TolC-dependent efflux pumps singly did not reproduce the tolC phenotypes.9–11
We have found that the AcrAB-TolC pump regulates the expression of the acrAB operon in response to cellular metabolism. It does so by affecting the expression or activity of specific acrAB transcriptional regulators.
Materials and methods
Growth conditions
Cultures were grown in lysogeny broth (LB) medium (per L: 10 g of tryptone, 5 g of yeast extract and 10 g of NaCl) at 37°C with agitation, except for those experiments described in Figure 1(a) to have been performed on M9 medium (per L: 6 g of Na2HPO4, 3 g of KH2PO4, 0.5 g of NaCl, 1 g of NH4Cl, 1 mM of MgSO4 and 0.2% glucose or glycerol). Antibiotics were used at 100 mg/L (ampicillin), 50 mg/L (kanamycin) and 25 mg/L (chloramphenicol).
Figure 1.

Effect of AcrAB-TolC inactivation or inhibition on acrAB expression. (a) acrABp-lacZ expression in the wild-type and ΔacrB strains measured by β-galactosidase assay using cells grown in different culture media. Statistically significant differences between both strains in each medium are shown as **P < 0.01 or ***P < 0.0001. The exact acrB−/acrB+ ratios—i.e. the induction of acrAB expression in the ΔacrB mutant compared with the wild-type—are shown above each pair of bars. (b) Effect of deletion of different components of the AcrAB-TolC pump on acrABp-lacZ expression measured using cells grown in LB. Significant differences between each mutant and the wild-type are shown as **P < 0.01 or ***P < 0.0001. (c) Cells were grown in LB medium in the presence of increasing concentrations of the efflux pump inhibitor PAβN to measure its effect on acrABp-lacZ expression. Statistically significant differences between the wild-type and the ΔacrB mutant both treated with the same concentration of PAβN were found at concentrations <100 µM (***P < 0.0001) but not ≥100 µM (P > 0.09). (a–c) All results are presented as average ± SEM (n = 3–4) and are shown normalized to acrABp-lacZ expression in the wild-type strain grown in LB. WT, wild-type.
Strains, plasmids and general genetic procedures
The bacterial strains and plasmids used in this study are listed in Table 1. PCR, phage P1 transduction to transfer KanR gene deletions between strains, and plasmid electroporation were performed according to standard procedures.12 KanR gene deletions were either obtained from the Keio collection13 via the E. coli Genetic Stock Center at Yale University (CGSC), or constructed using the λ Red recombinase method,14 plasmid pKD4 and the corresponding primers described in the Keio collection webpage (http://www.shigen.nig.ac.jp/ecoli/strain/top/top.jsp) for each gene to be deleted. Briefly, these primers were used to generate a PCR product of the kan gene of pKD4 with sequences flanking the desired gene at both ends. This product was used to replace the desired wild-type gene in the strain of interest, which was confirmed by PCR amplification and sequencing using specific primers flanking the deleted gene. When necessary, removal of the kanamycin cassette of constructed deletion mutants or Keio collection strains was performed using plasmid pCP20 as previously described,14 and confirmed by PCR amplification and sequencing using specific primers flanking the deleted genes. DNA sequencing was performed at the Tufts University Core Facility.
Table 1.
Bacterial strains and plasmids
| Name | Genotype/relevant characteristics | Reference/source |
|---|---|---|
| E. coli strains | ||
| BW25113 | (wild-type) F−, λ−, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), rph-1, Δ(rhaD-rhaB)568, hsdR514 | CGSC (Keio)13 |
| CR5000 | BW25113 ΔacrB | this studya |
| CR7000 | BW25113 ΔacrA | this studya |
| JW5503 | BW25113 ΔtolC::kan | CGSC (Keio)13 |
| CR7061 | BW25113 ΔacrB ΔtolC::kan | P1 JW5503 × CR5000 |
| CR7010 | BW25113 ΔacrR | this studya |
| CR7021 | BW25113 ΔacrB ΔacrR | this studyb |
| CR1000 | BW25113 ΔmarR | 15,33 |
| CR7035 | BW25113 ΔacrB ΔmarR | this studyb |
| JW5249 | BW25113 ΔmarA::kan | CGSC (Keio)13 |
| JW4359 | BW25113 Δrob::kan | CGSC (Keio)13 |
| JW4024 | BW25113 ΔsoxR::kan | CGSC (Keio)13 |
| JW4023 | BW25113 ΔsoxS::kan | CGSC (Keio)13 |
| CR7015 | BW25113 ΔacrR ΔsoxS::kan | P1 JW4023 × CR7010 |
| CR7025 | BW25113 ΔacrB ΔacrR ΔsoxS::kan | P1 JW5503 × CR7021 |
| Other ΔacrB regulatory gene mutants (ΔmarA::kan, Δrob::kan, ΔsoxR::kan or ΔsoxS::kan derivatives of CR5000; Figure 2), KanR | P1 Keio mutant × CR5000 | |
| Metabolic gene mutants (BW25113 derivatives; Figure 4), KanR | CGSC (Keio)13 | |
| ΔacrB metabolic gene mutants (CR5000 derivatives; Figure 4), KanR | P1 Keio mutant × CR5000 | |
| Plasmids | ||
| pKD4 | template for amplifying the kan gene, KanR | 14 |
| pCP20 | plasmid for excision of kan markers by FLP-mediated site-specific recombination, AmpR, ChlR | 14 |
| pNN608 | single copy, acrABp-lacZ, ChlR | 6 |
aThese strains were obtained by excising the kanamycin cassette from the corresponding Keio collection mutants13 obtained from the CGSC.
bDeletion of acrR in CR7021 or of marR in CR7035 were constructed using CR5000 as the parental strain and the λ Red recombinase method to create ΔacrR::kan or ΔmarR::kan derivatives whose kanamycin cassette was then removed using plasmid pCP20. Maintenance of the acrB deletion and the in-frame deletion of acrR or marR were confirmed by PCR amplification and sequencing using specific primers flanking the deleted genes.
RNA experiments
The expression levels of marA, soxS and acrR were studied by reverse transcription (RT) followed by real-time quantitative PCR (qPCR) as previously described.15 Briefly, cells were grown overnight, diluted 1 : 1000 in fresh LB and grown for 3 h to about 0.3 OD600. Then, the total RNA in each culture was stabilized using RNAprotect Bacteria Reagent (Qiagen), isolated by using an RNeasy Mini Kit (Qiagen) and two DNA removal steps, and its purity and concentration determined in a NanoDrop® ND-1000 spectrophotometer. The RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen). The obtained cDNA was then quantified in an Mx3000P detection system (Stratagene) using QuantiTect SYBR Green qPCR Master Mix from Qiagen and gene-specific standard plots. For each gene studied, the specific primers used for the RT and qPCR reactions are described elsewhere.16 gapA, which encodes the GAPDH enzyme, was used as a control gene according to Viveiros et al.16
β-Galactosidase assays
The expression of the acrABp-lacZ transcriptional fusion in single-copy plasmid pNN608 was determined by measuring β-galactosidase activity from exponential phase or stationary (overnight) phase cultures as previously described.17 All experiments were performed using cells grown in LB medium except for those samples specified in Figure 1(a).
Motility experiments
Migration through (swimming) or across (swarming) semisolid agar was determined using LB plates supplemented with 0.3% or 0.6% (w/v) agar, respectively. Plates were inoculated with each strain by stabbing them with a sterile toothpick. The zones of migration through the agar were measured after 16, 24 and 48 h incubation at 30°C. Data were obtained in at least three separate experiments, each performed in duplicate.
Statistical analysis
Statistically significant differences in RT–qPCR, β-galactosidase and motility experiments were identified with the t-test (two independent samples with equal variance, two-tailed distribution) using Microsoft® Excel 2010 software.
Microarray experiments
Cells were grown in LB medium and the total RNA in each culture was extracted as described in the ‘RNA experiments’ section. Three independent cultures of the wild-type (BW25113) and ΔacrB (CR5000) strains were used. RNA quality and concentration were determined in an Agilent 2100 BioAnalyzer. RNA samples were then processed, hybridized to GeneChip® E. coli Genome 2.0 arrays (Affymetrix) and analysed in the Keck Biotechnology Resource Laboratory at Yale School of Medicine. Determination of statistically differentially expressed genes was performed on the microarray data in the Computational Genomics Core at Tufts University. The full lists of genes up- or down-regulated in the ΔacrB mutant compared with the wild-type are given in Table S1 (available as Supplementary data at JAC Online).
Results
Inactivation or inhibition of AcrAB-TolC activates the acrAB promoter
Using an acrABp-lacZ fusion and measuring β-galactosidase activity from cells grown in LB medium, we found that the deletion of acrB increased the activity of the acrAB promoter. The increase was similar in cells in the exponential phase (2.2-fold, data not shown) and the stationary phase (2.3-fold; Figure 1a). These findings are in agreement with a previous result showing 1.5-fold induction of the acrAB promoter in exponential phase cells grown in L-broth in a strain lacking acrAB.18 When the cells were grown in 0.2% glucose- or 0.2% glycerol-minimal medium, the basal level of acrAB expression was lower but the induction of the acrAB promoter in the acrB mutant was even larger (3- to 4-fold) than in cells grown in LB (Figure 1a). This finding indicates that the induction of the acrAB promoter found in the ΔacrB mutant was not caused by lack of efflux of any toxic compound specifically present in LB medium. For this reason, and in agreement with earlier studies on the role of efflux on gene expression,11,15,19 LB medium was used for all subsequent experiments.
Deletion of the other components of the AcrAB-TolC pump also induced the acrAB promoter (Figure 1b). The acrA mutant showed a smaller level of induction than the acrB mutant, suggesting that another periplasmic adaptor protein might partially compensate for the lack of AcrA. The tolC and the double acrB tolC mutant showed a level of induction similar to that of the acrB mutant (Figure 1b), suggesting that most of the observed induction of the acrAB promoter is caused by lack of the AcrAB-TolC pump.
Induction of the acrAB promoter was also found when the pump function was inhibited using the efflux pump inhibitor phenylalanine-arginine-β-naphthylamide20 (PAβN) (Figure 1c). When wild-type cells were grown in the presence of increasing concentrations of PAβN, we observed dose-dependent induction of acrAB expression. At 250 μM PAβN, the expression of the acrAB promoter in wild-type cells increased to a level similar to that of the ΔacrB mutant grown without PAβN. Of note, this concentration of PAβN is similar to those used by Kern et al.21 and Coldham et al.22 (192 μM and 333 μM, respectively) to study the effect of this efflux pump inhibitor on drug accumulation and susceptibility to antibiotics. In ΔacrB cells, however, addition of PAβN had no effect on the acrAB promoter (Figure 1c). These results show that it is not its physical presence but its function that makes AcrAB-TolC influence its own expression.
AcrR, SoxRS and MarRA mediate feedback regulation of acrAB expression
Known regulators of acrAB were each deleted in both the wild-type (acrB+) and ΔacrB (acrB−) strains to determine their effect on ΔacrB-mediated induction of acrAB, which we measured as the ratio of acrAB expression between the acrB− and acrB+ strains (Figure 2). Deletion of acrR or soxS eliminated most of the ΔacrB-mediated induction of acrAB. Deletion of marA, and of the soxS regulator soxR, produced a smaller reduction in ΔacrB-mediated induction of acrAB, and the deletion of rob and the marA regulator marR produced no effect (Figure 2). The lack of induction of acrAB in the double ΔacrR ΔacrB mutant compared with the ΔacrR mutant is not because acrAB expression is already maximal in the ΔacrR strain, since we have found 2-fold further activation of the acrAB promoter in both mutants grown in the presence of external inducers (C. Ruiz and S. B. Levy, unpublished results).
Figure 2.

Effect of deletion of acrAB regulators on ΔacrB-mediated induction of acrABp-lacZ expression. Known regulators of acrAB were each deleted in both the wild-type (acrB+, light grey) and ΔacrB (acrB−, dark grey) parental strains to assess their role in ΔacrB-mediated induction of acrABp-lacZ expression; i.e. their effect on the acrB−/acrB+ ratio, which is shown above each corresponding pair of bars. The experiments were performed using cells grown in LB medium. The results are presented as average ± SEM (n = 4) and are shown normalized to acrABp-lacZ expression in the wild-type (acrB+ parental) strain. Statistically significant differences between the acrB−/acrB+ acrABp-lacZ ratio in each mutant compared with the ratio (2.3) of the parental strains are shown as *P < 0.02 or ***P < 0.0001.
We then studied whether the induction of acrAB expression in the ΔacrB strain was accompanied by and could possibly be the result of a change in the expression of these regulators. We found a ∼2-fold increase in the expression of soxS and marA in the ΔacrB strain compared with the wild-type (Figure 3). We also found that the induction of soxS expression by the deletion of acrB did not occur in strains in which soxR was also deleted (data not shown). We have previously shown that the inactivation of acrB does not induce marA expression in strains also inactivated for marR.15 Finally, we found no change in the expression of acrR in the ΔacrB strain compared with the wild-type (Figure 3). Combined, these results suggest that ΔacrB-mediated induction of acrAB expression is caused by a decrease in AcrR activity, not in acrR expression, and by an increase in soxS and marA expression mostly mediated by soxR and marR, respectively.
Figure 3.
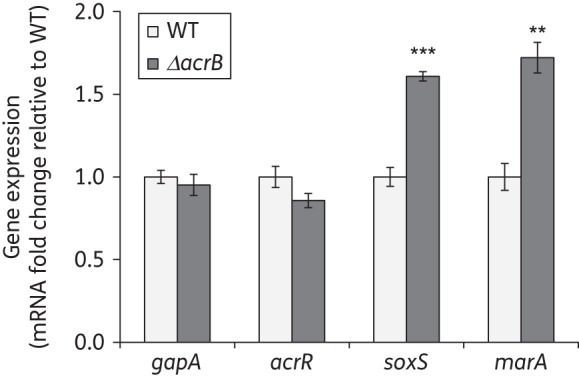
Effect of deletion of acrB on the expression of the acrAB regulators acrR, soxS and marA. The expression in exponential phase cultures grown in LB of the regulators involved in the feedback regulation of acrAB expression (soxS, marA and acrR), and of the control gene gapA, was measured by RT–qPCR in the wild-type and ΔacrB strains to assess whether it was affected by a lack of AcrAB-TolC. The results are presented as average ± SEM (n = 4) and are shown normalized to the expression of each gene in the wild-type strain. Statistically significant differences between the wild-type and ΔacrB strains are shown as **P < 0.01 or ***P < 0.0001. WT, wild-type.
Deletion of five different metabolic genes prevents feedback regulation of acrAB expression
Given that acrR, soxRS and marRA are known to respond to specific chemicals, we hypothesized that a change in their activity/expression, and thus the ΔacrB-mediated induction of acrAB expression, was ultimately caused by the accumulation of cellular metabolites normally effluxed by the AcrAB-TolC pump. To obtain data on the nature of these metabolites, we studied the effect on ΔacrB-mediated acrAB induction of the deletion of genes from metabolic pathways known to affect the expression or activity of acrAB, acrR, soxRS or marRA.18,19 We also studied metabolic genes whose expression was dramatically altered in the ΔacrB mutant compared with the wild-type in microarray experiments (Table 2).
Table 2.
Summary of the most significantly up- or down-regulated genes in the ΔacrB mutant relative to the wild-type found by microarray experiments
| Gene/operon/function | Fold changea |
|---|---|
| Activated | |
| motility and flagellar biosynthesisb | |
| fliA/Z | 58.5/22.9 |
| flgBCDEFGHIJ | 56.3–12.5 |
| fliFGHIJK | 43.6–15.4 |
| fliLMNOPQR | 40.9–4.6 |
| flgAMN | 31.7–7.9 |
| fliE | 15.7 |
| flgK/L | 8.0/9.1 |
| fliD/S | 8.5/6.6 |
| flhBAE | 6.8–5.1 |
| predicted protein | |
| ymdA | 6.2 |
| Repressed | |
| predicted conserved protein | |
| yifE | 16.8 |
| Fe-S cluster assembly scaffold complex | |
| sufABCDS | 3.7–2.2 |
Full lists of up- and down-regulated genes are given in Table S1.
aFold activation or repression in gene expression in the ΔacrB mutant relative to the wild-type. Only genes having a 3-fold change or larger that was also statistically significant (P < 0.05) are shown. For genes belonging to the same operon, the range between the gene showing the greatest change (usually the first gene in the operon) and the gene showing the smallest change (usually the last gene in the operon) is provided. When an operon has only two genes, the fold change for both genes is shown separated by ‘/’. Of note, glycerol and glycerol 3-phosphate transport and metabolism genes (glpABC, glpD, glpFK and glpTQ) were strongly activated (13.8-fold to 3.8-fold) and dipeptide and haem transporter genes (dppABCDF) were strongly repressed (5.7-fold to 3.3-fold) in the ΔacrB mutant relative to the wild-type, although neither of these changes was statistically significant (P > 0.05).
bThe acrB mutant also showed overexpression of the flagellar biosynthesis and motility34 genes flhDC (2.5-fold to 2.2-fold) and fliC (1.4-fold), although not statistically significant.
Of the 29 genes selected for testing (Figure 4), the deletion of eight of them induced acrAB expression in the wild-type (acrB+) background. Deletion of entA, trpE, metE, acnB, glpB and glpC produced a small induction of acrAB expression (1.2-fold to 1.5-fold). Deletion of entE or glpX induced acrAB expression 2.2-fold (Figure 4). This induction was similar to that found in the strain deleted for acrB (Figure 1a).
Figure 4.

Effect of deletion of 29 metabolic genes on ΔacrB-mediated induction of acrAB expression. Twenty-nine metabolic genes were each deleted in both the wild-type (acrB+, light grey) and ΔacrB (acrB−, dark grey) parental strains to assess their role in ΔacrB-mediated induction of acrABp-lacZ expression; i.e. their effect on the acrB−/acrB+ ratio, which is shown above each corresponding pair of bars. The experiments were performed using cells grown in LB medium. The results are presented as average ± SEM (n = 4) and are shown normalized to acrABp-lacZ expression in the wild-type (acrB+ parental) strain. Data are shown only for the five metabolic genes (entA, entE, cysH, purA and glpX) whose inactivation had a significant effect (P < 0.02) on ΔacrB-mediated induction of acrAB expression (on the acrB−/acrB+ ratio). The full list of genes tested was: fes, entA, entB, entC, entE and entF (enterobactin metabolism); trpA, trpC, trpD and trpE (tryptophan biosynthesis); aroA (chorismate metabolism); idcA, acnA and acnB (tricarboxylic acid cycle); cysH (cysteine biosynthesis); metE (methionine biosynthesis); purA, purC and purH (purine biosynthesis); glpA, glpB, glpC, glpD, glpF, glpK, glpQ, glpR and glpT (glycerol and glycerol 3-phosphate metabolism, transport and regulation); and glpX (gluconeogenesis). WT, wild-type.
Interestingly, the deletion of only three of these genes (entA, entE and glpX), and of two genes whose deletion did not significantly affect acrAB expression in the wild-type background (cysH and purA), decreased the ΔacrB-mediated induction of acrAB expression, i.e. reduced the acrB−/acrB+ ratio (Figure 4). Such reduction was partial for entA, cysH and purA, and nearly complete for entE and glpX (Figure 4). The fact that these five genes belong to four different metabolic pathways: enterobactin (entA and entE), cysteine (cysH) and purine (purA) biosynthesis, and gluconeogenesis (glpX), strongly suggests that ΔacrB-mediated induction of acrAB is caused by the accumulation of cellular metabolites from different metabolic pathways.
Addition of 2,3-dihydroxybenzoate (DHB), but not of fructose 1,6-biphosphate (F1,6P), induces acrAB expression via marRA
The two metabolic mutants with the strongest effect on acrAB regulation—entE and glpX—would be expected to accumulate DHB and F1,6P, respectively, based on the step of enterobactin biosynthesis or gluconeogenesis, respectively, that is blocked in these mutants (i.e. EntE uses ATP to catalyse the conversion of DHB into DHB-AMP, which is subsequently used by the enterobactin synthase complex to synthesize enterobactin,23 and GlpX catalyses the hydrolysis of F1,6P to fructose 6-phosphate,24 which is later converted into glucose 6-phosphate). We studied whether these two cellular metabolites added to cultures would induce acrAB expression. DHB is a small, weak acid/salt similar to salicylate, which diffuses spontaneously into the cytosol. F1,6P, like fructose 6-phosphate, is rapidly taken up by E. coli and its concentration within the cell becomes equal to that in the medium in <5 min.25 Such uptake is likely to be mediated by UhpT, which transports fructose 6-phosphate and other phosphate sugars into the cells.26
When the wild-type strain was grown in the presence of DHB, we found a statistically significant 1.6-fold induction of acrAB expression, whereas no significant increase was found for F1,6P (Figure 5a). Similar results were obtained when wild-type cells were grown to exponential phase in the absence of these compounds, followed by the addition of the metabolites and the measurement after 45 min of their effect on acrAB expression (data not shown). We also tested which regulator(s) were involved in the DHB-mediated induction of acrAB expression. Inactivation of acrR, soxS or soxR had little effect (data not shown), whereas inactivation of marA (completely) and marR (partially) prevented acrAB induction by DHB (Figure 5b).
Figure 5.

Effect of externally added cellular metabolites on acrAB expression. (a) Wild-type cells were grown in LB supplemented with the metabolites DHB (4 mM; a similar concentration to that used in Chubiz and Rao19) or F1,6P (5 mM) to measure their effect on acrABp-lacZ expression. (b) The effect of DHB was also measured in strains deleted for marA or marR. The results are presented as average ± SEM (n = 4) and are shown normalized to acrABp-lacZ expression in each strain grown without metabolite. Statistically significant differences for each strain between cells grown with and without metabolite are shown as *P < 0.02 or ***P < 0.0001. WT, wild-type.
Inactivation of the AcrAB-TolC pump affects the expression of many different genes and motility
Given that AcrAB-TolC affects its own expression, potentially via the efflux of multiple cellular metabolites, we asked whether deletion of the pump also affects the expression of other genes and other cellular functions. Microarray experiments comparing gene expression in the ΔacrB mutant with that in the wild-type strain revealed that inactivation of the AcrAB-TolC pump caused strong induction of genes involved in motility and flagellum biosynthesis, and repression of Fe-S cluster assembly scaffold complex genes (see Table 2 for a summary and see Table S1 for full data). Despite these many changes, we did not find differences between the ΔacrB and wild-type strains in growth rate (μ = 1.46 h−1 for both strains) or yield, and we did not observe defects in cell morphology or division by light microscopy in the ΔacrB mutant (data not shown; experiments performed in LB medium). However, in agreement with the strong induction of flagellum biosynthesis genes found in the acrB mutant, it showed increased motility in 0.3% (w/v) LB agar plates (swimming) (60% larger diameter after 16 h), but not in 0.6% (w/v) LB agar plates (swarming), as compared with the wild-type (Table 3). Inactivation of soxS, but not of marA, prevented the ΔacrB-mediated increase in motility in 0.3% (w/v) LB agar plates (Table 3).
Table 3.
Bacterial motility in LB medium supplemented with 0.3% agar (swimming)
| Strain | 16 h | 24 h | 48 h |
|---|---|---|---|
| Average diameter ± SEM (mm) | |||
| wild-type | 7.0 ± 0.2 | 17.1 ± 1.4 | 50.2 ± 1.4 |
| ΔacrB | 11.0 ± 0.6 | 21.7 ± 1.3 | 58.6 ± 1.6 |
| ΔmarA | 8.3 ± 0.3 | 16.8 ± 1.4 | 47.7 ± 2.3 |
| ΔacrB ΔmarA | 12.0 ± 0.6 | 22.5 ± 1.5 | 56.4 ± 4.1 |
| ΔsoxS | 12.0 ± 0.7 | 21.3 ± 1.1 | 57.3 ± 1.1 |
| ΔacrB ΔsoxS | 12.4 ± 0.9 | 21.6 ± 2.0 | 61.2 ± 3.8 |
| Ratio acrB−/acrB+ | |||
| ΔacrB/wild-type | 1.6a | 1.3a | 1.2a |
| ΔacrB ΔmarA/ΔmarA | 1.4a | 1.3a | 1.2 |
| ΔacrB ΔsoxS/ΔsoxS | 1.0 | 1.0 | 1.1 |
aSignificantly different motility between the pair of acrB− and acrB+ strains (P < 0.05; n = 3–6).
Discussion
Beyond their role in the efflux of antibiotics and other exogenous toxic compounds, there is an increasingly recognized role for TolC-dependent efflux pumps in cellular processes such as cell division and growth,9 metabolism10 and regulation of gene expression.11 However, the mechanisms and individual efflux pumps involved are still poorly understood. Here, we have focused on AcrAB-TolC, the main multidrug efflux pump that mediates multidrug resistance in E. coli, and whose overexpression has recently been suggested to precede and facilitate high-level resistance development mediated by target site mutations.27
We found that inactivation or inhibition of AcrAB-TolC activated the acrAB promoter (Figure 1). Such activation was mediated by three loci known to regulate acrAB: acrR, soxRS and marRA (Figure 2). Expression of soxS and marA was increased in the absence of the AcrAB-TolC pump (Figure 3). The role of acrR appears to be related to a decrease in the activity of the AcrR protein, since acrR expression was not altered in the ΔacrB mutant. Since AcrR also represses its own expression,6 it seems that the acrR and acrAB promoters differ in their response to the potentially less active AcrR protein, or to lower amounts of active AcrR. This different response might be related to the combined effect in the acrAB promoter of altered AcrR activity and increased levels of MarA and SoxS, which are not known to affect the acrR promoter.
Activation of the acrAB promoter seemed to be ultimately caused by the accumulation of cellular metabolites from at least four different metabolic pathways: enterobactin, cysteine and purine biosynthesis, and gluconeogenesis (Figure 4). These findings suggest a feedback regulatory circuit involved in the regulation of acrAB expression in response to AcrAB-TolC function. We propose a model (see Figure 6) in which the absence of a functional AcrAB-TolC pump leads to the accumulation of cellular metabolites produced by these four—and maybe other—metabolic pathways, which are usually effluxed by this pump. Such accumulation would then result in the inactivation of AcrR and the induction of soxS and marA expression, ultimately triggering up-regulation of acrAB expression to restore homeostasis.
Figure 6.

Proposed model for the regulation of acrAB expression by cellular metabolites. As detailed more extensively in the Discussion section, our results suggest that DHB and other unknown cellular metabolites normally excreted by the AcrAB-TolC pump accumulate in the ΔacrB strain, inactivating AcrR and inducing the expression of soxS and marA, ultimately up-regulating the expression of acrAB to restore homeostasis. Functional interactions are represented as arrows for activation/induction or as ‘┴’ for repression. Continuous lines indicate known interactions and broken lines indicate hypothetical interactions. DHB and two other putative metabolites are depicted as small shapes.
A role of AcrAB-TolC in the efflux of cellular metabolites was first proposed by Helling et al.18 They found that four different metabolic mutants (icdA, purB, cysH and metE) showed low-level resistance to nalidixic acid and increased levels of acrAB. For icdA and purB, nalidixic acid resistance required soxS plus either marA or rob. The accumulation of unknown cellular metabolites usually effluxed by TolC-dependent pumps was later suggested by Rosner and Martin11 to explain why tolC mutants show an increase in Rob activity and in the expression of soxS, marA and MarA/SoxS/Rob-regulated genes such as inaA. However, they concluded that AcrAB-TolC played no relevant role in the observed effects, at least when other pumps were present, because deletion of acrAB—or other TolC-dependent pumps singly—did not affect inaA expression. It was concluded that such effects were either mediated by an as yet unknown TolC-dependent pump or alternatively by more than one TolC-dependent pump. The second hypothesis appears to be the correct one according to a later manuscript published while this article was being written. In that manuscript, Rosner and Martin28 reported increased levels of the MarA/SoxS/Rob-regulated gene inaA in an acrB emrB mdtF triple mutant, although to a lesser degree than in the tolC mutant. By contrast, our results strongly support an important role for the AcrAB-TolC pump, even when other efflux pumps are present, in effluxing cellular metabolites from different metabolic pathways and in regulating soxS and marA expression. Such a discrepancy about the individual role of AcrAB-TolC in the regulation of soxS and marA expression may be related to the fact that we measured the effect of single deletion of this pump on soxS and marA expression directly by RT–qPCR, whereas Rosner and Martin11,28 used an indirect reporter (inaA-lacZ). Their observed increase in inaA expression in tolC mutants but not in acrB mutants may be additionally dependent on regulators of inaA other than marA and soxS, in the same way that AcrR is important for the regulation of acrAB. In fact, our microarray studies showed no significant increase in inaA expression in the ΔacrB mutant, despite it showing increased expression of soxS and marA.
Alternatively, oxidative stress has been proposed to be responsible for some of the effects found in tolC mutants, i.e. overexpression of the membrane stress-induced protein PspA, and metabolic changes leading to defects in cell division and growth in cells grown in minimal glucose medium but not those grown in LB.9,10 Since we found increased soxS expression and an important role for SoxRS in the induction of acrAB expression in the ΔacrB mutant, we cannot discard the possibility that oxidative stress also contributed to our results. The two hypotheses are not mutually exclusive because the accumulation of certain metabolites may lead to oxidative stress. However, our results seem to support the metabolite hypothesis as the more likely explanation for most of the phenotypes we observed. In fact, we have found that at least one cellular metabolite, DHB, induces the acrAB operon. Moreover, we found no altered expression of pspA in the ΔacrB mutant (Table S1); and we even found a 2-fold to 4-fold repression of sufABCDS (Table 2), an operon known to be induced by superoxide generators and hydrogen peroxide.29
Our studies have revealed several metabolic pathways whose inactivation induced acrAB expression and/or affected acrAB induction in the ΔacrB mutant (Figure 4), suggesting that the observed phenotypes are the result of the cumulative effect of more than one metabolite. Two of these pathways, gluconeogenesis and enterobactin biosynthesis, seem to play a particularly important role. Blocking these two pathways by using gene deletions builds up precursors that we hypothesize are capable of inducing acrAB expression. We also hypothesize that such precursors accumulate in the ΔacrB strain, which would explain why this strain also showed increased activation of the acrAB promoter.
Deletion of the gluconeogenesis gene glpX strongly increased acrAB expression in the wild-type strain and also prevented ΔacrB-mediated activation of the acrAB promoter (Figure 4). However, when we tested whether this effect was caused by the accumulation of the GlpX substrate F1,6P, by adding this metabolite to the culture medium, we found almost no induction of the acrAB operon (Figure 5a). As we did for the enterobactin pathway, it will be necessary to study additional gene deletions of the other upstream and downstream enzymes in the pathway to find out whether the induction of acrAB expression found in the glpX mutant is caused by the accumulation of a precursor of F1,6P, by the lack of an F1,6P downstream product or by any inactivation of the pathway itself.
Inactivation of the enterobactin biosynthetic genes entA or entE also strongly increased acrAB expression in the wild-type strain and prevented ΔacrB-mediated activation of the acrAB promoter; however, inactivation of the other genes of the enterobactin pathway had no effect (Figure 4). This finding allowed us to identify the two cellular metabolites whose conversion these two genes mediate—2,3-dihydro-2,3-dihydroxybenzoate and especially its immediate product DHB—as likely to be responsible for the induction of acrAB. DHB is already known to be a MarR ligand capable of inducing marA expression in vivo by inactivating MarR.19 By adding DHB to the culture medium, we confirmed that this metabolite induces acrAB expression and that such induction is mediated by marA and partially by marR (Figure 5). Salicylate, another MarR ligand structurally very similar to DHB, induces marA expression via both MarR and an unknown MarR-independent regulator.30 A similar situation may explain why lack of acrAB induction by DHB was less severe in the ΔmarR mutant than in the ΔmarA mutant. Because DHB diffuses freely across the membrane, its intra- and extracellular concentrations tend to equilibrate regardless of the origin of such DHB; i.e. externally added to the culture or internally overproduced because of the inactivation of entE. In either case, marA-mediated induction of acrAB expression by DHB may result in increased efflux of this compound (or its precursors) outside the cell via the AcrAB-TolC pump. An increased efflux rate of DHB that is higher than its diffusion rate across the cell membrane would result in a decrease in the intracellular concentration of this compound compared with its extracellular concentration, thus reducing its toxicity. A similar scenario might also apply to other metabolites that induce acrAB expression.
acrAB seems to be regulated by the cumulative effect of different metabolites. Some of these as yet unknown metabolites may affect AcrR and SoxRS, the two acrAB regulatory systems found to play the strongest role in the induction of acrAB expression in the ΔacrB mutant. In fact, ethidium bromide and other substrates of the AcrAB-TolC pump have been shown to bind to and inactivate AcrR in vitro.31 soxS expression is up-regulated by redox cycling compounds that directly oxidize and activate the soxS activator SoxR.32 Therefore, it seems plausible that some cellular metabolites can inactivate AcrR or activate SoxR.
It remains to be determined whether acrAB induction and the other changes observed in the ΔacrB mutant are a response to DHB and other as yet unknown metabolites being toxic, or whether these metabolites have a signalling role. Since we found no defects in the cell morphology or growth of the ΔacrB mutant, it seems that even if these metabolites are toxic, the gene expression changes that they produce and other changes yet to be found, e.g. the induction of other pumps, may allow the cells to cope with their accumulation. Either way, our findings suggest a strong interconnection between AcrAB-TolC function and gene regulation and metabolism.
Funding
This work was supported by United States Public Health Service grant number AI56021 from the National Institutes of Health.
Transparency declarations
None to declare.
Supplementary data
Table S1 is available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
Acknowledgements
We thank Kimberly Foster for helping with the motility experiments, and Laura McMurry for helping with the editing of the manuscript.
References
- 1.Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs. 2004;64:159–204. doi: 10.2165/00003495-200464020-00004. [DOI] [PubMed] [Google Scholar]
- 2.Blair JM, Piddock LJ. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr Opin Microbiol. 2009;12:512–9. doi: 10.1016/j.mib.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Warner DM, Levy SB. Different effects of transcriptional regulators MarA, SoxS and Rob on susceptibility of Escherichia coli to cationic antimicrobial peptides (CAMPs): Rob-dependent CAMP induction of the marRAB operon. Microbiology. 2010;156:570–8. doi: 10.1099/mic.0.033415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sulavik MC, Houseweart C, Cramer C, et al. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother. 2001;45:1126–36. doi: 10.1128/AAC.45.4.1126-1136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–12. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma D, Alberti M, Lynch C, et al. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol. 1996;19:101–12. doi: 10.1046/j.1365-2958.1996.357881.x. [DOI] [PubMed] [Google Scholar]
- 7.Webber MA, Bailey AM, Blair JM, et al. The global consequence of disruption of the AcrAB-TolC efflux pump in Salmonella enterica includes reduced expression of SPI-1 and other attributes required to infect the host. J Bacteriol. 2009;191:4276–85. doi: 10.1128/JB.00363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoki SK, Malinverni JC, Jacoby K, et al. Contact-dependent growth inhibition requires the essential outer membrane protein BamA (YaeT) as the receptor and the inner membrane transport protein AcrB. Mol Microbiol. 2008;70:323–40. doi: 10.1111/j.1365-2958.2008.06404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhamdhere G, Zgurskaya HI. Metabolic shutdown in Escherichia coli cells lacking the outer membrane channel TolC. Mol Microbiol. 2010;77:743–54. doi: 10.1111/j.1365-2958.2010.07245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zgurskaya HI, Krishnamoorthy G, Ntreh A, et al. Mechanism and function of the outer membrane channel TolC in multidrug resistance and physiology of enterobacteria. Front Microbiol. 2011;2:189. doi: 10.3389/fmicb.2011.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosner JL, Martin RG. An excretory function for the Escherichia coli outer membrane pore TolC: upregulation of marA and soxS transcription and Rob activity due to metabolites accumulated in tolC mutants. J Bacteriol. 2009;191:5283–92. doi: 10.1128/JB.00507-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 13.Baba T, Ara T, Hasegawa M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruiz C, Levy SB. Many chromosomal genes modulate MarA-mediated multidrug resistance in Escherichia coli. Antimicrob Agents Chemother. 2010;54:2125–34. doi: 10.1128/AAC.01420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viveiros M, Dupont M, Rodrigues L, et al. Antibiotic stress, genetic response and altered permeability of E. coli. PLoS One. 2007;2:e365. doi: 10.1371/journal.pone.0000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz C, McMurry LM, Levy SB. Role of the multidrug resistance regulator MarA in global regulation of the hdeAB acid resistance operon in Escherichia coli. J Bacteriol. 2008;190:1290–7. doi: 10.1128/JB.01729-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helling RB, Janes BK, Kimball H, et al. Toxic waste disposal in Escherichia coli. J Bacteriol. 2002;184:3699–703. doi: 10.1128/JB.184.13.3699-3703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chubiz LM, Rao CV. Aromatic acid metabolites of Escherichia coli K-12 can induce the marRAB operon. J Bacteriol. 2010;192:4786–9. doi: 10.1128/JB.00371-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomovskaya O, Warren MS, Lee A, et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother. 2001;45:105–16. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kern WV, Steinke P, Schumacher A, et al. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J Antimicrob Chemother. 2006;57:339–43. doi: 10.1093/jac/dki445. [DOI] [PubMed] [Google Scholar]
- 22.Coldham NG, Webber M, Woodward MJ, et al. A 96-well plate fluorescence assay for assessment of cellular permeability and active efflux in Salmonella enterica serovar Typhimurium and Escherichia coli. J Antimicrob Chemother. 2010;65:1655–63. doi: 10.1093/jac/dkq169. [DOI] [PubMed] [Google Scholar]
- 23.Khalil S, Pawelek PD. Enzymatic adenylation of 2,3-dihydroxybenzoate is enhanced by a protein-protein interaction between Escherichia coli 2,3-dihydro-2,3-dihydroxybenzoate dehydrogenase (EntA) and 2,3-dihydroxybenzoate-AMP ligase (EntE) Biochemistry. 2011;50:533–45. doi: 10.1021/bi101558v. [DOI] [PubMed] [Google Scholar]
- 24.Donahue JL, Bownas JL, Niehaus WG, et al. Purification and characterization of glpX-encoded fructose 1,6-bisphosphatase, a new enzyme of the glycerol 3-phosphate regulon of Escherichia coli. J Bacteriol. 2000;182:5624–7. doi: 10.1128/jb.182.19.5624-5627.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts IZ, Wolffe EL. Utilization of labeled fructose-6-phosphate and fructose-1,6-diphosphate by Escherichia coli. Arch Biochem Biophys. 1951;33:165–6. doi: 10.1016/0003-9861(51)90090-2. [DOI] [PubMed] [Google Scholar]
- 26.Hall JA, Maloney PC. Transmembrane segment 11 of UhpT, the sugar phosphate carrier of Escherichia coli, is an α-helix that carries determinants of substrate selectivity. J Biol Chem. 2001;276:25107–13. doi: 10.1074/jbc.M102017200. [DOI] [PubMed] [Google Scholar]
- 27.Singh R, Swick MC, Ledesma KR, et al. Temporal interplay between efflux pumps and target mutations in development of antibiotic resistance in Escherichia coli. Antimicrob Agents Chemother. 2012;56:1680–5. doi: 10.1128/AAC.05693-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosner JL, Martin RG. Reduction of cellular stress by TolC-dependent efflux pumps in Escherichia coli indicated by BaeSR and CpxARP activation of spy in efflux mutants. J Bacteriol. 2013;195:1042–50. doi: 10.1128/JB.01996-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JH, Yeo WS, Roe JH. Induction of the sufA operon encoding Fe-S assembly proteins by superoxide generators and hydrogen peroxide: involvement of OxyR, IHF and an unidentified oxidant-responsive factor. Mol Microbiol. 2004;51:1745–55. doi: 10.1111/j.1365-2958.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 30.Cohen SP, Levy SB, Foulds J, et al. Salicylate induction of antibiotic resistance in Escherichia coli: activation of the mar operon and a mar-independent pathway. J Bacteriol. 1993;175:7856–62. doi: 10.1128/jb.175.24.7856-7862.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su CC, Rutherford DJ, Yu EW. Characterization of the multidrug efflux regulator AcrR from Escherichia coli. Biochem Biophys Res Commun. 2007;361:85–90. doi: 10.1016/j.bbrc.2007.06.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu M, Imlay JA. The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol Microbiol. 2011;79:1136–50. doi: 10.1111/j.1365-2958.2010.07520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz C, Levy SB. Use of functional interactions with MarA to discover chromosomal genes affecting antibiotic susceptibility in Escherichia coli. Int J Antimicrob Agents. 2011;37:177–8. doi: 10.1016/j.ijantimicag.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith TG, Hoover TR. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv Appl Microbiol. 2009;67:257–95. doi: 10.1016/S0065-2164(08)01008-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.