

Background: E6AP/UBE3A is a Hect ligase implicated in neurodevelopment and cell transformation.
Results: Kinetic/biophysical analyses demonstrate that E6AP oligomerization is required for polyubiquitin degradation signal assembly and that changes in oligomerization regulates such activity.
Conclusion: E6AP oligomerization accounts for opposing effects of mutation and HPV16/18 E6 protein.
Significance: These findings explain in part the etiology of specific Angelman syndrome mutations and E6-mediated cell transformation.
Keywords: E3 Ubiquitin Ligase, Enzyme Kinetics, Enzyme Mechanisms, Kinetics, Ubiquitin, Hect Ligase
Abstract
Employing 125I-polyubiquitin chain formation as a functional readout of ligase activity, biochemical and biophysical evidence demonstrates that catalytically active E6-associated protein (E6AP)/UBE3A is an oligomer. Based on an extant structure previously discounted as an artifact of crystal packing forces, we propose that the fully active form of E6AP is a trimer, analysis of which reveals a buried surface of 7508 Å2 and radially symmetric interacting residues that are conserved within the Hect (homologous to E6AP C terminus) ligase superfamily. An absolutely conserved interaction between Phe727 and a hydrophobic pocket present on the adjacent subunit is critical for trimer stabilization because mutation disrupts the oligomer and decreases kcat 62-fold but fails to affect E2∼ubiquitin binding or subsequent formation of the Hect domain Cys820∼ubiquitin thioester catalytic intermediate. Exogenous N-acetylphenylalanylamide reversibly antagonizes Phe727-dependent trimer formation and catalytic activity (Ki = 12 mm), as does a conserved α-helical peptide corresponding to residues 474–490 of E6AP isoform 1 (Ki = 22 μm) reported to bind the hydrophobic pocket of other Hect ligases, presumably blocking Phe727 intercalation and trimer formation. Conversely, oncogenic human papillomavirus-16/18 E6 protein significantly enhances E6AP catalytic activity by promoting trimer formation (Kactivation = 1.5 nm) through the ability of E6 to form homodimers. Recombinant E6 protein additionally rescues the kcat defect of the Phe727 mutation and that of a specific loss-of-function Angelman syndrome mutation that promotes trimer destabilization. The present findings codify otherwise disparate observations regarding the mechanism of E6AP and related Hect ligases in addition to suggesting therapeutic approaches for modulating ligase activity.
Introduction
E6AP2/UBE3A is the founding member of the Hect (homologous to E6AP C terminus) ubiquitin ligase family and is defined by a highly conserved 350-residue C-terminal catalytic domain (1, 2). The Hect domain is characterized by the presence of an active site cysteine that forms an obligatory high energy thioester bond with ubiquitin prior to transfer of the latter to specific substrate proteins, the identity of which is defined by the N-terminal targeting domain (2). The E6AP Hect domain assembles Lys48-linked polyubiquitin degradation signals that are recognized by the 26 S proteasome (3, 4). Both activation and loss of E6AP function are implicated in various human diseases, as discussed elsewhere (5–7). Abrogation of E6AP function by deletion, imprinting defects, or mutation of the UBE3A gene locus within the 15q11–13 chromosome region is associated with the neurological disorder Angelman syndrome (8–10). Patients affected with Angelman syndrome are characterized by severe intellectual and developmental disability, speech impairment, behavioral uniqueness, epilepsy, and severely abnormal electroencephalography, among other symptoms (11–14). Most of the naturally occurring mutations within the UBE3A gene introduce deletions that generate truncated forms of E6AP lacking the intact Hect domain; however, ∼10% of the genetic alterations correspond to point mutations within the E6AP coding region (15, 16). Although many of the point mutants represent loss-of-function alterations and are not able to ubiquitinate their substrate, paradoxically, many retain the ability to form a thioester bond with ubiquitin (17). In contrast, duplication of the corresponding UBE3A gene is thought to result in some cases of autism spectrum disorder (18–21). These observations suggest a narrow range of E6AP activity for normal neurological development because UBE3A-deficient mouse models or those expressing higher levels of the ligase show phenotypes similar to Angelman syndrome or autism, respectively (18, 22–24). In general, the clinical symptoms associated with the neurological disorders and the identified targets of E6AP-catalyzed ubiquitination localize to regulatory pathways required for synaptic plasticity (25–29). The identification of Arc and Ephexin 5 as targets of E6AP, both of which function to mediate synaptic remodeling, provide a framework for reconciling the loss-of-function mutations in the E6AP maternal copy and the neurological and developmental defects present in affected individuals (25, 26, 30), although more recent evidence questions a role for E6AP in targeting Arc degradation (30).
The E6AP protein was initially identified by its interaction with the E6 viral protein encoded by human papilloma virus 16 (HPV16) (31–33). Although p53 degradation is normally mediated by the Mdm2 ubiquitin ligase (34), E6 protein is proposed to bind to E6AP and redirect its specificity to p53 (33, 35). Enhanced degradation of p53 in epithelial cells by the HPV16 and -18 viral strains induces cell transformation and development of cervical and oral cancers, depending on the site of infection (5, 36, 37). Similarly, hepatitis C virus encodes the NS5B protein that binds E6AP and induces degradation of the retinoblastoma protein tumor suppressor, increasing the risk of liver cirrhosis and hepatocellular carcinoma (38, 39). The E6AP sequence contains a leucine-rich motif (LXXLL) in the N-terminal region to which the E6 viral protein binds (40–42). Biophysical and structural analyses of the E6-E6AP interaction show that the N-terminal E6 Zn2+-binding domain primarily interacts with E6AP, whereas the C-terminal Zn2+-binding domain interacts with p53 (43–47). Although the interaction of E6 with E6AP is necessary for degradation of p53, the effect of such interaction on the catalytic activity of E6AP has not been adequately addressed; Kao et al. (48) have shown that ectopic E6 expression increases E6AP autoubiquitination and intracellular turnover.
Full-length E6AP is a protein of 100 kDa; however, only the structure of the truncated E6AP Hect domain in association with its cognate UbcH7 ubiquitin carrier protein has been reported (49). The Hect domain architecture displays an L-shape with distinct N-terminal and C-terminal subdomains connected by a flexible hinge region (49). The N-terminal subdomain can be further divided into large and small N-terminal subdomains also connected by flexible hinge segments (49). The active site Cys820 to which ubiquitin forms a thioester intermediate is contained in the C-terminal subdomain (49, 50). The UbcH7 carrier protein binds to a pocket in the small N-terminal subdomain, but the geometry of the bound UbcH7∼ubiquitin thioester relative to Cys820 has been a challenge to understanding within the context of a coherent mechanism for Cys820∼ubiquitin thioester formation and subsequent polyubiquitin chain formation, discussed recently (51). However, we have used kinetic analysis of 125I-polyubiquitin chain assembly to demonstrate for the first time the presence of two functionally distinct E2∼ubiquitin binding sites on the E6AP Hect domain, providing a mechanistically tractable resolution to the problem of active site thioester formation (51). Other kinetic evidence indicates that the canonical UbcH7 binding site presented in the original crystal structure (49) functions in chain elongation from the ubiquitin thioester formed at Cys820 (51), providing an entre into reconciling a potential mechanism for polyubiquitin chain assembly.
In the present work, we use kinetic and biophysical methods to demonstrate for the first time that an E6AP oligomer is the catalytically competent form of the enzyme. Based on an earlier structure for E6AP initially discounted as an artifact of crystal packing forces (49), we propose that the fully functional form of the oligomer is a trimer, computational analysis of which allowed us to identify conserved residues located at the subunit interfaces. Using rates of 125I-polyubiquitin chain assembly as a functional readout, we have identified a subset of residues essential for stabilizing the active trimer. Furthermore, a small molecule mimic of a key stabilizing interaction is sufficient to dissociate the trimer and abrogate E6AP-catalyzed chain assembly but not Cys820∼ubiquitin thioester formation. In contrast, E6 viral protein enhances E6AP activity by promoting oligomerization as a consequence of the ability of the former to dimerize through its N-terminal Zn2+ binding domain (51, 52). Remarkably, E6-induced oligomerization rescues synthetic and Angelman syndrome loss-of-function mutations contributing to subunit association and stabilization. The current results explain previously unresolved roles for a cohort of point mutations in the neurological pathology of Angelman syndrome, reveal new strategies for regulating E6AP function by modulating subunit assembly, and provide insights into the role of oligomerization in polyubiquitin chain formation by the Hect ligase superfamily.
MATERIALS AND METHODS
Bovine ubiquitin and creatine phosphokinase were purchased from Sigma. Ubiquitin was further purified to apparent homogeneity by FPLC and quantitated spectrophotometrically (53). Ubiquitin was radioiodinated by the Chloramine-T procedure to yield a specific radioactivity of ∼15,000 cpm/pmol using carrier-free Na125I purchased from either GE Healthcare or PerkinElmer Life Sciences (54). Human erythrocyte Uba1 (UBA1) was purified to apparent homogeneity from outdated human blood (54). Active Uba1 was quantitated by the stoichiometric formation of 125I-ubiquitin thioester (54–56). Human recombinant UbcH7 (UBE2L3) was that described previously (57, 58), and active E2 concentration was quantitated by the Uba1-dependent stoichiometric formation of UbcH7∼125I-ubiquitin thioester (51, 59). The E2 protein was stored at −80 °C in small aliquots and was stable for more than 6 months although subject to activity loss with successive freeze-thaw cycles (59). The N-acetyl-l-phenylalanylamide (Ac-PheNH2; E-1160) was obtained from Bachem Americas. The N-acetyl-NRIRMYSERRITVLYSL peptide (purity >95%) was obtained from PEPTIDE 2.0 Inc.
Generation and Purification of Recombinant E6AP
Human E6AP isoform 3 (UBE3A; IMAGE clone NM00046.2) was subcloned into pGEX4T1 to yield pGEX4T1-E6AP as described previously (51).3 The E6APF727D, E6APY533A, E6APD543A, E6APR626A, and E6APK688A mutants were generated from pGEX4T1-E6AP using the QuikChange protocol of Stratagene to yield pGEX4T1-E6APF727D, pGEX4T1-E6APY533A, pGEX4T1-E6APD543A, pGEX4T1-E6APR626A, and pGEX4T1-E6APK688A, respectively. The E6APΔ495 truncation lacking the C-terminal Hect domain was generated by inserting a STOP codon after codon 495 of pGEX4T1-E6AP to yield pGEX4T1-E6APΔ495. Residues 450–852 of full-length E6AP were subcloned by PCR into the BamHI/NotI sites of pGEX4T1 to yield pGEX4T1-E6AP-Hect, from which was expressed the GST-E6AP-Hect domain fusion protein. The active site Cys820 was similarly mutated to alanine by the QuikChange protocol to yield GST-E6AP-HectC820A protein. The coding regions for all E6AP clones were sequenced to preclude cloning artifacts and to verify the desired mutation. Wild type and mutant GST-E6AP proteins were expressed and purified as described previously (51, 58). The activities of GST-E6AP and its mutants were quantitated by their stoichiometric formation of 125I-ubiquitin thioester (51, 59, 60). Unless otherwise noted, the GST moiety was not processed from the fusion proteins by thrombin digestion (51).
The full-length E6AP sequence was subsequently cloned by PCR into the BamHI/HindIII sites of pFastBac Htb (Invitrogen) for baculoviral expression of the corresponding His6-E6AP. After bacmid amplification, the complete insert was sequenced to confirm the absence of cloning errors. The bacmid was transfected into Sf9 insect cells, and then the P1 virus was isolated and amplified as a P2 stock. The P2 stock was then used to transfect Sf9 cells (3.6 × 107 cells/T45 flask, 105 virus particles/T45flask), and protein expression was allowed to proceed for 5 days as recommended by the Bac-to-Bac Baculovirus Expression System manual (Invitrogen). Full-length recombinant His6-E6AP was isolated from the medium by affinity purification using a 1.5 × 3.0-cm HisTrap HP column (Amersham Biosciences). The column was equilibrated in 50 mm Tris-HCl (pH 8.0) and 20 mm imidazole, and the bound protein was eluted in 50 mm Tris-HCl (pH 8.0) containing 300 mm imidazole. After elution, the protein was equilibrated by dialysis into 50 mm Tris-HCl (pH 7.5) containing 300 mm NaCl and 1 mm DTT. Typically 1.6–2 mg of affinity-purified His6-E6AP protein could be isolated from 13 T150 flasks, of which 10–40% was active by the Uba1-dependent formation of E6AP∼125I-ubiquitin thioester (51, 58).
Expression and Purification of Recombinant HPV16 E6 Protein
HPV16 E6 protein coding sequence (GenBankTM EF122273.1; GI 119710759) was subcloned into BamHI/XhoI sites of pGEX4T3 to yield pGEX4T3-E6(HPV16). The E6(HPV16)Δ91 recombinant protein was obtained by inserting a STOP codon after codon 91 by the QuikChange protocol of Stratagene to yield pGEX4T3-E6(HPV16)Δ91. The HPV16 E6 C-terminal domain corresponded to residues 89–158 and was subcloned into BamHI/XhoI sites of pGEXT4T1 by PCR to yield pGEX4T1-E6Ct. Escherichia coli BL21 (DE3) cells harboring pGEX-E6(HPV16), pGEX4T3-E6(HPV16)Δ91, or pGEX4T1-E6(HPV16)E6Ct were grown at 37 °C and then induced at A600 of 0.6 by the addition of isopropyl-1-thio-β-d-galactopyranoside to a final concentration of 0.4 mm. After 3 h at 37 °C, cells were harvested by centrifugation at 6000 × g for 30 min and then resuspended in 50 mm Tris-HCl (pH 7.5) containing 150 mm NaCl and 1 mm DTT (59). Cells were lysed by Emulsiflex (Avestin) and then centrifuged at 30,000 × g for 30 min (59). Recombinant GST-E6(HPV16) fusion proteins were purified on glutathione-Sepharose, processed with thrombin, and then passed through a second glutathione-Sepharose column to remove free GST (59). The total protein content calculated by the theoretical 280 nm extinction coefficient yielded 2.4 mg/liter of culture except for E6(HPV16)Δ91, which yielded 0.5 mg/liter of culture. The proteins were flash frozen in small aliquots and stored at −80 °C.
E6AP-catalyzed 125I-Ubiquitin Conjugation Assay
The E3 ligase activity of recombinant E6AP was quantitated in kinetic assays under initial velocity conditions (51, 58). Rates of E6AP-catalyzed 125I-polyubiquitin chain formation were measured at 37 °C in incubations of 25-μl final volume containing 50 mm Tris-HCl (pH 7.5), 1 mm ATP, 10 mm MgCl2, 1 mm DTT, 10 mm creatine phosphate, 1 IU of creatine phosphokinase, 5 μm 125I-ubiquitin (∼1.5 × 104 cpm/pmol), 50 nm human Uba1, and the indicated concentrations of UbcH7 and E6AP (59, 61). Reactions were initiated by the addition of 125I-ubiquitin. After 10–30 min, the reactions were quenched by the addition of 25 μl of 2× SDS sample buffer containing 0.3% (v/v) β-mercaptoethanol, and then the samples were heated to 100 °C for 3 min. The polyubiquitin conjugates were resolved from free 125I-ubiquitin by 12% (w/v) SDS-PAGE under reducing conditions at 4 °C and visualized by autoradiography of the dried gels (59, 61). Polyubiquitin chain formation was measured by excising lanes between the top of the resolving gel and the top of the stacker gel, representing anchored and free 125I-polyubiquitin chains, respectively (62), and quantitating associated 125I-ubiquitin by γ-counting (51, 59, 61). Absolute rates of 125I-polyubiquitin chain formation were calculated from the associated radioactivity and the corrected specific radioactivity for 125I-ubiquitin (51, 59, 61). Datum points represent single assay determinations, and complete data sets were evaluated by non-linear regression analysis using GraFit version 5.0 (Erithicus). Active Uba1, E2, and E6AP were independently determined in parallel by their stoichiometric formation of 125I-ubiquitin thioester (59).
Static Light-scattering Measurements
His6-E6AP after the His trap affinity purification step was further purified by FPLC using a Mono Q 5/50 column (Amersham Biosciences) equilibrated in 50 mm Tris-HCl (pH 7.5) and 1 mm DTT. Recombinant His6-E6AP eluted at 300 mm NaCl in a 0–500 mm gradient (16 mm/ml, 1 ml/min). The molecular weight of His6-E6AP was determined in solution at 37 °C by static light scattering using a 235-DynaPro NanoStar laser light scattering spectrometer at 663 nm wavelength (Wyatt Technology Corp.). Spectra were collected for 18 μm His6-E6AP in 50 mm Tris-HCl (pH 7.5) containing 200 mm NaCl. The effect of Ac-PheNH2 on the molecular weight of His6-E6AP was determined in 50 mm Tris-HCl (pH 7.5) containing 200 mm NaCl, 16 μm His6-E6AP total protein, 61 mm Ac-PheNH2, and 8% (v/v) carrier methanol. The molecular weight of E6AP was also evaluated in reactions containing 0–350 μm Ac-NRIRMYSERRITVLYSL in 50 mm Tris-HCl (pH 7.5), 250 mm NaCl, 1 mm DTT, and 17 μm His6-E6AP. Data were analyzed with Dynamics® software at both default and maximum sensitivity to detect the presence of low concentrations of E6AP oligomeric forms.
Analysis of E6AP-Hect Interface Structure
The E6AP Hect domain structure (PDB code 1D5F) corresponding to the E6AP trimer reported by Huang et al. (49) was analyzed using PISA (PDBePISA-EMBL-EBI (Protein Interfaces, Surfaces, and Assemblies)) (63). The Assemblies analysis predicted a strong interaction between subunits, and default conditions detected an average of 38 residues/subunit involved in surface interactions between adjacent subunits (63). All structural representations were generated with PyMOL (Schrödinger, LLC).
RESULTS
E6AP-catalyzed Polyubiquitin Chain Formation Requires Oligomerization
Early in our studies, we examined the effect of the GST moiety on E6AP activity and found that processing of GST-E6AP fusion protein with thrombin resulted in a consistent 15–30% decrease in activity that resulted from a proportionately lower kcat but without effect on the Km for UbcH7∼125I-ubiquitin binding (not shown). Because GST protein is known to dimerize (64–66), one interpretation posits that GST promotes oligomerization of E6AP that results in the increased kcat. Consistent with this model, the addition of free GST to disrupt the putative oligomerization of GST-E6AP showed a biphasic concentration dependence (Fig. 1A). At free GST concentrations less than 5 μm, the abrupt concentration-dependent decrease in the initial rate of polyubiquitin chain formation exhibited a K½ of ∼1 μm, whereas at higher concentrations, there was a linear reduction in the initial rate with [GST]o (Fig. 1A). The linear phase at higher GST concentrations coincided with the appearance of a band of monoubiquitinated GST and probably results from competition of this reaction with polyubiquitin chain elongation. In contrast, the decrease in rate occurring at lower GST concentrations was consistent with disruption of GST-E6AP homodimers by free GST because the Kd for GST dimerization is 0.6 μm (66).
FIGURE 1.

E6AP catalyzed polyubiquitin chain formation requires oligomerization. A, initial rates of E6AP-catalyzed free polyubiquitin chain formation were determined as described under “Materials and Methods” in the presence of 8 nm human Uba1, 200 nm UbcH7, 0.2 nm GST-E6AP, 5 μm 125I-ubiquitin, and the indicated concentrations of recombinant free GST. B, autoradiogram of 125I-ubiquitin conjugation assays performed under initial velocity conditions in the presence of 60 nm human Uba1, 400 nm UbcH7, 6 nm GST-E6AP, 4 μm 125I-ubiquitin, and either 54 μm GST-E6APΔ495 or 66 μm E6APΔ495, as indicated. C, initial rates of 125I-ubiquitin conjugation were determined as in B in the presence of 8 nm GST-E6AP and the indicated concentrations of GST-E6APΔ495. As described under “Materials and Methods,” radioactivity associated with the stacker gel representing free and unanchored 125I-polyubiquitin chains was quantitated to calculate the resulting initial velocities. D, initial rates of 125I-ubiquitin conjugation were determined as in C but with the indicated concentrations of E6APΔ495. E, initial rate assays of 125I-ubiquitin conjugation determined as in C in the presence of the indicated concentrations of GST-HectC820A (open circles) or HectC820A (closed circles). F, schematic diagram depicting the model for inhibition of polyubiquitin chain formation by the Δ495 truncation or free Hect domain. For C–E, solid lines represent nonlinear inverse hyperbolic regression fits of the data using GraFit version 5.0.
To directly evaluate the potential contribution of E6AP oligomerization to catalytic activity, we introduced a STOP codon in the GST-E6AP coding region to truncate the Hect domain from E6AP, generating GST-E6APΔ495, to test whether the truncated protein could disrupt oligomerization in a manner similar to the free GST moiety. The addition of either 54 μm GST-E6APΔ495 or 66 μm thrombin-processed E6APΔ495 to wild type enzyme decreased the initial rate of GST-E6AP-catalyzed polyubiquitin chain formation (Fig. 1B). When the experiment of Fig. 1B was repeated at different concentrations of either GST-E6APΔ495 or E6APΔ495, hyperbolic concentration-dependent decreases in the initial rates of polyubiquitin chain formation resulted (Fig. 1, C and D, respectively). At higher concentrations than shown in Fig. 1, C and D, GST-E6APΔ495 and E6APΔ495 quantitatively blocked wild type GST-E6AP-catalyzed polyubiquitin chain formation (not shown). Nonlinear hyperbolic regression analysis yielded Ki values of 12 ± 3 and 19 ± 8 μm for GST-E6APΔ495 and E6APΔ495, corresponding to ΔGbinding values of −6.7 and −6.4 kcal/mol, respectively. Although the individual Ki values are not statistically different, the corresponding ΔΔGbinding for contribution of the GST moiety to GST-E6AP dimerization predicts a Kd for GST association of 1.6 μm, in good agreement with the published value of 0.6 μm (66). Results identical to those of Fig. 1, C and D, were obtained when the experiments were repeated with catalytically inactive GST-E6AP-HectC820A or E6AP-HectC820A in which the active site Cys820 of the Hect domain was mutated to alanine, yielding Ki values of 31 ± 4 and 27 ± 9 μm, respectively (Fig. 1E). Inhibition by E6APΔ495 is consistent with a model in which the truncation competitively disrupts an intrinsic E6AP wild type oligomer that is required for activity, schematically illustrated in Fig. 1F. Overall, the data reveal that oligomerization of E6AP is required for catalytic competence in polyubiquitin chain assembly. The similarities in Ki values between the E6APΔ495 and E6AP-HectC820A proteins suggest that the Hect domain and the targeting regions both contribute to oligomerization.
Active E6AP Is an Oligomer
Resolution of bacterially expressed protein by SDS-PAGE and visualization by Coomassie staining revealed a series of GST-associated bands in our preparations ranging from a relative mobility of ∼130 kDa for full-length protein, in good agreement with the expected size of 125 kDa, to 25 kDa, representing the free GST moiety (Fig. 2A, left lane); however, only the band of highest relative molecular weight, corresponding to full-length GST-E6AP, formed a 125I-ubiquitin thioester (51). Because bacterially expressed recombinant GST-E6AP is isolated as a mixture of full-length and C-terminal truncations (Fig. 2A, left), the results of Fig. 1 suggest that our kinetic studies probably underestimate the intrinsic kcat for the ligase due to the presence of endogenous GST-E6AP fragments. To test this, we cloned full-length E6AP into a baculovirus expression system to yield >95% full-length ligase. The resulting N-terminal His6-E6AP could be isolated from the medium without the marked fragmentation observed for bacterially expressed GST-E6AP and exhibited a relative molecular mass of 100 kDa, corresponding to the predicted value for full-length protein (Fig. 2A, right). After purification, maintaining the protein in a low concentration of NaCl was essential to prevent protein precipitation upon removal of imidazole. On a lower percentage SDS-polyacrylamide gel, the full-length affinity-purified band could be resolved into two closely migrating bands, only the slower migrating band of which could be detected on Western blots using anti-His6 antibody compared with detection by anti-E6AP antibody (Fig. 2B). A trace fragment band lacking the His6 tag could also be detected by anti-E6AP Western blot (Fig. 2B). Co-purification of E6AP lacking the affinity tag with His6-E6AP is consistent with oligomerization of the enzyme indicated by the results of Fig. 1.
FIGURE 2.
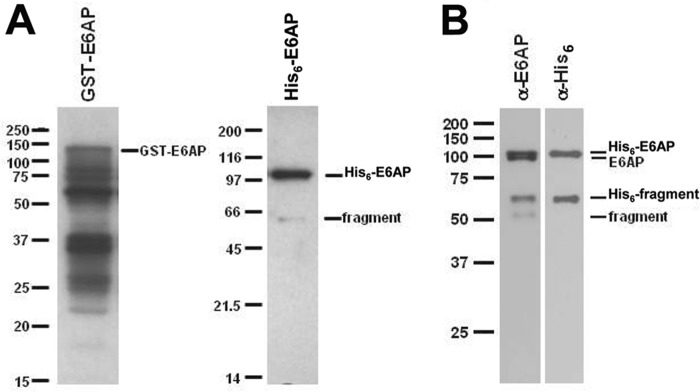
SDS-PAGE of selected E6AP preparations. A, Coomassie-stained 10% (w/v) SDS-PAGE resolution of affinity-purified recombinant GST-E6AP expressed in E. coli (left) versus His6-E6AP expressed in baculovirus (right). B, Western blot of 12% (w/v) SDS-PAGE resolution of baculovirus-expressed His6-E6AP stained with anti-E6AP antibody (left) and then stripped and restrained with anti-His6 antibody (right). Mobility markers are shown to the left of the corresponding panels. Mobilities of selected E6AP species are shown to the right of the corresponding panels.
We have previously demonstrated that the recombinant GST-E6AP in preparations identical to that analyzed in Fig. 2A (left) exhibited hyperbolic kinetics for 125I-polyubiquitin chain formation with respect to [UbcH7]o, from which values of Km and kcat, the latter defined as Vmax/[GST-E6AP]o, could be determined (51). Baculovirally expressed His6-E6AP also yielded hyperbolic kinetics for 125I-polyubiquitin chain formation with respect to [UbcH7]o from which Km (46 ± 7 nm) and kcat (0.63 ± 0.03 s−1) could be determined by nonlinear hyperbolic regression analysis (51) (Table 1). That homogeneous full-length E6AP exhibits a Km indistinguishable from that of the bacterially expressed heterogeneous GST-E6AP preparations, but a 20-fold higher kcat is consistent with our prediction based on a model for oligomerization. Similarly, GST-E6AP Hect domain protein composed of residues 450–872 is capable of modest polyubiquitin chain formation and has an affinity for UbcH7∼ubiquitin thioester (Km = 89 ± 11 nm) that is slightly less than that of full-length E6AP but a kcat that is 2000-fold lower (Table 1). In contrast, removing the GST moiety by in situ processing with thrombin prior to assay abrogates chain formation (not shown); however, the resulting Hect domain moiety catalyzes a slow rate of monoubiquitination, as noted previously (51). The kinetics of Hect domain monoubiquitination follows hyperbolic kinetics with respect to [UbcH7]o and yields Km and kcat values similar to those of the unprocessed GST-Hect domain (Table 1). The results of Table 1 are not a consequence of differences in the amounts of active protein in the various preparations because in each case the ligase is quantitated by the functional assay of stoichiometric Hect domain∼125I-ubiquitin thioester formation. Therefore, polyubiquitin chain formation kinetics for the various forms of E6AP (Table 1) and the effect of N-terminal truncation (Fig. 1) are consistent with the catalytically active form of the enzyme existing as an oligomer.
TABLE 1.
Effect of E6AP length on chain formation kinetics
| Km | kcat | kcat/Km | |
|---|---|---|---|
| nm | s−1 | m−1 s−1 | |
| Heterogeneous GST-E6AP | 58 ± 6 | 3.1 ± 0.9 × 10−2 | 5.4 × 105 |
| E6AP Hect domaina | 91 ± 25 | 7.0 ± 0.1 × 10−4 | 8.5 × 103 |
| GST-E6AP Hect domain | 89 ± 11 | 3.2 ± 0.1 × 10−4 | 3.6 × 103 |
| Homogeneous His6-E6AP | 46 ± 7 | 6.3 ± 0.3 × 10−1 | 1.3 × 107 |
a Determined by E6AP Hect domain monoubiquitination.
A Trimer Is the Likely Fully Active Form of E6AP
To test directly the ability of E6AP to oligomerize, we determined the solution molecular weight of wild type His6-E6AP by gel filtration chromatography (Fig. 3A). The relative molecular weight for E6AP, monitored by 125I-polyubiquitin chain formation under E3-limiting conditions, encompasses a peak centered at 190 kDa. This molecular weight is consistent with a stable dimer of 100-kDa subunits or less stable higher oligomers subject to dissociation by dilution as the complex passes through the column. To distinguish between the latter, we analyzed a parallel sample by static light scattering, which does not involve sample dilution. Following Mono Q anion exchange chromatography to remove inactive high molecular weight aggregates, freshly prepared active His6-E6AP displayed a molecular mass of 283 kDa by static light scattering that was consistent with a trimer (Fig. 3A). The low abundance higher molecular weight species of ∼50-nm radius represents aggregates not completely removed by the Mono Q FPLC step. In parallel experiments, the molecular mass ranged from ∼200 to 400 kDa depending on protein concentration, pH, and ionic strength, consistent with equilibrium oligomerization of the 100-kDa monomer (not shown).
FIGURE 3.

Functional E6AP is an oligomer. A, 150 μl of 75 μm full-length His6-E6AP was analyzed in a 1× 30-cm Superose 12 FPLC gel filtration column equilibrated in 50 mm Tris-HCl (pH 7.5) and 50 mm NaCl. Protein was monitored by 280 nm absorbance (filled circles), and enzyme activity was monitored by E3-limiting 125I-polyubiquitin chain formation (open circles). Inset, calibration plot with the elution position of the peak E6AP activity shown with an open circle. B, static light scattering analysis of 18 μm full-length His6-E6AP in 50 mm Tris-HCl (pH 7.5) containing 200 mm NaCl at 37 °C as described under “Materials and Methods.” The main peak of 283 kDa exhibits a polydispersity of 17%. The higher molecular weight low abundance peak of 50-nm radius represents residual aggregates not removed by Mono Q FPLC. C, static light scattering analysis identical to A in the presence of 16 μm His6-E6AP and 8% (v/v) methanol in the absence (solid line; 23% polydispersity) or presence (dashed line; 14% polydispersity) of 61 mm Ac-PheNH2.
Wild type E6AP is thought to exist as a monomer; however, the original publication reporting the structure of the isolated Hect domain also noted a trimeric structure that was proposed to arise from crystal packing interactions (49) (Fig. 4A). Based on the data of Fig. 3 and the requirement that E6AP-catalyzed polyubiquitin chain formation requires oligomerization, we propose that the fully active form of the ligase is a trimer. The symmetric trimer (PDB code 1D5F) reported by Huang et al. (49) buries an extensive combined surface area of 7508 Å2 comprising a large fraction of apolar residues at the subunit interfaces that represents a solvation free energy of −4.2 kcal/mol by PISA analysis (63) (Fig. 4A), corresponding to an apparent Kd of 25 μm for E6AP Hect subunit interactions without considering additional hydrogen bond or salt bridge interactions (supplemental Table 1), which are difficult mathematically to model due to uncertainties related to the actual microenvironments of the individual bonds. However, the predicted Kd based on desolvation agrees well with the empirical Ki obtained with GST-HectC820A (31 ± 4 μm) and HectC820A (27 ± 9 μm) (Fig. 1D). Sequence comparison among known Hect domain sequences reveals marked conservation (37% identity, 69% conservation of side chain polarity) among the residues at the subunit interfaces of the E6AP trimer (supplemental Fig. 1 and Table 2). Collectively, these features are characteristic of protein-protein binding interfaces rather than crystal packing interactions (67, 68). Moreover, the separation distance between N-terminal residues of the Hect domain subunits suggests that the N-terminal targeting domains of the subunits present on the full-length molecule would not hinder trimer formation (Fig. 4B).
FIGURE 4.

Active E6AP is a trimer stabilized by Phe727 interactions. A, structure of the E6AP Hect domain trimer (PDB code 1D5F) reported by Huang et al. (49). Each chain is colored differently to emphasize subunit packing, whereas the forward subunit is rendered as a ribbon and colored to distinguish the large N-terminal subdomain (marine), the small N-terminal subdomain (cyan), and the C-terminal subdomain (dark blue). The active site Cys820 and N-terminal residues are rendered in CPK and colored yellow and red, respectively. B, the structure of A is rotated 90º into the plane of the page to emphasize subunit packing. Residues of the hydrophobic pocket are rendered in white for the gold subunit, and Phe727 of the blue subunit is rendered in orange. C, closer view of Phe727 intercalated into the hydrophobic pocket of the adjacent subunit. D, the concentration dependence of Ac-PheNH2 on the initial rate of 125I-polyubiquitin chain assembly under E6AP-limiting conditions, with the solid line representing a nonlinear inverse regression analysis fit to a hyperbolic kinetics; inset, semireciprocal plot of the resulting data. Incubations contained 400 nm Uba1, 200 nm UbcH7, 8.8 nm His6-E6AP, 5 μm 125I-ubiquitin, and the indicated concentrations of Ac-PheNH2. E, double reciprocal plot of the dependence of [UbcH7]o on the initial rate of polyubiquitin chain formation conducted as in D in the absence (solid circles) or presence (open circles) of 44 mm Ac-PheNH2. All incubations contained 5% (v/v) methanol as a carrier for the Ac-PheNH2.
As a trimer, the 1DF5 structure reveals a number of radially symmetric subunit interactions. For example, the large N-terminal subdomain of the Hect domain contains a subset of conserved apolar residues at the subunit interface (Ile600, Tyr601, Leu723, Leu726, Ile732, and Leu735) that forms a hydrophobic pocket into which intercalates Phe727 of the adjacent subunit in the trimer (49) (Fig. 4C), providing a stabilization of −2.13 kcal/mol by PISA analysis. Although there are other residues and surfaces contributing to stabilization of the trimer, intercalation of Phe727 into the hydrophobic pocket appears to be of special importance because mutation of Phe727 to alanine destabilizes trimeric E6AP in favor of the monomer, the latter of which retains the ability to form a ubiquitin thioester intermediate (49). Retention of the ability to form the thioester intermediate was originally interpreted as evidence that the monomer represented the biological relevant structure (49). However, we have recently demonstrated that the E6AP catalytic cycle comprises a two-step mechanism involving rapid transthiolation of activated ubiquitin from the cognate E2∼ubiquitin co-substrate to the Hect Cys820 active site residue followed by rate-limiting chain elongation to form the polyubiquitin degradation signal (51). Because of this, Cys820 thioester formation is an insensitive measure of E6AP catalytic competence (51), accounting for the reported inconsistencies between known loss-of-function mutations in Angelman syndrome and retention of the ability of the enzyme to form an active Cys820-linked thioester to ubiquitin (17). To re-examine this point, we mutated Phe727 to aspartic acid in order to more efficiently disrupt trimer formation. The E6APF727D mutation significantly ablates the ability of the enzyme to form polyubiquitin chains, as shown by the 62-fold decrease in kcat without significant effect on the Km for UbcH7∼125I-ubiquitin thioester binding (Table 2) or end point Cys820 thioester formation (not shown), the latter as noted previously (49).
TABLE 2.
Summary of kinetic constants
| Km | kcat | kcat/Km | |
|---|---|---|---|
| nm | s−1 | m−1 s−1 | |
| GST-E6AP-UbcH7a | 58 ± 8 | 3.1 ± 0.9 × 10−2 | 5.4 × 105 |
| GST-E6APF727D-UbcH7b | 72 ± 25 | 5.0 ± 0.7 × 10−4 | 7.6 × 103 |
| GST-E6APY533A-UbcH7 | 40 ± 12 | 3.1 ± 0.2 × 10−3 | 7.9 × 104 |
| GST-E6APD543A-UbcH7b | 540 ± 400 | 4.0 ± 1.0 × 10−3 | 7.5 × 103 |
| GST-E6APR626A-UbcH7b | 150 ± 50 | 2.0 ± 0.2 × 10−4 | 1.3 × 103 |
| GST-E6APK688A-UbcH7 | 39 ± 10 | 5.2 ± 0.4 × 10−2 | 1.3 × 105 |
a From Table 1.
b The observed rates of these mutants approach our limit of detection for 125I-polyubiquitin chain formation and result in a substantial increase in the S.E. of the measurements.
As additional evidence that trimeric E6AP represents the functional form of the enzyme, we examined the effect of Ac-PheNH2 as a mimic of Phe727. The addition of Ac-PheNH2 to His6-E6AP results in quantitative dissociation of the oligomer to a 104 kDa peak in good agreement with the expected molecular weight of the monomer (Fig. 3C). This observation is consistent with substitution of Ac-PheNH2 into the conserved hydrophobic pocket to disrupt the radially symmetric subunit interactions stabilizing the trimer and is accompanied by quantitative inhibition of 125I-polyubiquitin chain formation (Fig. 4D). The dependence of initial velocity on [UbcH7]o in the absence or presence of 44 mm Ac-PheNH2 shows the amino acid derivative to be a non-competitive inhibitor with a Ki = 12 ± 3 mm, which is in good agreement with the calculated Kd of 27 mm predicted from the −2.13 kcal/mol stabilization predicted by PISA (see above). In addition, non-competitive inhibition by Ac-PheNH2 is consistent with that predicted for an effect on subunit dissociation (Fig. 4E) and the observed consequence of the E6APF727D mutation (Table 2). Although Ac-PheNH2 can potentially interact with other regions comprising the interface of full-length His6-E6AP, the linearity of the double reciprocal plots for Fig. 4, D (inset) and E, are consistent with binding at a single homogeneous site, presumably the buried conserved hydrophobic pocket.
A Conserved α-Helix Blocks E6AP Trimer Formation
Examination of the extant structures for other Hect domains, including WWP1 (PDB code 1ND7), Smurf2 (PDB code 1ZVD), HuWE1 (PDB code 3H1D), Nedd4-1 (PDB code 2XBB), Nedd4-2 (PDB code 2XBF), and yeast RSP5 (PDB codes 3OLM and 4LCD), provides no other instance of trimer formation (69–73). However, the other Hect domain structures contain additional sequence N-terminal to the truncation site for E6AP at Asn497 that is not present in the E6AP structure (49). Immediately N-terminal to Asn497 is an extensive amphipathic α-helix corresponding to residues 474–490 that is relatively conserved among the Hect ligases, contributes to domain stability, and correlates with reduced autoubiquitination and target protein conjugation (71, 74). In all of the Hect domain structures containing the additional segment, the hydrophobic face of the amphipathic N-terminal α-helix binds to the hydrophobic pocket normally occupied by Phe727 in the trimeric E6AP structure (69–73). We speculate that the latter interaction of the α-helix might block oligomerization, accounting for the observed reduction in ligase activity because we here demonstrate that trimer formation is required for polyubiquitin chain assembly. To address the role of the N-terminal α-helix, the initial rate of polyubiquitin chain formation was analyzed in the absence or presence of an N-terminal blocked Ac-NRIRMYSERRITVLYSL peptide corresponding to residues 474–490 of E6AP isoform 1 to mimic the proposed effect of this segment (Fig. 5). With increasing concentrations of the peptide, the initial rate of E6AP-catalyzed chain formation decreased with a hyperbolic dependence (Fig. 5A), as shown by the linearity of the corresponding semireciprocal plot, yielding an apparent Ki of 22 ± 2 μm (Fig. 5A, inset). When the concentration dependence of the initial rate for polyubiquitin chain formation on [UbcH7]o was examined in the absence or presence of 32 μm peptide, the isolated α-helix was a classic non-competitive inhibitor (Fig. 5B). The empirical Ki probably underestimates the intrinsic affinity of the wild type N-terminal α-helix because the isolated peptide is unlikely quantitatively to maintain the secondary structure of the intact protein. Nonetheless, observation of non-competitive inhibition is consistent with the peptide blocking trimerization of E6AP in a manner analogous to Ac-PheNH2 (Fig. 4, D and E) and mutation of Phe727 (Table 2). Further, the peptide promotes dissociation of the E6AP oligomer when analyzed by static light scattering (Fig. 5C).
FIGURE 5.

The N-terminal α-helical peptide inhibits E6AP ligase function. A, initial rates of 125I-polyubiquitin chain assembly under E6AP-limiting conditions were analyzed in the absence or presence of the indicated concentrations of N-terminal peptide and evaluated by nonlinear regression fit to an inverse hyperbolic equation. Reactions contained 34 nm Uba1, 390 nm UbcH7, 1.3 nm His6-E6AP, and 4 μm 125I-ubiquitin. Inset, semireciprocal plot of the data. B, double reciprocal plot of initial rates of 125I-polyubiquitin chain assembly under E6AP-limiting conditions in the absence (closed circles) or presence (open circles) of 32 μm N-terminal peptide. Reactions contained 33 nm Uba1, 0.6 nm His6-E6AP, 4 μm 125I-ubiquitin, and the corresponding [UbcH7]o concentrations. C, the static light scattering spectrum of 17 μm full-length His6-E6AP was determined as described in the legend to Fig. 3C in the absence (solid line; 10 ± 3% polydispersity) or presence (dashed line; 10 ± 2% polydispersity) of 88 μm Ac-NRIRMYSERRITVLYSL peptide.
Identification of Additional Subunit Interface Residues Affecting E6AP Catalytic Activity
The present data and recent insights into the properties of protein subunit interfaces support the E6AP trimer as the catalytically relevant structure (67, 68). The interaction surfaces of the trimer were analyzed by PISA to define other side chain interactions contributing to stability beyond that of Phe727 (supplemental Table 1). A comprehensive sequence alignment of human Hect ligases cross-referenced to the results of PISA identified a number of conserved positions that appear potentially critical, including the hydrophobic pocket residues into which Phe727 intercalates, as discussed earlier. Interestingly, several of these residues represent or are adjacent to documented Angelman syndrome point mutations. We were particularly interested in Tyr533, which is an Angelman syndrome mutation site and which participates in a pattern of side chain interactions between subunits (75). Tyrosine 533 also exists in the region of subunit interface recently suggested by Chan et al. to be sensitive to cAbl-dependent regulation by phosphorylation of Tyr636 (76).
For two of the subunits in the trimer structure, the side chain phenolic group of Tyr533 hydrogen bonds with the amide hydrogen of Asp543 present on the same polypeptide chain (Fig. 6B), whereas for the third subunit, Tyr533 is rotated 60º and hydrogen-bonds to the amide hydrogens of Arg626 and Thr624 on the adjacent chain (Fig. 6C). More important, for all three subunits, the side chain carboxyl and amide carbonyl groups of Asp543 hydrogen-bond to the side chain guanidinium of Arg626 (Fig. 6, B and C). Tyrosine 533 and Asp543 reside within the Hect large N-terminal subdomain, whereas Arg626 is in the small N-terminal subdomain of the adjacent subunit, so that the three residues constitute a radial symmetric pattern of subunit interactions. We independently mutated each of the residues to alanine and examined the effect on the kinetics for 125I-polyubiquitin chain formation with respect to [UbcH7]o under rigorously E3-limiting conditions (Table 2). As a control, we similarly also mutated Lys688, a residue present on the same small N-terminal subdomain interface as Arg626, which PISA analysis indicates hydrogen-bonds to the side chain of Glu535 in the adjacent subunit. All four mutants formed 125I-ubiquitin thioesters to Cys820 with kinetics qualitatively similar to that of wild type enzyme in short term incubations described previously (51).
FIGURE 6.

Selected side chain interactions within the E6AP Hect domain trimer. A, structure of the E6AP Hect domain trimer portrayed in Fig. 4B illustrating additional residues stabilizing the structure. B and C, close-up views of the spatial orientation for Tyr533, Asp543, and Arg626. Subunit residues are color-coded like those in A. Residues in parentheses are those previously examined by Chan et al. (76).
Mutation of Lys688 did not have a significant effect on the kinetics of 125I-polyubiquitin chain assembly (Table 2). However, given the large number of predicted subunit interactions stabilizing the trimer (supplemental Table 1), abrogating a single site might not be expected a priori to have a large effect except for those interactions particularly critical for structural integrity. In contrast to the effect of mutating Tyr688, mutation of Tyr533 decreased kcat 10-fold but had no effect on the Km for UbcH7∼ubiquitin binding (Table 2). Similarly, mutation of Arg626 decreased kcat 155-fold but had little effect on Km (Table 2). Both of the latter mutants are consistent with an effect on oligomerization, given their position within the trimer structure and distance from the active site Cys820 as well as the fact that neither mutation alters the ability of E6AP to bind its UbcH7∼ubiquitin thioester substrate. Mutation of Asp543 similarly ablated kcat 8-fold but additionally increased Km significantly (Table 2). Together with the effects of Tyr636 phosphorylation (76), the consequences of these point mutants reveal complex interactions affecting the catalytic function of E6AP.
E6 Protein Enhances E6AP Polyubiquitin Chain Synthesis
HPV16 E6 protein is thought to promote viral replication and host epithelial cell transformation by serving as an E6AP adapter to target the p53 tumor suppressor protein for 26 S proteasome-dependent degradation (32, 77, 78). Recent structural work demonstrates that the 158-amino acid E6 viral protein contains structurally related N-terminal and C-terminal Zn2+ binding domains connected by a linker polypeptide (46). The C-terminal domain binds p53 but also shows limited interaction with E6AP (43, 79). In contrast, the N-terminal domain exhibits significant affinity for E6AP through binding to a canonical LXXLL motif on the ligase and is additionally responsible for E6 protein self-association (40–42, 46), accounting for the observation that the complex between E6 protein and p53 consists of a p53 tetramer and an E6 dimer (45, 80). As expected, disrupting the dimerization interface by mutation inhibits E6-dependent p53 conjugation by E6AP but also enhances E6 protein solubility (46), which probably relates to earlier observations that ectopic expression of E6 protein stimulates autoubiquitination and subsequent E6AP degradation in vivo (81).
Because the trimer is the presumed catalytically fully active form of E6AP, we speculated that the effect of E6 protein on E6AP turnover (82) and the stabilization of p53 when the E6 dimerization interface is disrupted might reflect the ability of the viral protein to promote E6AP oligomerization. Fig. 7A shows the effect of recombinant E6 protein on the [UbcH7]o dependence of E6AP-catalyzed 125I-polyubiquitin chain formation. Initial rates of chain formation were significantly enhanced in the presence of E6 protein, with the kcat increasing from 0.057 ± 0.029 s−1 in the absence of E6 protein to 0.81 ± 0.045 s−1 in its presence. The corresponding double reciprocal plot shows E6 protein to be a nonessential activator, exhibiting a kcat effect (inset), and with a Kd for E6 binding corresponding to 1.5 nm. These results are consistent with a mechanism in which the E6 dimer promotes oligomerization of E6AP to the catalytically competent trimer through binding to a site distinct from the catalytic site, presumably the leucine-rich LXXLL motif (40–42). Although the N-terminal domain contains both the dimerization interface and the LXXLL association motif (46), the recombinant E6(HPV16)Δ91 N-terminal domain encompassing residues 1–91 (46) inhibits E6AP-catalyzed polyubiquitin chain formation with a hyperbolic concentration dependence (apparent Ki = 4 μm; not shown) and displays non-competitive inhibition with respect to [UbcH7]o, corresponding to Ki = 6 μm (Fig. 7B). Similarly, the recombinant E6(HPV16)Ct C-terminal domain encompassing residues 89–158 is a non-competitive inhibitor of E6AP with respect to [UbcH7]o corresponding to Ki = 4 μm (not shown). Collectively, the results indicate that both E6 domains bind E6AP, presumably at different sites, to promote oligomerization of full-length E6AP. This conclusion supports earlier empirical binding evidence demonstrating interaction of each domain with E6AP (44, 46).
FIGURE 7.

E6 protein enhances E6AP ligase activity and rescues selected Angelman syndrome mutations. A, initial rates of 125I-ubiquitin polyubiquitin chain formation were determined under E6AP-limiting conditions in the absence (closed circles) or presence (open circles) of 20 nm E6 protein as described under “Material and Methods” and evaluated by nonlinear regression fit to the Michaelis-Menten equation. Assays contained 70 nm Uba1, 0.4 nm His-E6AP, 4 μm 125I-ubiquitin, and the indicated UbcH7 concentrations. Inset, double reciprocal plot of the data. B, double reciprocal plot of the initial rates of 125I-ubiquitin conjugation activity determined under E6AP-limiting conditions in the absence (closed circles) or presence (open circles) of 3.3 μm E6 (HPV16)Δ91, as described under “Materials and Methods” and in A. Assays contained 30 nm Uba1, 1 nm His6-E6AP, 4 μm 125I-ubiquitin, and the indicated UbcH7 concentrations. C, conjugation reactions similar to those of B were conducted with 110 nm Uba1, 480 nm UbcH7, 5 μm 125I-ubiquitin, and 1 nm wild type or mutant GST-E6AP in the absence (lanes 1–7) or presence (lanes 8–13) of 20 nm E6(HPV16). D, quantitation of product formation rates from C in the absence (light gray) or presence (dark gray) of E6(HPV16). Data for wild type GST-E6AP and GST-E6APY533A are plotted on the left axis, whereas data for GST-E6APR626A, -D543A, and -F727D are plotted on the right axis.
Because full-length E6 protein acts as a nonessential activator, presumably by promoting E6AP trimer formation, we asked whether the viral protein could rescue the loss-of-function phenotype displayed by the E6APF727D point mutant. The addition of 20 nm E6 protein significantly enhanced the initial rate of 125I-polyubiquitin chain formation for wild type protein (Fig. 7C, lanes 7 and 13) as seen in Fig. 7A and E6APF727D (Fig. 7C, lanes 6 and 12). Similar rescue of 125I-polyubiquitin chain formation was observed in an 8-fold increase in rate for E6APY533A (Fig. 7C, lanes 3 and 9) and a 13-fold increase in rate for E6APR626A (Fig. 7C, lanes 4 and 10). In contrast, no enhancement in rate was observed for the E6APD543A point mutant (Fig. 7C, lanes 5 and 11). These results are consistent with E6 rescuing the loss-of-function phenotype for selected point mutants by promoting oligomerization.
DISCUSSION
Homo- and hetero-oligomerization regulate many complex biochemical processes in the cell (83), and recent findings suggest that such protein interactions are critical for the mechanism(s) of assembling polyubiquitin signals during key events of cell regulation (52, 84–88). The present studies were prompted in part by our attempt to explicate the puzzling observation that the assembly of 125I-polyubiquitin chains by GST-E6AP is ablated when the affinity tag is removed following digestion with thrombin. Subsequent experiments indicated that the ability of GST to dimerize (64–66) promotes oligomerization of E6AP, confirmed by the quantitative inhibition of ligase-catalyzed chain formation by free GST, GST-E6APΔ495, and GST-E6APC820A (Fig. 1). The ability of thrombin-processed E6APΔ495 and E6APC820A to similarly inhibit wild type GST-E6AP polyubiquitin chain formation demonstrates that oligomerization is an intrinsic property of the Hect ligase, with the interaction interfaces probably spanning the N-terminal targeting and C-terminal Hect domains of the ligase (Fig. 1). In addition, co-purification of recombinant baculovirally expressed His6-E6AP with full-length E6AP lacking the His6 affinity tag is consistent with oligomerization of the ligase (Fig. 2B). The progressive increase in kcat for E6AP-catalyzed 125I-polyubiquitin chain assembly with the absence of competing degradative fragments is consistent with a role for oligomerization in the activity of E6AP (Table 1). Finally, static light scattering measurements are consistent with the homotrimer as the catalytically relevant form of E6AP (Fig. 3).
Trimeric E6AP was initially dismissed as an artifact of crystal packing forces, particularly because a F727A point mutant capable of disrupting the oligomer did not affect the ability of the enzyme to catalyze transthiolation from E2∼ubiquitin thioester (49); however, more comprehensive functional assays were unavailable for testing additional potential effects of trimer disruption. In the present studies, we demonstrate that a paralogous F727D mutation intended to more completely disrupt trimer formation significantly ablates the ability of the enzyme to catalyze elongation of polyubiquitin chains to <2% of wild type enzyme (Table 2), concomitant with dissociation of the oligomer (Fig. 3). As noted previously for the F727A mutant (89), mutation of Phe727 to aspartic acid has no effect on formation of the essential Cys820∼ubiquitin thioester intermediate (not shown). In silico PISA analysis of the subunit interfaces present within the E6AP trimer identifies a cohort of conserved residues within the 7508 Å2 buried by oligomerization (supplemental Fig. 1A). Of the significant number of hydrogen bond and salt bridge interactions identified by PISA (supplemental Table 1), the conserved radially symmetric network of interactions represented by the intersubunit intercalation of Phe727 into the hydrophobic pocket present on the adjacent subunit appears particularly important because mutation of Phe727 or antagonizing the interaction by the addition of Ac-PheNH2 disrupts trimer formation and significantly ablates activity (Fig. 4). The linearity of the double reciprocal plots in the presence of Ac-PheNH2 (Fig. 4, D and E) and the good agreement between the empirical Ki of 12 mm and the value of 27 mm predicted from the calculated binding energy of Phe727 (−2.13 kcal/mol) are consistent with the amino acid derivative binding at the unique hydrophobic pocket present in the subunit interface.
Among the extant structures for Hect domains, that of E6AP is unique in forming a trimer (69–73). The 1DF5 E6AP trimer is also notable in being the only such Hect domain structure lacking the additional N-terminal sequence segment present in the other paralogs. In the other structures, a conserved α-helix within this sequence binds to the hydrophobic pocket otherwise occupied by Phe727 in the E6AP trimer (69–73). We demonstrate that E6AP residues 474–490, corresponding to the α-helical region, act as a non-competitive inhibitor of 125I-polyubiquitin chain formation with an affinity (Ki = 22 ± 2 μm) considerably greater than that of Ac-PheNH2 (Ki = 12 ± 3 mm) (Fig. 5). The presence of the additional α-helical segment is reported to enhance the intrinsic stability of the Hect domain and to ablate autoubiquitination (71, 74). The latter has been interpreted as demonstrating a regulatory role for the interaction between the α-helical segment and the hydrophobic pocket in the native full-length structure. Although we cannot a priori rule out such a regulatory role for the α-helix, we propose that association of the α-helical segment with the hydrophobic pocket is a structural artifact of the position of the truncation for the other Hect domain constructs and predict that in the absence of this interaction, the other ligases would assume a trimeric structure analogous to that of E6AP. We note as well that in the absence of trimer formation, packing of the Hect domains within the unit cells influences the orientation of the C-terminal domain, altering their positions relative to the large N-terminal subdomains (69–73). The various positions observed for the C-terminal domains in different Hect domain structures have been previously interpreted to reflect the requirement for flexibility during the catalytic cycle of the enzyme, suggested by the ablation of activity accompanying mutations in the linker region connecting the C-terminal and large N-terminal subdomains (69, 73, 82). We suggest, instead, that these alternate orientations for the C-terminal domain may reflect artifactual crystal packing forces present in the absence of trimerization.
A second subunit interface identified as important for trimer activity localizes to interactions among Tyr533 and Asp543, within the large N-terminal subdomain, and the small N-terminal subdomain residue Arg626 (76) of the adjacent subunit (Fig. 6). These residues form a network of hydrogen-bonded interactions that appear to stabilize the trimer because individual mutation of each significantly affects kcat but with little consequence for UbcH7∼ubiquitin thioester binding (Table 2). The functional effects of these mutations collectively support the trimer as the catalytically relevant structure for E6AP. Mutation of Asp543 appears to be the single exception because its mutation affects both kcat and binding of the UbcH7∼ubiquitin thioester substrate (Table 2). The latter may reflect a structural contribution for the side chain interactions of Asp543 or the special role for the residue in bridging the effects of Tyr533 and Arg626 (Fig. 6), which makes its mutation equivalent to a double mutant. In contrast, disrupting the hydrogen bond between Lys688 in the small N-terminal subdomain and Glu535 in the adjacent large N-terminal subdomain by mutation of the former has no consequence, indicating that the effects of the previous mutations are specific to those residues rather than a general feature of the interface. Interestingly, this subunit interface harbors Tyr636, which Chan et al. (76) have identified as a substrate for c-Abl phosphorylation in the regulation of E6AP function. Chan et al. (76) have speculated that the inhibition of E6AP activity observed on phosphorylation of Tyr636 might result from blocking oligomerization of the enzyme. Our empirical observations and the effect of mutation on other residues within this region support a role for Tyr636 in regulating trimer formation and E6AP activity.
The ability of E6 protein to recruit p53 to E6AP for targeted degradation is the accepted paradigm by which HPV-16 transforms infected epithelial cells (32, 77, 78). Structural studies demonstrating dimerization of E6 protein have more recently accounted for the observed stoichiometry of the resulting E62-p534 complex (45, 80). Given our observation that oligomeric E6AP is the catalytically competent form of the ligase, we asked whether the ability of E6 protein to dimerize could promote E6AP-catalyzed polyubiquitin chain formation. The data of Fig. 7 demonstrate that E6 protein is a potent nonessential activator of E6AP catalytic activity within a low nanomolar concentration range (Kactivation = 1.5 nm). The Kactivation for ligase stimulation, representing the binding of E6 to E6AP, is significantly lower than the 4 μm Kd for binding of the viral protein to E6AP reported previously by surface plasmon resonance (44), probably reflecting the enhanced entropically coupled affinity associated with binding to linked sites (90). The latter indicates that E6 protein probably functions at intracellular concentrations significantly lower than suggested by the isolated in vitro equilibrium binding affinity. In the absence of substrate, E6AP catalyzes the assembly of free and anchored polyubiquitin chains, the latter attached to the ligase through autoubiquitination (51). This bifurcation of products is proposed to occur by partitioning of a Cys820∼polyubiquitin intermediate between transfer to water or a lysine present on the ligase, respectively (51). The addition of E6 protein uniformly enhances the formation of both 125I-polyubiquitin products, consistent with the partitioning model (Fig. 7C). The latter observation explains the earlier observation that ectopic expression of E6 protein stimulates the autoubiquitination and degradation of endogenous E6AP (48). More importantly, these observations anticipate that E6 protein-dependent epithelial cell transformation may arise in part through a general stimulation of ligase activity toward its normal cohort of endogenous targets rather than serving exclusively as a binding adapter to target a small subset of alternate targets, such as p53 (Fig. 7A). Such a proposal is consistent with earlier observations that an E6 SAT8–10 mutant defective in targeting E6AP-dependent p53 degradation (48) but retaining the ability to immortalize human epithelial cells, induce colony growth on soft agar, and stimulate telomerase activity (84, 86) also simulates E6AP autoubiquitination and degradation in vitro (48).
The 14-fold stimulation in E6AP activity at saturating E6 protein suggests that only 0.7% of the active ligase, determined by stoichiometric Cys820∼125I-ubiquitin thioester formation, is present as the active trimer under the conditions of Fig. 7A, although we cannot rule out an intermediate level of activity from potential dimeric species derived from the trimeric structure. Consistent with the remarkable effect of E6 in promoting oligomerization, the viral protein complements the kinetic phenotypes resulting from the F727D, R626A, and Y533A mutations, the latter being a documented Angelman syndrome mutation site (75). Because the F727D mutation results from defective trimer formation, functional complementation of the R626A and Y533A mutations suggests that they as well represent trimer formation defects.
The present observations significantly extend our understanding of the underlying mechanism of the E6AP catalytic cycle and that of the Hect domain superfamily. Identification of oligomeric E6AP, presumably as the trimer or multiples thereof, as the catalytically relevant form of the ligase explains a number of previous observations in the literature and reveals potential pharmacological approaches to modulate activity of the enzyme. Accordingly, Ac-PheNH2 serves as an important proof of principle lead compound for potential future E6AP antagonists targeted to the disruption of oligomerization. Similarly, E6 protein suggests as yet unidentified cellular mechanisms modulating E6AP oligomerization that could serve as additional therapeutic targets. Finally, these observations emphasize the importance of quantitative biochemically defined studies unambiguously defining the mechanism of the ubiquitin ligases.
This work was supported, in whole or in part, by National Institutes of Health Grant GM034009 (to A. L. H.).

This article contains supplemental Tables 1 and 2 and Fig. 1.
The sequence for E6AP isoform 3 differs from isoform 1, from which the original crystal structure was determined (49), by the presence of an additional 20 amino acids at the N terminus. To facilitate comparison with the crystal structure, residues for isoform 3 will be referenced to the paralogous position of isoform 1 (i.e. by subtracting 20 from the isoform 3 residue number).
- E6AP
- E6-associated protein (gene name UBE3A)
- E6
- human papilloma virus 16 E6 protein
- Hect
- homologous to E6AP C terminus
- HPV
- human papilloma virus
- Ac-PheNH2
- N-acetylphenylalanylamide
- E1
- generic term for activating enzymes of Class 1 ubiquitin-like proteins
- E2/Ubc
- generic name for ubiquitin carrier protein/ubiquitin-conjugating enzyme
- E3
- generic name for ubiquitin:protein isopeptide ligase
- UbcH7
- human E2 carrier protein (gene name UBE2L3)
- Uba1
- ubiquitin-activating enzyme (gene name UBE1).
REFERENCES
- 1. Huibregtse J. M., Scheffner M., Beaudenon S., Howley P. M. (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 2563–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rotin D., Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 [DOI] [PubMed] [Google Scholar]
- 3. Wang M., Pickart C. M. (2005) Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 24, 4324–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang M., Cheng D., Peng J., Pickart C. M. (2006) Molecular determinants of polyubiquitin linkage selection by an HECT ubiquitin ligase. EMBO J. 25, 1710–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beaudenon S., Huibregtse J. M. (2008) HPV E6, E6AP and cervical cancer. BMC Biochem. 9, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Flashner B. M., Russo M. E., Boileau J. E., Leong D. W., Gallicano G. I. (2013) Epigenetic factors and autism spectrum disorders. Neuromolecular Med. 15, 339–350 [DOI] [PubMed] [Google Scholar]
- 7. Nicholls R. D., Knepper J. L. (2001) Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet. 2, 153–175 [DOI] [PubMed] [Google Scholar]
- 8. Matentzoglu K., Scheffner M. (2008) Ubiquitin ligase E6-AP and its role in human disease. Biochem. Soc. Trans. 36, 797–801 [DOI] [PubMed] [Google Scholar]
- 9. Kishino T., Lalande M., Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73 [DOI] [PubMed] [Google Scholar]
- 10. Sutcliffe J. S., Jiang Y. H., Galijaard R. J., Matsuura T., Fang P., Kubota T., Christian S. L., Bressler J., Cattanach B., Ledbetter D. H., Beaudet A. L. (1997) The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 7, 368–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mabb A. M., Judson M. C., Zylka M. J., Philpot B. D. (2011) Angelman syndrome. Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsuura T., Sutcliffe J. S., Fang P., Galjaard R. J., Jiang Y. H., Benton C. S., Rommens J. M., Beaudet A. L. (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77 [DOI] [PubMed] [Google Scholar]
- 13. Lossie A. C., Whitney M. M., Amidon D., Dong H. J., Chen P., Theriaque D., Hutson A., Nicholls R. D., Zori R. T., Williams C. A., Driscoll D. J. (2001) Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 38, 834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams C. A., Beaudet A. L., Clayton-Smith J., Knoll J. H., Kyllerman M., Laan L. A., Magenis R. E., Moncla A., Schinzel A. A., Summers J. A., Wagstaff J. (2006) Angelman syndrome 2005. Updated consensus for diagnostic criteria. Am. J. Med. Genet. A 140, 413–418 [DOI] [PubMed] [Google Scholar]
- 15. Clayton-Smith J., Laan L. (2003) Angelman syndrome. A review of the clinical and genetic aspects. J. Med. Genet. 40, 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang Y., Lev-Lehman E., Bressler J., Tsai T. F., Beaudet A. L. (1999) Genetics of Angelman syndrome. Am. J. Hum. Genet. 65, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cooper E. M., Hudson A. W., Amos J., Wagstaff J., Howley P. M. (2004) Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem. 279, 41208–41217 [DOI] [PubMed] [Google Scholar]
- 18. Smith S. E., Zhou Y. D., Zhang G., Jin Z., Stoppel D. C., Anderson M. P. (2011) Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3, 103ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schaaf C. P., Sabo A., Sakai Y., Crosby J., Muzny D., Hawes A., Lewis L., Akbar H., Varghese R., Boerwinkle E., Gibbs R. A., Zoghbi H. Y. (2011) Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 20, 3366–3375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glessner J. T., Wang K., Cai G., Korvatska O., Kim C. E., Wood S., Zhang H., Estes A., Brune C. W., Bradfield J. P., Imielinski M., Frackelton E. C., Reichert J., Crawford E. L., Munson J., Sleiman P. M., Chiavacci R., Annaiah K., Thomas K., Hou C., Glaberson W., Flory J., Otieno F., Garris M., Soorya L., Klei L., Piven J., Meyer K. J., Anagnostou E., Sakurai T., Game R. M., Rudd D. S., Zurawiecki D., McDougle C. J., Davis L. K., Miller J., Posey D. J., Michaels S., Kolevzon A., Silverman J. M., Bernier R., Levy S. E., Schultz R. T., Dawson G., Owley T., McMahon W. M., Wassink T. H., Sweeney J. A., Nurnberger J. I., Coon H., Sutcliffe J. S., Minshew N. J., Grant S. F., Bucan M., Cook E. H., Buxbaum J. D., Devlin B., Schellenberg G. D., Hakonarson H. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Samaco R. C., Hogart A., LaSalle J. M. (2005) Epigenetic overlap in autism-spectrum neurodevelopmental disorders. MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yashiro K., Riday T. T., Condon K. H., Roberts A. C., Bernardo D. R., Prakash R., Weinberg R. J., Ehlers M. D., Philpot B. D. (2009) Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sato M., Stryker M. P. (2010) Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U.S.A. 107, 5611–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang H. S., Burns A. J., Nonneman R. J., Baker L. K., Riddick N. V., Nikolova V. D., Riday T. T., Yashiro K., Philpot B. D., Moy S. S. (2013) Behavioral deficits in an Angelman syndrome model. Effects of genetic background and age. Behav. Brain Res. 243, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greer P. L., Hanayama R., Bloodgood B. L., Mardinly A. R., Lipton D. M., Flavell S. W., Kim T. K., Griffith E. C., Waldon Z., Maehr R., Ploegh H. L., Chowdhury S., Worley P. F., Steen J., Greenberg M. E. (2010) The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Margolis S. S., Salogiannis J., Lipton D. M., Mandel-Brehm C., Wills Z. P., Mardinly A. R., Hu L., Greer P. L., Bikoff J. B., Ho H. Y., Soskis M. J., Sahin M., Greenberg M. E. (2010) EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 143, 442–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gregianin E., Vazza G., Scaramel E., Boaretto F., Vettori A., Leonardi E., Tosatto S. C., Manara R., Pegoraro E., Mostacciuolo M. L. (2013) A novel SACS mutation results in non-ataxic spastic paraplegia and peripheral neuropathy. Eur. J. Neurol. 20, 1486–1491 [DOI] [PubMed] [Google Scholar]
- 28. Dagli A., Buiting K., Williams C. A. (2012) Molecular and clinical aspects of Angelman syndrome. Mol. Syndromol. 2, 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Philpot B. D., Thompson C. E., Franco L., Williams C. A. (2011) Angelman syndrome. Advancing the research frontier of neurodevelopmental disorders. J. Neurodev. Disord. 3, 50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kühnle S., Mothes B., Matentzoglu K., Scheffner M. (2013) Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. U.S.A. 110, 8888–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheffner M., Huibregtse J. M., Vierstra R. D., Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 [DOI] [PubMed] [Google Scholar]
- 32. Huibregtse J. M., Scheffner M., Howley P. M. (1991) A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10, 4129–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beer-Romero P., Glass S., Rolfe M. (1997) Antisense targeting of E6AP elevates p53 in HPV-infected cells but not in normal cells. Oncogene 14, 595–602 [DOI] [PubMed] [Google Scholar]
- 34. Haupt Y., Maya R., Kazaz A., Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 35. Talis A. L., Huibregtse J. M., Howley P. M. (1998) The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J. Biol. Chem. 273, 6439–6445 [DOI] [PubMed] [Google Scholar]
- 36. Lawson J. S., Glenn W. K., Heng B., Ye Y., Tran B., Lutze-Mann L., Whitaker N. J. (2009) Koilocytes indicate a role for human papilloma virus in breast cancer. Br. J. Cancer 101, 1351–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muench P., Probst S., Schuetz J., Leiprecht N., Busch M., Wesselborg S., Stubenrauch F., Iftner T. (2010) Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 70, 6913–6924 [DOI] [PubMed] [Google Scholar]
- 38. Munakata T., Liang Y., Kim S., McGivern D. R., Huibregtse J., Nomoto A., Lemon S. M. (2007) Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 3, 1335–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shirakura M., Murakami K., Ichimura T., Suzuki R., Shimoji T., Fukuda K., Abe K., Sato S., Fukasawa M., Yamakawa Y., Nishijima M., Moriishi K., Matsuura Y., Wakita T., Suzuki T., Howley P. M., Miyamura T., Shoji I. (2007) E6AP ubiquitin ligase mediates ubiquitylation and degradation of hepatitis C virus core protein. J. Virol. 81, 1174–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen J. J., Hong Y., Rustamzadeh E., Baleja J. D., Androphy E. J. (1998) Identification of an α helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 273, 13537–13544 [DOI] [PubMed] [Google Scholar]
- 41. Elston R. C., Napthine S., Doorbar J. (1998) The identification of a conserved binding motif within human papillomavirus type 16 E6 binding peptides, E6AP and E6BP. J. Gen. Virol. 79, 371–374 [DOI] [PubMed] [Google Scholar]
- 42. Be X., Hong Y., Wei J., Androphy E. J., Chen J. J., Baleja J. D. (2001) Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP. Biochemistry 40, 1293–1299 [DOI] [PubMed] [Google Scholar]
- 43. Liu Y., Cherry J. J., Dineen J. V., Androphy E. J., Baleja J. D. (2009) Determinants of stability for the E6 protein of papillomavirus type 16. J. Mol. Biol. 386, 1123–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zanier K., Charbonnier S., Baltzinger M., Nominé Y., Altschuh D., Travé G. (2005) Kinetic analysis of the interactions of human papillomavirus E6 oncoproteins with the ubiquitin ligase E6AP using surface plasmon resonance. J. Mol. Biol. 349, 401–412 [DOI] [PubMed] [Google Scholar]
- 45. Zanier K., Ruhlmann C., Melin F., Masson M., Ould M'hamed Ould Sidi A., Bernard X., Fischer B., Brino L., Ristriani T., Rybin V., Baltzinger M., Vande Pol S., Hellwig P., Schultz P., Travé G. (2010) E6 proteins from diverse papillomaviruses self-associate both in vitro and in vivo. J. Mol. Biol. 396, 90–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zanier K., Charbonnier S., Sidi A. O., McEwen A. G., Ferrario M. G., Poussin-Courmontagne P., Cura V., Brimer N., Babah K. O., Ansari T., Muller I., Stote R. H., Cavarelli J., Vande Pol S., Travé G. (2013) Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 339, 694–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nominé Y., Charbonnier S., Ristriani T., Stier G., Masson M., Cavusoglu N., Van Dorsselaer A., Weiss E., Kieffer B., Travé G. (2003) Domain substructure of HPV E6 oncoprotein. Biophysical characterization of the E6 C-terminal DNA-binding domain. Biochemistry 42, 4909–4917 [DOI] [PubMed] [Google Scholar]
- 48. Kao W. H., Beaudenon S. L., Talis A. L., Huibregtse J. M., Howley P. M. (2000) Human papillomavirus type 16 E6 induces self-ubiquitination of the E6AP ubiquitin-protein ligase. J. Virol. 74, 6408–6417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., Pavletich N. P. (1999) Structure of an E6AP-UbcH7 complex. Insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 50. Scheffner M., Huibregtse J. M., Howley P. M. (1994) Identification of a human ubiquitin-conjugating enzyme that mediates the E6-AP-dependent ubiquitination of p53. Proc. Natl. Acad. Sci. U.S.A. 91, 8797–8801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ronchi V. P., Klein J. M., Haas A. L. (2013) E6AP/UBE3A ubiquitin ligase harbors two E2∼ubiquitin binding sites. J. Biol. Chem. 288, 10349–10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heer A., Alonso L. G., de Prat-Gay G. (2011) E6*, the 50 amino acid product of the most abundant spliced transcript of the e6 oncoprotein in high-risk human papillomavirus, is a promiscuous folder and binder. Biochemistry 50, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 53. Baboshina O. V., Haas A. L. (1996) Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2epf and Rad6 are recognized by the 26S proteasome subunit 5. J. Biol. Chem. 271, 2823–2831 [DOI] [PubMed] [Google Scholar]
- 54. Haas A. L. (2005) Purification of E1 and E1-like enzymes. Methods Mol. Biol. 301, 23–35 [DOI] [PubMed] [Google Scholar]
- 55. Haas A. L., Rose I. A. (1982) The mechanism of ubiquitin activating enzyme. A kinetic and equilibrium analysis. J. Biol. Chem. 257, 10329–10337 [PubMed] [Google Scholar]
- 56. Haas A. L., Bright P. M. (1988) The resolution and characterization of putative ubiquitin carrier protein isozymes from rabbit reticulocytes. J. Biol. Chem. 263, 13258–13267 [PubMed] [Google Scholar]
- 57. Tokgöz Z., Siepmann T. J., Streich F., Jr., Kumar B., Klein J. M., Haas A. L. (2012) E1-E2 interactions in ubiquitin and Nedd8 ligation pathways. J. Biol. Chem. 287, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ronchi V. P., Haas A. L. (2012) Measuring rates of ubiquitin chain formation as a functional readout of ligase activity. Methods Mol. Biol. 832, 197–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Siepmann T. J., Bohnsack R. N., Tokgöz Z., Baboshina O. V., Haas A. L. (2003) Protein interactions within the N-end rule ubiquitin ligation pathway. J. Biol. Chem. 278, 9448–9457 [DOI] [PubMed] [Google Scholar]
- 60. van Woerden G. M., Harris K. D., Hojjati M. R., Gustin R. M., Qiu S., de Avila Freire R., Jiang Y. H., Elgersma Y., Weeber E. J. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat. Neurosci. 10, 280–282 [DOI] [PubMed] [Google Scholar]
- 61. Baboshina O. V., Crinelli R., Siepmann T. J., Haas A. L. (2001) N-end rule specificity within the ubiquitin/proteasome pathway is not an affinity effect. J. Biol. Chem. 276, 39428–39437 [DOI] [PubMed] [Google Scholar]
- 62. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 63. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 64. Maru Y., Afar D. E., Witte O. N., Shibuya M. (1996) The dimerization property of glutathione S-transferase partially reactivates Bcr-Abl lacking the oligomerization domain. J. Biol. Chem. 271, 15353–15357 [DOI] [PubMed] [Google Scholar]
- 65. Vargo M. A., Nguyen L., Colman R. F. (2004) Subunit interface residues of glutathione S-transferase A1–1 that are important in the monomer-dimer equilibrium. Biochemistry 43, 3327–3335 [DOI] [PubMed] [Google Scholar]
- 66. Huang Y. C., Misquitta S., Blond S. Y., Adams E., Colman R. F. (2008) Catalytically active monomer of glutathione S-transferase π and key residues involved in the electrostatic interaction between subunits. J. Biol. Chem. 283, 32880–32888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Levy E. D., Teichmann S. (2013) Structural, evolutionary, and assembly principles of protein oligomerization. Prog. Mol. Biol. Transl. Sci. 117, 25–51 [DOI] [PubMed] [Google Scholar]
- 68. Perica T., Chothia C., Teichmann S. A. (2012) Evolution of oligomeric state through geometric coupling of protein interfaces. Proc. Natl. Acad. Sci. U.S.A. 109, 8127–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Verdecia M. A., Joazeiro C. A., Wells N. J., Ferrer J. L., Bowman M. E., Hunter T., Noel J. P. (2003) Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol. Cell 11, 249–259 [DOI] [PubMed] [Google Scholar]
- 70. Ogunjimi A. A., Wiesner S., Briant D. J., Varelas X., Sicheri F., Forman-Kay J., Wrana J. L. (2010) The ubiquitin binding region of the Smurf HECT domain facilitates polyubiquitylation and binding of ubiquitylated substrates. J. Biol. Chem. 285, 6308–6315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pandya R. K., Partridge J. R., Love K. R., Schwartz T. U., Ploegh H. L. (2010) A structural element within the HUWE1 HECT domain modulates self-ubiquitination and substrate ubiquitination activities. J. Biol. Chem. 285, 5664–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maspero E., Mari S., Valentini E., Musacchio A., Fish A., Pasqualato S., Polo S. (2011) Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 12, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kamadurai H. B., Souphron J., Scott D. C., Duda D. M., Miller D. J., Stringer D., Piper R. C., Schulman B. A. (2009) Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol. Cell 36, 1095–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ogunjimi A. A., Briant D. J., Pece-Barbara N., Le Roy C., Di Guglielmo G. M., Kavsak P., Rasmussen R. K., Seet B. T., Sicheri F., Wrana J. L. (2005) Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol. Cell 19, 297–308 [DOI] [PubMed] [Google Scholar]
- 75. Fang P., Lev-Lehman E., Tsai T. F., Matsuura T., Benton C. S., Sutcliffe J. S., Christian S. L., Kubota T., Halley D. J., Meijers-Heijboer H., Langlois S., Graham J. M., Jr., Beuten J., Willems P. J., Ledbetter D. H., Beaudet A. L. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet. 8, 129–135 [DOI] [PubMed] [Google Scholar]
- 76. Chan A. L., Grossman T., Zuckerman V., Campigli Di Giammartino D., Moshel O., Scheffner M., Monahan B., Pilling P., Jiang Y. H., Haupt S., Schueler-Furman O., Haupt Y. (2013) C-Abl phosphorylates E6AP and regulates its E3 ubiquitin ligase activity. Biochemistry 52, 3119–3129 [DOI] [PubMed] [Google Scholar]
- 77. Scheffner M., Werness B. A., Huibregtse J. M., Levine A. J., Howley P. M. (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63, 1129–1136 [DOI] [PubMed] [Google Scholar]
- 78. Huibregtse J. M., Scheffner M., Howley P. M. (1993) Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell Biol. 13, 4918–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lagrange M., Charbonnier S., Orfanoudakis G., Robinson P., Zanier K., Masson M., Lutz Y., Trave G., Weiss E., Deryckere F. (2005) Binding of human papillomavirus 16 E6 to p53 and E6AP is impaired by monoclonal antibodies directed against the second zinc-binding domain of E6. J. Gen. Virol. 86, 1001–1007 [DOI] [PubMed] [Google Scholar]
- 80. Medcalf E. A., Milner J. (1993) Targeting and degradation of p53 by E6 of human papillomavirus type 16 is preferential for the 1620+ p53 conformation. Oncogene 8, 2847–2851 [PubMed] [Google Scholar]
- 81. Thomas M., Tomaić V., Pim D., Myers M. P., Tommasino M., Banks L. (2013) Interactions between E6AP and E6 proteins from α and β HPV types. Virology 435, 357–362 [DOI] [PubMed] [Google Scholar]
- 82. Dai B., Pieper R. O., Li D., Wei P., Liu M., Woo S. Y., Aldape K. D., Sawaya R., Xie K., Huang S. (2010) FoxM1B regulates NEDD4-1 expression, leading to cellular transformation and full malignant phenotype in immortalized human astrocytes. Cancer Res. 70, 2951–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Marianayagam N. J., Sunde M., Matthews J. M. (2004) The power of two. Protein dimerization in biology. Trends Biochem. Sci. 29, 618–625 [DOI] [PubMed] [Google Scholar]
- 84. Spitkovsky D., Aengeneyndt F., Braspenning J., von Knebel Doeberitz M. (1996) p53-independent growth regulation of cervical cancer cells by the papillomavirus E6 oncogene. Oncogene 13, 1027–1035 [PubMed] [Google Scholar]
- 85. Streich F. C., Jr., Ronchi V. P., Connick J. P., Haas A. L. (2013) TRIM ligases catalyze polyubiquitin chain formation through a cooperative allosteric mechanism. J. Biol. Chem. 288, 8209–8221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kiyono T., Foster S. A., Koop J. I., McDougall J. K., Galloway D. A., Klingelhutz A. J. (1998) Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396, 84–88 [DOI] [PubMed] [Google Scholar]
- 87. Kentsis A., Gordon R. E., Borden K. L. (2002) Control of biochemical reactions through supramolecular RING domain self-assembly. Proc. Natl. Acad. Sci. U.S.A. 99, 15404–15409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun L., Deng L., Ea C. K., Xia Z. P., Chen Z. J. (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 [DOI] [PubMed] [Google Scholar]
- 89. Wang X., Herr R. A., Chua W. J., Lybarger L., Wiertz E. J., Hansen T. H. (2007) Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Page M. I., Jencks W. P. (1971) Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. U.S.A. 68, 1678–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]