

Background: Small heterodimer partner interacting leucine zipper protein (SMILE) is a nuclear corepressor of the nuclear receptor family.
Results: Ursodeoxycholic acid (UDCA) increases SMILE gene expression, which contributes to inhibiting liver X receptor α (LXRα)-mediated hepatic lipogenesis.
Conclusion: UDCA-induced SMILE inhibits LXRα-mediated hepatic lipogenic gene expression.
Significance: SMILE improves hepatic lipid metabolism.
Keywords: AMP-activated Kinase (AMPK), Bile Acid, Lipogenesis, Nuclear Receptors, Transcription Repressor, Corepressor, Liver X Receptor (LXR), Small Heterodimer Partner Interacting Leucine Zipper Protein (SMILE), Sterol Regulatory Element-binding Protein-1 (SREBP-1c)
Abstract
Small heterodimer partner interacting leucine zipper protein (SMILE) has been identified as a nuclear corepressor of the nuclear receptor (NRs) family. Here, we examined the role of SMILE in the regulation of nuclear receptor liver X receptor (LXRα)-mediated sterol regulatory element binding protein-1c (SREBP-1c) gene expression. We found that SMILE inhibited T0901317 (T7)-induced transcriptional activity of LXRα, which functions as a major regulator of lipid metabolism by inducing SREBP-1c, fatty acid synthase (FAS), and acetyl-CoA carboxylase (ACC) gene expression. Moreover, we demonstrated that SMILE physically interacts with LXRα and represses T7-induced LXRα transcriptional activity by competing with coactivator SRC-1. Adenoviral overexpression of SMILE (Ad-SMILE) attenuated fat accumulation and lipogenic gene induction in the liver of T7 administered or of high fat diet (HFD)-fed mice. Furthermore, we investigated the mechanism by which ursodeoxycholic acid (UDCA) inhibits LXRα-induced lipogenic gene expression. Interestingly, UDCA treatment significantly increased SMILE promoter activity and gene expression in an adenosine monophosphate-activated kinase-dependent manner. Furthermore, UDCA treatment repressed T7-induced SREBP-1c, FAS, and ACC protein levels, whereas knockdown of endogenous SMILE gene expression by adenovirus SMILE shRNA (Ad-shSMILE) significantly reversed UDCA-mediated repression of SREBP-1c, FAS, and ACC protein levels. Collectively, these results demonstrate that UDCA activates SMILE gene expression through adenosine monophosphate-activated kinase phosphorylation, which leads to repression of LXRα-mediated hepatic lipogenic enzyme gene expression.
Introduction
Liver X receptor α (LXRα)3 is a member of the nuclear receptor superfamily and heterodimerizes with retinoid X receptor (RXR). LXRα normally binds to the DR-4 motif known as the LXR response element (LXRE) in their target genes and is activated by specific cholesterol metabolites such as oxysterols or synthetic nonsteroidal LXR ligands (T0901317) (1–3). Two isoforms of LXR exist with different expression patterns. LXRβ is expressed ubiquitously, whereas LXRα is mainly expressed in liver, adipose tissue, and macrophages known to play an important role in lipid metabolism (1, 4, 5). LXRα has emerged as an important regulator of gene expression involved in lipid and cholesterol metabolism (6, 7). LXRs play an important role in fatty acid synthesis by directly or indirectly controlling lipogenic gene expression. LXRα-mediated fatty acids synthesis is controlled by sterol regulatory element-binding protein-1c (SREBP-1c), which regulates gene expression involved in the lipogenic pathway, and acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) in hepatocytes (8). Consistently, LXRα-deficient mice display markedly reduced hepatic fatty acid synthesis and triglycerides (9). LXRα activity is regulated by agonists, antagonists, SIRT1, NCoR, and SMRT (10, 11). We previously demonstrated that orphan nuclear receptor DAX-1 inhibits LXRα transcriptional activity and improves hepatic lipogenesis (12).
Small heterodimer partner interacting leucine zipper protein (SMILE) belongs to the basic leucine zipper family transcription factor. In contrast to other basic leucine zipper proteins, SMILE cannot bind to DNA as a homodimer, unlike other basic leucine zipper proteins (13). SMILE has also been proposed as a coactivator of activating transcription factor 4 (14). Moreover, we have revealed that SMILE functions as a nuclear co-repressor of the estrogen receptor (ER), glucocorticoid receptor (GR), constitutive androstane receptor (CAR), hepatocyte nuclear factor 4α, and estrogen receptor-related receptor γ (ERR γ) (15–17). Moreover, we reported that curcumin induces SMILE gene expression through a liver kinase B1/adenosine monophosphate-activated kinase (AMPK) pathway and represses endoplasmic reticulum (ER) stress-responsive gene transcription (18). A recent study demonstrated that SMILE activates tumor suppressor p53 and inhibits the function of BMP-6 by interacting with Smads (19, 20). However, the detailed mechanism by which SMILE regulates expression of genes involved in hepatic lipid metabolism remains unknown.
Bile acids affect triglyceride (TG) homeostasis and have recently emerged as a key metabolic regulator of glucose and lipid metabolism (21). Ursodeoxycholic acid (UDCA) is a bile acid and is used to treat several liver diseases such as primary biliary cirrhosis and hepatitis C (22, 23). UDCA induces hepatobiliary secretion and decreases retention of toxic hydrophobic bile acids, thereby rendering bile more hydrophilic and cytotoxic (23). Moreover, UDCA has beneficial effects on cholestatic disorders due to its anti-apoptotic, anti-fibrotic, and cytoprotective effects (24). UDCA treatment increases glutathione synthesis by activating the PI3K/Akt/Nrf2 pathway (25) and have beneficial effects on hepatic steatosis and insulin resistance (26, 27). Other bile acids such as cholic acid (CA) and chenodeoxycholic acid (CDCA) prevent hepatic TG accumulation (28). However, the molecular mechanism of UDCA in hepatic lipid metabolism remains largely unknown.
In the present study, we show that SMILE negatively regulates LXRα transcriptional activity by directly interacting with LXRα and competes with the LXRα coactivator SRC-1. Moreover, we observed that SMILE overexpression inhibited LXRα-mediated Srebp-1c gene expression and decreased LXRα agonist-induced hepatic TG level and lipid accumulation. Moreover, UDCA treatment attenuated LXRα-mediated lipogenic gene expression, whereas SMILE knockdown released UDCA-mediated gene expression of Srebp-1c, Fas, and Acc. Overall, our results suggest that SMILE acts as a novel corepressor of LXRα and UDCA-induced SMILE gene expression, which leads to inhibition of LXRα-mediated hepatic lipogenic gene expression.
EXPERIMENTAL PROCEDURES
Animals and Treatment
C57BL/6J male mice were obtained from Korea Research Institute of Bioscience and Biotechnology (Daejeon, South Korea). Age matched mice (weight, 25–28 g) were maintained on a standard diet under a 12-h light/dark cycle at 22 ± 2 °C for 2 weeks with free access to food and water in a pathogen-free facility. UDCA was administered daily orally at a dose of 2 mg, after which the mice had free access to food and water. All animal experiments were approved by the Institutional Animal Care and Use Committee of Chonnam National University.
Materials and Plasmids
The synthetic LXR agonist TO901317 (T7) was purchased from Cayman Chemicals (Ann Arbor, MI). Bile acids (UDCA and CDCA) were purchased from Sigma. The AMPK inhibitor, compound C, was purchased from Calbiochem (San Diego, CA). The reporter plasmids LXRE-Luc, SREBP-1c-Luc, and SMILE-Luc were described previously (12, 17). pCMV-β-gal, pcDNA3-HA-LXRα, pcDNA3-HA-SRC-1, pcDNA3-ERRγ, pcDNA3-FLAG-SMILE, pGEX4T-1, pGEX4T-1-SMILE, pEBG-empty, pEBG-SMILE, pSUPER-si-empty, and pSUPER-si-SMILE, FLAG-SMILE LXXLL mutants (m1, m2, m3, m4, and m5) were described previously (12, 15, 17). pEBG-LXRα WT, pEBG-LXRα C, pEBG-LXRα DE, and pEBG-LXRα AB were subcloned into the BamHI/KpnI sites of the pEBG vector.
Cell Culture and Transient Transfection Assay
HepG2 and 293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Cambrex Bio Science, Walkersville, MD) and antibiotics (Invitrogen). The cells were split in 24-well plates at densities of 2–8 × 104 cells/well the day before transfection. Transient transfections were performed using the Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Total DNA used in each transfection was adjusted to 1 μg/well by adding the appropriate amount of empty vector, and CMV-β-galactosidase plasmids were cotransfected as an internal control. Cells were harvested 40–48 h after the transfection for luciferase and β-galactosidase assays. Luciferase activity was normalized to β-galactosidase activity.
Preparation of Recombinant Adenovirus
The recombinant adenovirus encoding human SMILE has been described previously (15). The shSMILE (AAGGCGTCGTCGTCTCTTAAA) constructs were constructed with a 21-mer double-stranded oligonucleotide containing +1053 to 1074 of the SMILE cDNA sequence into the pBS/U6 vector. The cDNA encoding shSMILE was cloned into the pAdTrack-CMV vector. pAdTrack-CMV-shSMILE was recombined with adenoviral gene carrier vector by transformation in pretransformed adEasy-BJ21-competent cells. The shAMPKα2 (ATCATCTTATCATTGACAATCGGAGAA) were constructed using double-stranded oligonucleotides containing +1071 to 1094 of the AMPKα2 cDNA sequence into the pBS/U6 vector and then adenoviral vector systems were used as previously described (12).
Isolation and Culture of Primary Mouse Hepatocytes
Mouse primary hepatocytes were isolated from the livers of 7-week-old male C57BL6 mice. The mice were anesthetized with Zoletile and their livers were exposed surgically. The liver was first perfused with resuspension buffer and then perfused with collagenase solution. Subsequently, the liver was finely chopped in a Petri dish and then filtered through 85-μm pore mesh. Hepatocytes were collected by centrifugation at 800 × g for 2–5 min at 4 °C. Hepatocyte viability was assessed by trypan blue exclusion assay and was consistently in excess of 85%. Hepatocytes were then seeded onto collagen type 1-coated 60-mm dishes as described previously (12).
Triglyceride and Cholesterol Measurements
Once the animals were sacrificed, blood was immediately collected, and serum levels of TG and cholesterol were measured using chemistry analyzer kits (Hitachi 7150; Tokyo, Japan).
Histomorphological Analysis
Liver tissues were processed in paraffin after fixation in 10% neutral-buffered formalin. Tissue sections (3–4 μm thick) were deparaffininzed, rehydrated, and stained with hematoxylin and eosin (H&E) for histological examination. The histological evaluation was carried out using a BX51 light microscope (Olympus, Tokyo, Japan). Histological damage in H&E sections was examined under a microscope at ×200 magnification.
Oil Red-O Staining
Fresh liver tissues were embedded carefully in OCT in a plastic mold after freezing at −80 °C. Tissue sections (8–10 μm thick) were incubated for 30 min at room temperature on a slide. The tissues were fixed with 10% formalin for 1 h, and then washed 60% isopropyl alcohol. Then 0.6% Oil Red-O working solution (w/v, 60% isopropyl alcohol and 40% water) was added to each slide for 30 min at room temperature, the solution was removed, and deionized water was added to wash the tissue. Fat droplets in the liver were stained red.
RNA Interference
SMILE knockdown was performed using the pSuper vector system (15). HepG2 cells were transfected with siRNA constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The siRNA-transfected cells were subjected to the second transfection as indicated in the figure legends.
GST Pulldown Assay
Briefly, LXRα was labeled with [35S]methionine using the TnT-coupled reticulocyte lysate system (Promega Corp., Madison, WI) according to the manufacturer's instructions. GST alone and GST-fused SMILE (GST-SMILE) proteins were prepared for in vitro GST pulldown assays as previously described (17). Briefly, 293T cells were transfected with 1 μg of each of the indicated plasmids using the Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instruction. The cells were collected 24 h after transfection and solubilized. The in vivo GST pulldown was performed as described previously (17).
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed as described previously (17). In brief, HepG2 cells in 60-mm culture dishes were fixed with 1% formaldehyde, washed with ice-cold phosphate-buffered saline, harvested, and solicited. The soluble chromatin was then subjected to immunoprecipitation using anti-LXRα, anti-SMILE (sc-49329, Santa Cruz Biotechnology, Santa Cruz, CA), anti-glucocorticoid receptor (GR)-interacting protein 1, anti-SRC-1, or anti-HA antibodies followed by treatment with Protein A-agarose/salmon sperm DNA (Upstate Biotechnology, Upstate, NY). Unrelated IgG was used a negative control for immunoprecipitation. Precipitated DNA was recovered via phenol/chloroform extraction, and DNA samples were quantified by quantitative real time-polymerase chain reaction (PCR) using two pairs of primers encompassing the proximal (−300/−10 bp) or distal (−1800/−1500 bp) region of the mouse Srebp-1c promoter. The primers used for PCR were as follows: proximal, forward, 5′-TGGTTGCCTGTGCGGCAG-3′ and reverse, 5′-TCAGGCCCCGCCAGGCTTTAA-3′; distal, forward, 5′-GCTGGATGTCCAGGCTGAG-3′ and reverse, 5′-CCAGAGGTATGCAAGCAGA-3′.
Quantitative RT-PCR
Total RNA was extracted from either tissue samples or rat primary hepatocytes under various conditions using TRIzol reagent (Invitrogen), according to the manufacturer's protocol. SMILE, Srebp-1c, Fas, and Acc gene expression was analyzed by quantitative RT-PCR as described previously (17). The primers used for human/rat SMILE, SREBP-1c, FAS, ACC1α, and β-actin PCR were as follows: human/mouse SMILE, forward, 5′-AAAAGAGGCGGAGAAAGTCC-3′ and reverse, 5′-CTCTGAAGAGCGAGGTGGTC-3′; SREBP-1c, forward, 5′-TGAGAAGCGCTACCGGGCTGCTATCAATGACAAGATTGT-3′ and reverse, 5′-CTCCACTGCCACAAGCTGCCACCAGGTCCTTCAGTG-3′; FAS, forward, 5′-GCTGCGGAAACTTCAGGAAAT-3′ and reverse, 5′-AGAGACGTGTCACTCCTGGACT-3′; ACC1α, forward, 5′-GCGGGAGGAGTTCCTAATTC-3′ and reverse, 5′-TGTCCCAGACGTAAGCCTTC-3′; and β-actin, forward, 5′-GTCATCACCATTGGCAATGAG-3′ and reverse, 5′-CGTCATACTCCTGCTTGCTG-3′.
Western Blot Analysis and Co-immunoprecipitation (Co-IP)
Western blot and co-IP analyses were performed as described previously (17). Briefly, HepG2 cells were transfected with the indicated expression vectors or small interfering RNA oligonucleotides and treated with T7. Cell lysates were prepared 48 h after transfection and separated on 10% SDS-PAGE gels. The proteins were transferred to a nitrocellulose membrane (Amersham Biosciences), and the membranes were probed with anti-HA, FLAG, SMILE, LXRα, SREBP1, FAS, ACC, or β-actin antibodies and developed after incubation with a secondary antibody using an enhanced chemiluminescence kit (Amersham Biosciences) according to the manufacturer's instruction.
Statistical Analysis
Data are expressed as mean ± S.E. The statistical analysis was performed using the two-tailed Student's t test or one-way analysis of variance and results were considered to be statistically significant at p < 0.05 or p < 0.001.
RESULTS
SMILE Decreases LXRα-mediated Lipogenic Enzyme Gene Expression
To investigate the role of SMILE in T7-induced lipogenic gene expression, we infected adenovirus expressing SMILE (Ad-SMILE) in T7-treated mouse primary hepatocytes and HepG2 cells. We found that adenoviral overexpression of SMILE significantly decreased T7-induced protein levels of LXRα target genes Srebp-1c, Fas, and Acc (Fig. 1, A and B). In addition, adenoviral overexpression of SMILE significantly decreased mRNA levels of SREBP-1c, FAS, and ACC in both primary mouse hepatocytes and HepG2 cells (Fig. 1, C and D). Taken together, these results indicate that SMILE inhibits LXRα-mediated lipogenic enzyme gene expression.
FIGURE 1.

SMILE decreases T7-mediated lipogenic gene expression. A and B, mouse primary hepatocytes and HepG2 cells were infected with Ad-SMILE or Ad-GFP and then exposed to T7. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. Protein levels were normalized to α-tubulin level. Each data point represents mean ± S.E. C and D, quantitative PCR analysis was performed using total RNA from Ad-SMILE or Ad-GFP-infected mouse primary hepatocytes or HepG2 cells after T7 treatment. The Srebp-1c, Fas, Acc, SMILE, and β-actin genes were amplified using specific primers for Srebp-1c, Fas, Acc, SMILE, and β-actin. mRNA levels were normalized to β-actin expression. Results are representative of three independently performed experiments.
SMILE Competes with SRC-1 to Decrease LXRα Transcriptional Activity
To confirm whether SMILE inhibits LXRα transcriptional activity, 293T and HepG2 cells were co-transfected with SMILE, the LXRα expression vector, and the LXRα-specific reporter in the presence or absence of the LXRα agonist T7. Overexpression of SMILE strongly inhibited the activity of the LXRα-mediated reporter containing the LXR binding site (LXRE-luc) (Fig. 2, A and B). It has been reported that SRC-1 acts as a co-activator of LXRα to activate LXRα transcriptional activity (29). To define the functional mechanism of LXRα repression by SMILE, we employed competition experiments using transient transfection of HepG2 cells. We observed that SMILE-mediated repression of LXRE-luc and Srebp-1c promoter activity were significantly released by SRC-1 co-transfection (Fig. 2, C and D). These results suggest that SMILE competes with SRC-1 to decrease LXRα-mediated transcriptional activity. Next, we performed co-IP assays using LXRα, SRC-1, and FLAG-SMILE antibodies to confirm whether SMILE inhibits the protein interaction between LXRα and SRC-1. Overexpression of SMILE decreased the endogenous interaction of SRC-1 with LXRα. In contrast, overexpressed SMILE strongly interacted with LXRα (Fig. 2E). Moreover, we performed ChIP assay to examine whether SMILE also inhibited recruitment of LXRα on the Srebp-1c promoter. We found that SMILE abolished recruitment of LXRα on the Srebp-1c promoter (Fig. 2F). On the basis of inhibition of LXRα DNA binding by SMILE, we hypothesized that the effect of SMILE on LXRα DNA binding was mediated by alterations in the interaction between LXRα and RXR. To investigate this possibility, we performed co-IP assays to test if SMILE affects LXRα/RXRα dimerization. The effect of SMILE on heterodimerization of LXRα/RXRα was determined by co-immunoprecipitation in vitro. The interaction between LXRα and RXRα was suppressed by SMILE expression in a dose-dependent manner (supplemental Fig. S1). These results suggest that SMILE impairs the interaction between LXRα and RXRα, accounting for SMILE-mediated inhibition of LXRα DNA binding, at least in part. Taken together, these results indicate that SMILE physically competes with SRC-1 to repress LXRα transcriptional activity and leads to the blocking of LXRα DNA binding on the target gene promoter.
FIGURE 2.

SMILE competes with SRC-1 to modulate LXRα transcriptional activity. A and B, 293T or HepG2 cells were co-transfected with 200 ng of LXRE-luc reporter with 200 ng of pcDNA3-Flag-SMILE or pcDNA3 empty vector, respectively, and 24 h later treated with vehicle (dimethyl sulfoxide, DMSO) or 10 μm T7 for 12 h. The cells were harvested and lysates were utilized for the luciferase and β-galactosidase assay. Western blot (WB) analysis shows overexpression of LXRα and SMILE (B, bottom). C and D, HepG2 cells were co-transfected with 200 ng of LXRE-luc reporter (C), Srepb-1c-luc (D), pcDNA3-HA-LXRα, pcDNA3-HA-SRC-1, pcDNA3-Flag-SMILE, or pcDNA3 empty vector, respectively, for 24 h and then treated with vehicle (DMSO) or 10 μm T7 for 12 h. The cells were harvested, and lysates were utilized for luciferase and β-galactosidase assays. Western blot analysis shows overexpression of LXRα, SRC-1, and SMILE (C, bottom). E and F, HepG2 cells were co-transfected with pcDNA3-Flag-SMILE, and the cells were treated with vehicle (DMSO) or T7 (10 μm). After 6 h, protein extracts were co-immunoprecipitated using LXRα or IgG antibody and Western blotted with the indicated antibody (E). Chromatin immunoprecipitation analysis was performed using LXRα antibody. PCR amplification of immunoprecipitated (IP) chromatin fragments was conducted using primer pairs specific for proximal, regulatory region, and a distal, nonregulatory region of the Srebp-1c gene promoter (F).
SMILE Directly Interacts with LXRα
To determine whether the repression of LXRα transcriptional activity by SMILE is mediated through a direct physical interaction, we performed co-IP assays using LXRα- and SMILE-specific antibodies. Co-IP assays demonstrated that SMILE directly interacted with LXRα in HepG2 cells (Fig. 3A). Next, to confirm whether the interaction between the two proteins is direct or not, we performed in vitro GST pulldown assays. GST alone and GST-fused SMILE were bacterially expressed and incubated with in vitro translated 35S-labeled LXRα protein. We found that in vitro translated 35S-labeled LXRα protein directly interacted with bacterially expressed SMILE, but not with GST alone (Fig. 3B). To further confirm the interaction between SMILE and LXRα in vivo, we performed in vivo GST pulldown assays upon transfecting cells with GST-alone (pEBG) or GST-SMILE (pEBG-SMILE) with HA-LXRα. After GST purification, HA-LXRα was detected in the coprecipitates only when co-expressed with GST-SMILE but not with GST alone in the presence or absence of the LXRα agonist T7 (Fig. 3C). Previous reports demonstrated that DAX-1 directly interacts with the ligand binding domain of LXRα to compete with SRC-1 (12). To identify the LXRα interacting domain with SMILE, we performed in vivo GST pulldown experiments using a series of GST-tagged LXRα mutants (Fig. 3D). We found that SMILE interacted with the LXRα C-terminal, which contains the ligand binding domain and the activation function-2 domain (Fig. 3E). In addition, all LXRα and SMILE GST fusion proteins used in the assays were expressed at comparable levels (Fig. 3E, middle and lower panels). Because the LXXLL motif plays an important role in the regulation of nuclear receptor (30), transfection analyses were carried out using LXXLL mutant constructs to examine whether SMILE-dependent inhibition of LXRα activity is mediated by the LXXLL motif of SMILE. The transient transfection study showed that third (m3) or fourth (m4) single LXXLL motif mutants of SMILE failed to inhibit LXRα transactivation to the level comparable with wild type SMILE. Therefore, these results suggest that the LXXLL motifs are essential for SMILE to inhibit LXRα activity (Fig. 3F). Taken together, these results demonstrate that SMILE directly interacts with LXRα, and the LXXLL motif of SMILE is required to inhibit LXRα activity.
FIGURE 3.

Interaction between SMILE and LXRα. A, endogenous interaction between LXRα and SMILE. Protein extracts from HepG2 cells were subject to co-immunoprecipitated (IP) with LXRα or SMILE and the interaction between LXRα and SMILE was determined by Western blotting using the SMILE antibody (left panel) or LXRα antibody (right panel). LXRα and SMILE expression (lower two panels) from the 10% lysate was analyzed by Western blotting (WB) with the indicated antibodies. B, in vitro GST pulldown assay. 35S radiolabeled LXRα protein was incubated with GST-only or GST-SMILE fusion proteins. The input lane represents 10% of total volume of in vitro translated proteins used for binding assay. Protein interactions were detected via autoradiography. C, HepG2 cells were co-transfected with expression vectors for HA-LXRα with pEBG-SMILE (GST-SMILE) or pEBG alone (GST-only) and then treated with dimethyl sulfoxide or T7. Complex formation (upper two panels, GST purification) and the amount of HA-LXRα (lower panel, lysate) used for the in vivo binding assay determined the interaction with the anti-HA antibody. The same blot was stripped and re-probed with an anti-GST antibody (middle panel) to confirm expression levels of the GST fusion protein (GST-SMILE) and the GST control (GST). D, schematic representation of the structures of the LXRα deletion mutants. AB, N-terminal domain; C, DNA binding domain; DE, hinge and ligand binding domain; AF2, activation function-2 domain; Δ, deletion region. E, HepG2 cells were co-transfected with expression vectors for HA-SMILE and indicated pEBG-LXRα (GST-LXRα). The interaction was determined via Western blot using anti-HA. The same blot was stripped and reprobed with anti-GST antibody to confirm the expression levels of the GST fusion protein (GST-LXRα mutants) and the GST control (GST-only). F, effects of SMILE LXXLL mutants on LXRα-mediated transcriptional activity. HepG2 cells were cotransfected with reporter vector LXRE-luc, together with indicated expression vector for LXRα, wild-type (WT) FLAG-SMILE or FLAG-SMILE LXXLL mutants (m1, m2, m3, m4, and m5). Luciferase activity was measured 48 h after transfection.
UDCA Induces SMILE Gene Expression
A previous report demonstrated that the bile acid CDCA inhibits T7-induced gene expression of ACCα and other lipogenic enzyme (31) and also reduces hypertriglyceridemia (28). Another natural bile acid, UDCA, also decreases TG level and improves cholestasis (24, 32). To test if UDCA improved hepatic lipid metabolism is caused by SMILE, we assessed levels of SMILE mRNA and protein following UDC treatment. UDCA treatment increased SMILE protein levels in a time-dependent manner both in mouse primary hepatocytes and HepG2 cells (Fig. 4, A and B). UDCA was administered to mice to further determine the effects of UDCA on SMILE gene expression in vivo. UDCA treatment (80 mg/kg/day) for 1 week significantly increased SMILE protein levels (Fig. 4C). In addition, we also observed that UDCA strongly increased the SMILE protein level in a dose-dependent manner (Fig. 4D). We next asked whether UDCA administration elicits hepatotoxicity. H&E staining revealed similar liver morphology between normal and UDCA-treated mice. Moreover, UDCA treatment did not alter AST (aspartate aminotransferase) and ALT (alanine aminotransferase) levels compared with control (supplemental Fig. S2, A–F). Previous reports demonstrated that CDCA suppress hepatic lipogenesis by inducing SHP gene expression (31). Next, we compared the effects of different bile acids on SMILE expression in HepG2 cells. Treatment with UDCA time dependently increased the SMILE mRNA level but not SHP mRNA (Fig. 4E). In contrast, CDCA significantly increased the SHP mRNA level. However, no effect on SMILE gene expression was observed following CDCA treatment (Fig. 4F). Taken together, these results show that UDCA has a positive role in SMILE gene expression and suggest that UDCA-mediated inhibition of LXRα-mediated hepatic lipogenesis may be elicited through the induction of SMILE gene expression.
FIGURE 4.

UDCA induces SMILE gene expression in mice. A and B, mouse primary hepatocytes and HepG2 cells were treated with UDCA (200 μm) for various periods and then the cells were harvested for Western blot analysis using SMILE antibody. Protein levels were normalized to those of α-tubulin. C, 80 mg/kg of UDCA was administered daily to mice for 1 week. Then, the mice were sacrificed and the isolated liver was processed for Western blot analysis with SMILE antibody. D, the indicated dose of UDCA (0–200 mg/kg) was administered to mice for 4 days. Mice were sacrificed and Western blot analysis was performed using SMILE antibody from the mouse liver tissue. Each data point represents mean ± S.E.
UDCA-induced SMILE Gene Expression Depends on AMPK Signaling
We have reported that AMPK increases SMILE gene expression (18), which prompted us to evaluate the effect of UDCA on AMPK signaling. First, we analyzed AMPK phosphorylation and subsequent changes in SMILE gene expression in primary mouse primary hepatocytes following UDCA treatment. Consistent with our hypothesis, UDCA time dependently increased AMPK phosphorylation and SMILE gene expression in mouse primary hepatocytes (Fig. 5A). Next, to investigate whether AMPK signaling is involved in UDCA-induced SMILE gene expression, we employed compound C, AMPK inhibitor, and adenovirus expressing short hairpin RNA for AMPKα2 (Ad-shAMPKα2). UDCA-induced SMILE gene expression was significantly attenuated by compound C treatment, which is consistent with abolished phosphorylation of AMPK (Fig. 5B). Consistent with the effect of compound C, knockdown of AMPKα2, a liver-enriched isoform of AMPKα, significantly decreased induction of SMILE protein and mRNA level by UDCA treatment (Fig. 5, C and D). Moreover, we found that UDCA also activated SMILE promoter activity, whereas compound C treatment dose dependently inhibited UDCA-induced SMILE promoter activity (Fig. 5E). Consistent with the compound C effect, dominant-negative AMPK (dn-AMPK) overexpression strongly inhibited UDCA-induced SMILE promoter activity (Fig. 5F). To further investigate the role of AMPK, we tested if AMPK has any direct role on the stability of SMILE proteins. We overexpressed FLAG-tagged SMILE and infected AMPK-CA (constitutively active form) or AMPK-DN (dominant-negative form) virus to confirm that AMPK affects SMILE protein stabilization. However, overexpressed SMILE protein level was not significantly changed by adenoviral overexpression of AMPK, indicating that AMPK plays a role on SMILE expression at transcriptional level (supplemental Fig. S3). These results suggest that the AMPK pathway is mainly involved in the UDCA-induced expression of SMILE at the transcriptional level.
FIGURE 5.

UDCA-induced AMPK signaling elicits SMILE gene expression. A, mouse primary hepatocytes were cultured for 12 h under serum starvation. The cells were treated with 200 μm UDCA for various time periods. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. Protein levels were normalized to those of α-tubulin. B, mouse primary hepatocytes were co-treated for 12 h with UDCA (200 μm), compound C (AMPK inhibitor), or dimethyl sulfoxide, and then the cells were harvested for Western blot analysis. C and D, mouse primary hepatocytes cells were infected with adenovirus US (Ad-US) or adenovirus sh-AMPKα2 (Ad-shAMPKα2), and the cells treated with vehicle (dimethyl sulfoxide) and UDCA (200 μm). After 12 h, the cells were harvested for Western blot analysis and quantitative RT-PCR. Data represent mean ± S.D. of three individual experiments. E, HepG2 cells were transfected with 200 ng of SMILE-luc reporter vector. After 24 h, they were treated with 200 μm UDCA for 12 h with the indicated amounts of compound C (com.C). F, HepG2 cells were co-transfected with 200 ng of the SMILE-luc reporter vector and the dominant-negative AMPK expression vector. After 24 h, the cells were treated with vehicle (DMSO) or 200 μm UDCA. The cells were harvested and lysates were utilized for luciferase and β-galactosidase assays. Data in A and B are represented as mean ± S.E.
UDCA Decreases LXRα-mediated Lipogenic Gene Expression by Inducing SMILE
To test if UDCA also inhibits lipogenic gene expression and whether the ability of UDCA to inhibit lipogenic gene expression is associated with SMILE, we performed an adenovirus-mediated knockdown experiment with adenovirus expressing short hairpin RNA for SMILE (Ad-shSMILE). UDCA strongly inhibited the T7-mediated increase in SREBP-1c, FAS, and ACC protein levels, which were significantly reversed by knockdown of endogenous SMILE expression in mouse primary hepatocytes and HepG2 cells (Fig. 6, A and B). However, knockdown of basal SMILE expression did not display significant changes in lipogenic gene expression. Therefore, uninduced levels of SMILE may not be involved in the regulation of the basal hepatic lipogenic gene expression (supplemental Fig. S4). Taken together, these results indicate that SMILE is involved in UDCA-mediated repression of lipogenic gene expression. Next, we performed knockdown of endogenous SMILE using a SMILE small interfering RNA expressing plasmid to investigate whether UDCA inhibits LXRα target gene promoter activity by inducing SMILE. As expected, knockdown of endogenous SMILE significantly abolished UDCA-mediated inhibition of T7-induced Srebp-1c promoter activity (Fig. 6C). Furthermore, we performed ChIP assay experiments to define the molecular mechanism of UDCA-induced SMILE on the regulation of LXRα. UDCA treatment significantly decreased recruitment of LXRα to the Srebp-1c promoter. However, SMILE knockdown significantly released UDCA-mediated suppression of LXRα recruitment at the Srebp-1c promoter (Fig. 6D). Overall, these results suggest that UDCA inhibits LXRα-mediated lipogenic gene expression by inducing SMILE gene expression, which decreases LXRα recruitment on the target gene promoter.
FIGURE 6.

UDCA decreases LXRα target gene expression via induction of SMILE. A and B, mouse primary hepatocytes and HepG2 cells were infected with adenovirus US (Ad-US) or adenovirus sh-SMILE (Ad-shSMILE), and the cells treated with vehicle (dimethyl sulfoxide), T7 (10 μm), and UDCA (200 μm). After 12 h, the cells were harvested for Western blot analysis using the indicated antibodies. Protein levels were normalized to those of α-tubulin. Data represent mean ± S.D. of three individual experiments. C, HepG2 cells were co-transfected with pSUPER-empty or pSUPER-siSMILE together with the LXRα expression vector and the Srebp-1c-luc promoter vectors. After 24 h, the cells were treated with vehicle (dimethyl sulfoxide), 10 μm T7, and 200 μm UDCA for 12 h, and luciferase activity was quantified. D, HepG2 cells were co-transfected with pSUPER-empty and pSUPER-siSMILE. After 24 h, the cells treated with vehicle (dimethyl sulfoxide), 10 μm T7, and 200 μm UDCA for 6 h. Chromatin immunoprecipitation analysis was performed using LXRα antibody. PCR amplification of immunoprecipitated chromatin fragments was conducted using primer pairs specific for the proximal, regulatory, and a distal, nonregulatory region of the Srebp-1c gene promoter.
SMILE Improves T7-induced Hepatic TG Level and Lipid Accumulation in Mice
To assess the functional effects of SMILE on T7-induced hepatic lipogenesis, we analyzed hepatic fat accumulation and lipogenic gene expression in normal and T7-treated mice. A 1-week T7 treatment caused severe fat accumulation in the liver of mice, as indicated by increases in the intensity of Oil Red-O staining and hepatic TG levels. As expected, adenovirus injection of SMILE significantly improved hepatic lipid accumulation (Fig. 7A) and TG levels (Fig. 7B) compared with those in adenovirus GFP-injected mice. However, hepatic cholesterol accumulation and hepatic cholesterol levels remained unchanged (Fig. 7, C and D). In parallel with the improved hepatic fat accumulation and TG levels by SMILE, the increased Srebp-1c, Fas, and Acc gene expression were significantly decreased by SMILE (Fig. 7E). Next, we assessed the inhibitory effect of SMILE on high fat diet (HFD)-induced hepatic lipogenic gene expression in mice. Consistent with changes in T7-induced lipogenic gene expression by SMILE, adenoviral overexpression of SMILE significantly improved HFD-induced hepatic lipogenic gene expression (Fig. 7F). Taken together, these results indicate that activating SMILE considerably improved T7- or diet-induced hepatic fat accumulation by regulating of lipogenic enzyme gene expression.
FIGURE 7.

SMILE improves T7-induced hepatic lipid accumulation. A, male 7-week-old C57BL6 mice were provided with a standard rodent diet. T0901317 (LXR agonist, 50 mg/kg body weight) or vehicle (1% methylcellulose and 1% Tween 80) were administered by oral gavage each day for 1 week. An aliquot of 0.5 × 109 plaque-forming units of GFP or SMILE adenovirus were delivered by tail vein injection on day 4 of oral gavage. Three days after injection, the mice were sacrificed and Oil Red-O staining was performed on the liver samples. B, liver triglyceride levels were analyzed from the mouse liver tissue as in A. C and D, liver cholesterol staining (C) and hepatic cholesterol levels (D) were analyzed as in A and B. E, Srebp-1c, Fas, and Acc mRNA levels in mouse liver were analyzed by real time quantitative RT-PCR. F, real time-quantitative RT-PCR analysis of hepatic Srebp-1c, Fas, and Acc in adenovirus GFP or SMILE injected mice that were fed chow or HFD for 12 weeks. All data were normalized to those of β-actin and ribosomal L32 expression. Data in B and D–F are represented as mean ± S.D.
DISCUSSION
We demonstrated previously that SMILE acts as a corepressor of nuclear receptors (16). In this study, we found that SMILE decreased LXRα-mediated lipogenic target gene expression such as SREBP-1c, FAS, and ACC in hepatocytes. Moreover, we demonstrated that SMILE improved hepatic lipid accumulation, TG levels, and inhibited hepatic lipogenic gene expression using an in vivo model of T7-treated and HFD-fed mice. SMILE-mediated LXRα target gene transcription was regulated by competition with SRC-1 and the LXRα DNA binding block. Moreover, we showed that inhibiting lipogenic gene expression through UDCA-induced AMPK signaling depended on inducing SMILE gene expression.
LXRα is crucial for hepatic lipogenic gene expression and fatty acids synthesis in hepatocytes (9). In this study, we determined that SMILE is a corepressor of LXRα. In the transient transfection assay, SMILE overexpression significantly repressed LXRα transcriptional activity. Therefore, the repression of LXRα transcriptional activity by SMILE suggests that SMILE could improve LXRα-mediated fatty liver disease by regulating LXRα transcriptional activity. This hypothesis is corroborated by the observation that SMILE improved T7-induced hepatic lipid accumulation and TG levels. Moreover, SMILE inhibited hepatic lipogenic gene expression in HFD-fed mice. Overall, these findings strongly suggest that SMILE is a potent modulator of hepatic lipogenesis by regulating LXRα. Moreover, a previous report demonstrated that SMILE represses GR, CAR, and hepatocyte nuclear factor 4α (16). GR, CAR, and hepatocyte nuclear factor 4α are important for hepatic regulation and processing of glucose, lipids, drugs, and bile acids (33–35). Based on previous reports, we suggest that SMILE may be involved in diverse liver metabolic activities. SMILE knock-out and a transgenic animal model would be useful to better understand the role of SMILE in liver metabolism. Similar to SMILE-mediated repression of LXR, DAX-1 and SHP also decrease LXRα transcriptional activity (12, 36, 37). Consistent with previous reports, we found that SMILE physically interacted with LXRα and significantly inhibited recruitment of LXRα on the Srebp-1c promoter. Moreover, a domain mapping analysis using an in vivo GST pulldown assay showed that the LBD/AF2 domain of LXRα was essential for interaction with SMILE (Fig. 3E). In addition, it has been demonstrated that SRC-1 activates LXRα transcriptional activity (29). Here, we demonstrated that SMILE competes with the coactivator SRC-1. Moreover, we also found that SMILE inhibits LXRα binding on the Srebp-1c promoter in part by blocking the interaction between LXRα and RXRα (supplemental Fig. S1). These data suggest that SMILE regulates LXRα activity via multiple inhibitory mechanisms.
UDCA has been used as a therapeutic agent for fatty liver disease (27, 38) and cholestatic disorder diseases (24, 39). UDCA also has beneficial effects on liver regeneration in rats with non-alcoholic fatty liver disease (38). However, the effect of UDCA on lipid metabolism has remained largely unknown. Here, we showed that UDCA significantly activated SMILE gene expression depends on AMPK activation. In contrast, UDCA strongly decreased SREBP-1c, FAS, and ACC gene expression by inducing the SMILE gene (Fig. 6, A and B). These results demonstrate that SMILE is a critical transcriptional regulator of genes involved in UDCA-mediated regulation of lipid metabolism. Moreover, UDCA improves hepatic ER stress and insulin sensitivity. Notably, UDCA, a side chain-shortened homologue of UDCA, improves fatty liver and atherosclerosis (21). Consistent with the efficacy of UDCA in ER stress, our previous study showed that SMILE plays critical roles in regulating ER stress (18). Similar to the UDCA effect on lipogenic gene expression, a previous report demonstrated that cholic acid decreases hepatic expression of SREBP-1c, FAS, and ACC by inducing SHP gene expression (28). In addition, CDCA also suppresses T7-induced lipogenic gene expression (31). In the present study, we observed that UDCA particularly activated SMILE expression, whereas CDCA had no effect on SMILE gene expression (Fig. 4, E and F). UDCA is also reported to be a FXR ligand that activates SHP gene expression. However, UDCA-mediated FXR activation is much less than frequent CDCD (41). Consistent with these observations, SHP gene expression was unchanged following UDCA treatment in our study. Therefore, UDCA may improve hepatic lipid metabolism through a SHP-independent mechanism. Moreover, the use of CDCA and cholic acid is limited in humans because they can cause hepatotoxicity and increase low density lipoprotein cholesterol (42). UDCA decreases hepatocyte sensitivity to hydrophobic bile acid-induced oxidative stress in the fatty liver (43). Therefore, these observations suggest that UDCA-induced SMILE expression has a beneficial effect on the regulation of hepatic lipogenic gene expression without side effects as with CDAC and CA.
Previous reports have demonstrated that UDCA activates p38 MAPK, extracellular signal-regulated protein kinase (ERK), and PI3K pathways (25, 40). In addition, CDCA also activates the p38 MAPK, c-Jun N-terminal kinase, and ERK pathways but not the PI3K pathway (31). However, the molecular signaling to repress lipogenic gene expression by UDCA has remained unclear. In this study, UDCA strongly increased AMPK phosphorylation, and UDCA-mediated SMILE gene expression was blocked by repression of AMPK signaling. However, the downstream effectors of AMPK signaling to induce SMILE gene expression remain unknown. We investigated the underlying mechanism of UDCA, particularly focusing on gene expression involved in lipid metabolism in hepatocytes. However, whether the AMPK pathway plays a major role in UDCA-induced gene expression of SMILE and UDCA-mediated improvement of fatty liver needs to be examined in an animal study. Future studies will reveal the extent to which UDCA-induced SMILE mediates various liver metabolism disorders.
In summary, we found that SMILE acts as a novel corepressor of LXRα by competing with coactivator SRC-1 to inhibit hepatic lipogenic gene expression. Moreover, UDCA also inhibited lipogenic gene expression depending on SMILE gene expression. Based on these findings, we suggest that the UDCA-mediated AMPK signaling pathway induces SMILE gene expression. Moreover, SMILE decreased LXRα activity via competition with SRC-1 and subsequently decreased LXRα-mediated lipogenic enzyme gene expression in the liver (Fig. 8E). Thus, activating SMILE gene expression represents a potential therapeutic approach to improve hepatic lipid metabolism.
FIGURE 8.
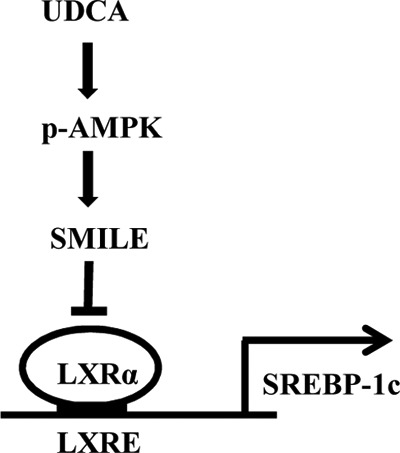
Schematic diagram of the regulation of hepatic lipogenesis by the UDCA-AMPK-SMILE signaling pathway. UDCA activates AMPK, which leads to the induction of SMILE gene expression. LXRα-mediated lipogenic gene expression is subsequently repressed by the UDCA-AMPK-SMILE pathway through inhibition of LXRα transcriptional activity.
Acknowledgment
We are grateful to S. Y. Choi for critical reading of the manuscript.
This work was supported in part by National Creative Research Initiatives Grant 20110018305 and Future-based Technology Development Program (BIO Fields) Grant 20100019512 through the National Research Foundation of Korea (NRF) funded by the Korean government (Ministry of Science, ICT & Future Planning).

This article contains supplemental Figs. S1–S4.
- LXRα
- liver X receptor α
- SMILE
- small heterodimer partner interacting leucine zipper protein
- FAS
- fatty acid synthase
- ACC
- acetyl-CoA carboxylase
- SREBP1
- sterol-regulatory element binding protein-1
- SRC-1
- steroid receptor coactivator 1
- GR
- glucocorticoid receptor
- CAR
- constitutive androstane receptor
- ERR γ
- estrogen receptor-related receptor γ
- AMPK
- adenosine monophosphate-activated kinase
- UDCA
- ursodeoxycholic acid
- CDCA
- chenodeoxycholic acid
- HFD
- high fat diet
- T7
- T0901317
- TG
- triglyceride
- ER
- estrogen receptor
- LXRE
- LXR response element
- RXR
- retinoid X receptor
- CA
- cholic acid
- co-IP
- co-immunoprecipitation.
REFERENCES
- 1. Willy P. J., Umesono K., Ong E. S., Evans R. M., Heyman R. A., Mangelsdorf D. J. (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 9, 1033–1045 [DOI] [PubMed] [Google Scholar]
- 2. Janowski B. A., Grogan M. J., Jones S. A., Wisely G. B., Kliewer S. A., Corey E. J., Mangelsdorf D. J. (1999) Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. U.S.A. 96, 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schultz J. R., Tu H., Luk A., Repa J. J., Medina J. C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D. J., Lustig K. D., Shan B. (2000) Role of LXRs in control of lipogenesis. Genes Dev. 14, 2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teboul M., Enmark E., Li Q., Wikström A. C., Pelto-Huikko M., Gustafsson J. A. (1995) OR-1, a member of the nuclear receptor superfamily that interacts with the 9-cis-retinoic acid receptor. Proc. Natl. Acad. Sci. U.S.A. 92, 2096–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Auboeuf D., Rieusset J., Fajas L., Vallier P., Frering V., Riou J. P., Staels B., Auwerx J., Laville M., Vidal H. (1997) Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-α in humans. No alteration in adipose tissue of obese and NIDDM patients. Diabetes. 46, 1319–1327 [DOI] [PubMed] [Google Scholar]
- 6. Grefhorst A., Elzinga B. M., Voshol P. J., Plösch T., Kok T., Bloks V. W., van der Sluijs F. H., Havekes L. M., Romijn J. A., Verkade H. J., Kuipers F. (2002) Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem. 277, 34182–34190 [DOI] [PubMed] [Google Scholar]
- 7. Gbaguidi G. F., Agellon L. B. (2004) The inhibition of the human cholesterol 7α-hydroxylase gene (CYP7A1) promoter by fibrates in cultured cells is mediated via the liver X receptor α and peroxisome proliferator-activated receptor α heterodimer. Nucleic Acids Res. 32, 1113–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eberlé D., Hegarty B., Bossard P., Ferré P., Foufelle F. (2004) SREBP transcription factors. Master regulators of lipid homeostasis. Biochimie 86, 839–848 [DOI] [PubMed] [Google Scholar]
- 9. Repa J. J., Liang G., Ou J. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP1) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 14, 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y., Bolten C., Bhat B. G. (2002) Induction of human liver X receptor α gene expression via an autoregulatory loop mechanism. Mol. Endocrinol. 16, 506–514 [DOI] [PubMed] [Google Scholar]
- 11. Li X., Zhang S., Blander G., Tse J. G., Krieger M., Guarente L. (2007) SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. 28, 91–106 [DOI] [PubMed] [Google Scholar]
- 12. Nedumaran B., Kim G. S., Hong S., Yoon Y. S., Kim Y. H., Lee C. H., Lee Y. C., Koo S. H., Choi H. S. (2010) Orphan nuclear receptor DAX-1 acts as a novel corepressor of liver X receptor α and inhibits hepatic lipogenesis. J. Biol. Chem. 285, 9221–9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu R., Misra V. (2000) Zhangfei. A second cellular protein interacts with herpes simplex virus accessory factor HCF in a manner similar to Luman and VP16. Nucleic Acids Res. 28, 2446–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hogan M. R., Cockram G. P., Lu R. (2006) Cooperative interaction of Zhangfei and ATF4 in transactivation of the cyclic AMP response element. FEBS Lett. 580, 58–62 [DOI] [PubMed] [Google Scholar]
- 15. Xie Y. B., Lee O. H., Nedumaran B., Seong H. A., Lee K. M., Ha H., Lee I. K., Yun Y., Choi H. S. (2008) SMILE, a new orphan nuclear receptor SHP-interacting protein, regulates SHP-repressed estrogen receptor transactivation. Biochem. J. 416, 463–473 [DOI] [PubMed] [Google Scholar]
- 16. Xie Y. B., Nedumaran B., Choi H. S. (2009) Molecular characterization of SMILE as a novel corepressor of nuclear receptors. Nucleic Acids Res. 37, 4100–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie Y. B., Park J. H., Kim D. K., Hwang J. H., Oh S., Park S. B., Shong M., Lee I. K., Choi H. S. (2009) Transcriptional corepressor SMILE recruits SIRT1 to inhibit nuclear receptor estrogen receptor-related receptor γ transactivation. J. Biol. Chem. 284, 28762–28774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Misra J., Chanda D., Kim D. K., Li T., Koo S. H., Back S. H., Chiang J. Y., Choi H. S. (2011) Curcumin differentially regulates endoplasmic reticulum stress through transcriptional corepressor SMILE (small heterodimer partner-interacting leucine zipper protein)-mediated inhibition of CREBH (cAMP responsive element-binding protein H). J. Biol. Chem. 286, 41972–41984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. López-Mateo I., Villaronga M. Á., Llanos S., Belandia B. (2012) The transcription factor CREBZF is a novel positive regulator of p53. Cell Cycle 11, 3887–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee J. H., Lee G. T., Kwon S. J., Jeong J., Ha Y. S., Kim W. J., Kim I. Y. (2012) CREBZF, a novel Smad8-binding protein. Mol. Cell. Biochem. 368, 147–153 [DOI] [PubMed] [Google Scholar]
- 21. Trauner M., Claudel T., Fickert P., Moustafa T., Wagner M. (2010) Bile acids as regulators of hepatic lipid and glucose metabolism. Dig. Dis. 28, 220–224 [DOI] [PubMed] [Google Scholar]
- 22. Omata M., Yoshida H., Toyota J., Tomita E., Nishiguchi S., Hayashi N., Iino S., Makino I., Okita K., Toda G., Tankkawa K., Kumada H., and Japanese C-Viral Hepatitis Network (2007) A large-scale, multicentre, double-blind trial of ursodeoxycholic acid in patients with chronic hepatitis C. Gut 56, 1747–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Copaci I., Micu L., Iliescu L., Voiculescu M. (2005) New therapeutical indications of ursodeoxycholic acid. Rom. J. Gastroenterol. 14, 259–266 [PubMed] [Google Scholar]
- 24. Beuers U., Boyer J. L., Paumgartner G. (1998) Ursodeoxycholic acid in cholestasis. Potential mechanisms of action and therapeutic applications. Hepatology 28, 1449–1453 [DOI] [PubMed] [Google Scholar]
- 25. Arisawa S., Ishida K., Kameyama N., Ueyama J., Hattori A., Tatsumi Y., Hayashi H., Yano M., Hayashi K., Katano Y., Goto H., Takagi K., Wakusawa S. (2009) Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem. Pharmacol. 77, 858–866 [DOI] [PubMed] [Google Scholar]
- 26. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ratziu V., de Ledinghen V., Oberti F., Mathurin P., Wartelle-Bladou C., Renou C., Sogni P., Maynard M., Larrey D., Serfaty L., Bonnefont-Rousselot D., Bastard J. P., Rivière M., Spénard J., FRESGUN (2011) A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J. Hepatol. 54, 1011–1019 [DOI] [PubMed] [Google Scholar]
- 28. Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP1. J. Clin. Invest. 113, 1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huuskonen J., Fielding P. E., Fielding C. J. (2004) Role of p160 coactivator complex in the activation of liver X receptor. Arterioscler. Thromb. Vasc. Biol. 24, 703–708 [DOI] [PubMed] [Google Scholar]
- 30. Heery D. M., Hoare S., Hussain S., Parker M. G., Sheppard H. (2001) Core LXXLL motif sequences in CREB-binding protein, SRC1, and RIP140 define affinity and selectivity for steroid and retinoid receptors. J. Biol. Chem. 276, 6695–6702 [DOI] [PubMed] [Google Scholar]
- 31. Talukdar S., Bhatnagar S., Dridi S., Hillgartner F. B. (2007) Chenodeoxycholic acid suppresses the activation of acetyl-coenzyme A carboxylase-α gene transcription by the liver X receptor agonist T0–901317. J. Lipid Res. 48, 2647–2663 [DOI] [PubMed] [Google Scholar]
- 32. Lepage G., Paradis K., Lacaille F., Sénéchal L., Ronco N., Champagne J., Lenaerts C., Roy C. C., Rasquin-Weber A. (1997) Ursodeoxycholic acid improves the hepatic metabolism of essential fatty acids and retinol in children with cystic fibrosis. J. Pediatr. 130, 52–58 [DOI] [PubMed] [Google Scholar]
- 33. Timsit Y. E., Negishi M. (2007) CAR and PXR. The xenobiotic-sensing receptors. Steroids 72, 231–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watt A. J., Garrison W. D., Duncan S. A. (2003) HNF4. A central regulator of hepatocyte differentiation and function. Hepatology 37, 1249–1253 [DOI] [PubMed] [Google Scholar]
- 35. Heitzer M. D., Wolf I. M., Sanchez E. R., Witchel S. F., DeFranco D. B. (2007) Glucocorticoid receptor physiology. Rev. Endocr. Metab. Disord. 8, 321–330 [DOI] [PubMed] [Google Scholar]
- 36. Kay H. Y., Kim W. D., Hwang S. J., Choi H. S., Gilroy R. K., Wan Y. J., Kim S. G. (2011) Nrf2 inhibits LXRα-dependent hepatic lipogenesis by competing with FXR for acetylase binding. Antioxid. Redox Signal. 15, 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brendel C., Schoonjans K., Botrugno O. A., Treuter E., Auwerx J. (2002) The small heterodimer partner interacts with the liver X receptor α and represses its transcriptional activity. Mol. Endocrinol. 16, 2065–2076 [DOI] [PubMed] [Google Scholar]
- 38. Uzun M. A., Koksal N., Aktas S., Gunerhan Y., Kadioglu H., Dursun N., Sehirli A. O. (2009) The effect of ursodeoxycholic acid on liver regeneration after partial hepatectomy in rats with non-alcoholic fatty liver disease. Hepatol. Res. 39, 814–821 [DOI] [PubMed] [Google Scholar]
- 39. Poupon R., Poupon R. E. (1995) Ursodeoxycholic acid therapy of chronic cholestatic conditions in adults and children. Pharmacol. Ther. 66, 1–15 [DOI] [PubMed] [Google Scholar]
- 40. Schoemaker M. H., Conde de la Rosa L., Buist-Homan M., Vrenken T. E., Havinga R., Poelstra K., Haisma H. J., Jansen P. L., Moshage H. (2004) Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 39, 1563–1573 [DOI] [PubMed] [Google Scholar]
- 41. Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B. (1999) Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 42. Schoenfield L. J., Lachin J. M. (1981) Chenodiol (chenodeoxycholic acid) for dissolution of gallstones, the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann. Intern. Med. 95, 257–282 [DOI] [PubMed] [Google Scholar]
- 43. Angulo P. (2002) Use of ursodeoxycholic acid in patients with liver disease. Curr. Gastroenterol. Rep. 4, 37–44 [DOI] [PubMed] [Google Scholar]