

Background: The NLRP3 inflammasome is a critical component of the innate immune system, and its malfunction contributes to the pathogenesis of inflammatory diseases.
Results: Nitrostyrene specifically inhibits NLRP3 activation.
Conclusion: Nitrostyrene blocks the assembly of NLRP3 inflammasome by inhibiting NLRP3 ATPase activity.
Significance: We identify a novel chemical probe for studying the molecular mechanism of NLRP3 inflammasome activation.
Keywords: Caspase, Inflammation, Innate Immunity, Macrophages, NOD-like Receptor (NLR)
Abstract
The NLRP3 inflammasome is a critical component of the innate immune system. NLRP3 activation is induced by diverse stimuli associated with bacterial infection or tissue damage, but its inappropriate activation is involved in the pathogenesis of inherited and acquired inflammatory diseases. However, the mechanism by which NLRP3 is activated remains poorly understood. In this study, we explored the role of kinases in NLRP3 inflammasome activation by screening a kinase inhibitor library and identified 3,4-methylenedioxy-β-nitrostyrene (MNS) as an inhibitor for NLRP3 inflammasome activation. Notably, MNS did not affect the activation of the NLRC4 or AIM2 (absent in melanoma 2) inflammasome. Mechanistically, MNS specifically prevented NLRP3-mediated ASC speck formation and oligomerization without blocking potassium efflux induced by NLRP3 agonists. Surprisingly, Syk kinase, the reported target of MNS, did not mediate the inhibitory activity of MNS on NLRP3 inflammasome activation. We also found that the nitrovinyl group of MNS is essential for the inhibitory activity of MNS. Immunoprecipitation, mass spectrometry, and mutation studies suggest that both the nucleotide binding oligomerization domain and the leucine-rich repeat domain of NLRP3 were the intracellular targets of MNS. Administration of MNS also inhibited NLRP3 ATPase activity in vitro, suggesting that MNS blocks the NLRP3 inflammasome by directly targeting NLRP3 or NLRP3-associated complexes. These studies identified a novel chemical probe for studying the molecular mechanism of NLRP3 inflammasome activation which may advance the development of novel strategies to treat diseases associated with abnormal activation of NLRP3 inflammasome.
Introduction
The innate immune system senses infection or injury through pattern recognition receptors and responds by inducing the production of proinflammatory and antimicrobial molecules. A major molecule produced upon pattern recognition receptor activation is IL-1β, a potent proinflammatory cytokine that contributes to host defense and the pathogenesis of several inflammatory diseases (1). Consequently, IL-1β production is tightly regulated at both the translational and post-translational levels. IL-1β is synthesized by phagocytes as an inactive precursor and processed into biologically active IL-1β by the inflammasome, a large intracellular oligomeric protein complex (2). Inflammasome activation results in the recruitment and activation of caspase-1, which then cleaves pro-IL-1β and pro-IL-18 into their mature forms. Inflammasomes contain a sensor protein, which can be a Nod2-like receptor, such as NLRP3, NLRP1, and NLRC4, or the PYHIN family member AIM2 (absent in melanoma 2) (3). The NLRP3 inflammasome is particularly interesting among the inflammasomes because it responds to diverse stimuli, including bacteria, viruses, components released by dying cells, and particulate matter (4). NLRP3 has been implicated in the pathogenesis of several human diseases, such as gouty arthritis, silicosis, type 2 diabetes, atherosclerosis and Alzheimer disease (5–11). Furthermore, genetic studies indicate that a spectrum of inflammatory disorders designated as cryopyrin-associated periodic fever syndromes are caused by inherited NLRP3 mutations (12–14). Although the NLRP3 inflammasome has been intensively investigated using cell culture, mouse genetic models, and various infection models, the signaling mechanism leading to NLRP3 inflammasome activation is still unclear (15). A better understanding of the signaling mechanism of NLRP3 inflammasome activation will enable the development of novel therapeutic strategies to treat NLRP3-associated human diseases. In this study, we identified 3,4-methylenedioxy-β-nitrostyrene (MNS) as a potent and specific inhibitor of the NLRP3 inflammasome.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies against mouse caspase-1, Asc, Nlrp3, and Nlrc4 have been described previously (16, 17). Murine IL-1β antibody (AF-401-NA) was purchased from R&D Systems. IL-18 antibody (5180R-100) was purchased from BioVision. Antibodies against Syk, phosphotyrosine (Tyr(P)-100), and GST were from Cell Signaling. Antibodies against actin and GAPDH were from Genescript. The InhibitorSelect 384-well protein kinase Library I, MNS, Bay 11-7082, and nigericin were purchased from Millipore. Anti-FLAG antibody, ATP, 3,4-(methylenedioxy)cinnamic acid, 1,2-methylenedioxy-4-propenylbenzene, trans-β-nitrostyrene, benzoylnitromethane, and trans-4-hydroxy-3-methoxy-β-nitrostyrene were from Sigma. Biotinyl-6-aminohexanoic acid (C16H27N3O4S) was purchased from Chem-Implex (Wood Dale, IL). Biotinylation of trans-4-hydroxy-3-methoxy-β-nitrostyrene with biotinyl-6-aminohexanoic acid was performed as described (18). The purity of biotinylated product was 97.3% as determined by HPLC. Ultrapure LPS from Escherichia coli 0111:B4 and poly(dA·dT)/lyovec were purchased from Invivogen. Salmonella enterica sv. typhimurium strain SL1344 was a gift from Denise Monack (Stanford University, Stanford, CA). Streptavidin magnetic beads were from Pierce. Recombinant GST-NLRP3 was purchased from Abnova. Pi ColorLock Gold phosphate detection system was purchased from Innova Bioscience. All other reagents if not specified were from Sigma.
Cells and Treatments
Bone-marrow derived macrophages (BMDMs) were prepared and cultured as described previously (19). For screening with a kinase inhibitor library, 5 × 104 cells were plated on 96-well plates overnight. Cells were primed with 100 ng/ml LPS for 4 h in serum-free Iscove's modified Dulbecco's medium. Cells were incubated with each inhibitor (10 μm) for 15 min before being pulsed with 5 mm ATP for 30 min. The release of IL-1β in culture supernatants was determined by ELISA. For the detection of inflammasome activation by Western blotting, 1 × 106 cells were plated on 12-well plates overnight. Inhibitors were added to medium in the last 15 min of LPS priming. Inflammasome activation was induced by adding respective stimuli: 5 mm ATP (30 min), 10 μm nigericin (1 h), 500 μg/ml silica (4 h), 2 μg/ml poly(dA·dT) (4 h), and S. enterica sv. typhimurium (m.o.i. = 10, 1 h).
Western Blots
Cells were lysed in ice-cold PBS buffer containing 1% Nonidet P-40 supplemented with complete protease inhibitor mixture (Roche Applied Science). The proteins from cell-free supernatants were precipitated by choloform/methanol method. Protein samples were separated by SDS-PAGE and transferred to PVDF membranes by electroblotting (Bio-Rad), and membranes were immunoblotted with respective antibodies.
Measurement of Cytokines
Mouse IL-1β and TNF-α in culture supernatants were measured by ELISA kits (R&D Systems) according to manufacturer's manual. Mouse IL-18 in culture supernatants was measured with an ELISA kit from eBioscience. Assays were performed in triplicate for each independent experiment.
Generation of Syk−/− Chimeras
Fetal livers were harvested at day 16 of gestation from both WT and syk−/− embryos generated by intercrossing syk+/− transgenic mice (S129). 5 × 106 cells were injected into the lateral tail vein of 8-week-old recipients that had been lethally irradiated with two doses of 550 rads of x-irradiation separated by 3 h. The recipient animals received antibiotics (sulfamethoxazole and trimethoprim) for 8 weeks after reconstitution. Bone marrows were harvested from chimeras and differentiated for macrophages as described above.
Potassium Efflux Assay
Intracellular K+ measurements were performed as described previously (20). Briefly, macrophages were plated on 96-well plates. Culture medium was thoroughly aspirated after stimulation. Cells were lysed with 3% ultrapure HNO3. Intracellular K+ was then determined by inductively coupled plasma optical emission spectrometry with an Optima 2000 DV spectrometer (PerkinElmer Life Sciences) using yttrium as internal standard.
ASC Speck Staining and ASC Oligomer Cross-linking
BMDMs were plated on an 8-well chamber slide overnight. Cells were primed and pretreated with inhibitor or DMSO before being stimulated with ATP or nigericin, transfected with poly(dA·dT), or infected with S. enterica sv. typhimurium. After stimulation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with PBS buffer containing 3% BSA. Cells were stained with anti-Asc antibody and Alexa Fluor 488-conjugated secondary antibody. DAPI was used to stain nuclei. Cell images were taken with Zeiss fluorescent microscope.
For ASC oligomer cross-linking, cells were lysed with PBS buffer containing 0.5% Triton X-100, and the cell lysates were centrifuged at 8000 rpm for 15 min at 4 °C. The Triton X-100-insoluble pellets were washed with PBS twice and then suspended in 200 μl of PBS. The pellets were then cross-linked at room temperature for 30 min by adding disuccinimidyl suberate (2 mm). The cross-linked pellets were spun down at 8000 rpm for 15 min and dissolved directly in SDS sample buffer.
HEK293T Cell Transfection
2.5 × 106 HEK293T cells were plated on a 100-mm Petri dish a day before transfection. Cells were transfected for 16 h with triple FLAG-tagged full-length NLRP3 (FL, 1.25 μg), pyrin deletion mutant ΔPyrin (amino acids 94–1036, 1.25 μg), LRR deletion mutant ΔLRR (amino acids 1–741, 2.5 μg), pyrin domain (amino acids 1–93, 1.25 μg), NOD domain (amino acids 220–536, 5 μg), and LRR domain (amino acids 742–991, 2.5 μg) by Lipofectamine LTX (Invitrogen). Cell culture medium was replaced with prewarmed serum-free DMEM. Cells were incubated with biotinylated compound biotin-trans-4-hydroxy-3-methoxy-β-nitrostyrene (biotin-HMNS) (10 μm) for 30 min at 37 °C and then lysed in 1.5 ml of ice-cold lysis buffer (50 mm Tris, pH 7.4, 2 mm EDTA, 150 mm NaCl, 0.5% Nonidet P-40, 1× EDTA-free proteinase inhibitor mixture (Roche Applied Science)). Cell lysates were clarified by centrifugation (14,000 × g) at 4 °C for 10 min. Cell lysates were incubated with streptavidin magnetic beads (25-μl slurry) overnight. The proteins bound by biotin-HMNS were pulled down and subjected to Western blotting.
Pulldown Assay by Biotin-HMNS
LPS primed BMDMs were lysed in ice-cold lysis buffer (50 mm Tris, pH 7.4, 2 mm EDTA, 150 mm NaCl, 0.5% Nonidet P-40, 1×Roche EDTA-free proteinase inhibitor mixture). 1 ml of cell lysates with total 1 mg protein were incubated with biotin (1 μm) or biotin-HMNS (1 μm) at 4 °C for 1 h. 50 μl of streptavidin magnetic beads (slurry) was added to cell lysates, followed by further incubation at 4 °C for 1 h. Magnetic beads were pulled down with a magnetic stand and washed with lysis buffer four times. Bound proteins were dissolved in SDS sample buffer. Proteins pulled down by biotin-HMNS were fractionated by SDS-polyacrylamide gel and subjected to mass spectrometry with whole lane analysis. When cells were used for the binding assay, cells were pretreated with biotin-HMNS (10 μm) for 30 min before lysis. For binding assay of GST-NLRP3 by biotin-HMNS, glutathione was removed from the solution of recombinant GST-NLRP3 with Slide-A-Lyzer Mini Dialysis Devices (Pierce) before incubation. For competition binding assay, 5 × 106 LPS-primed BMDMs were lysed in 1 ml of lysis buffer. Cell lysates were pretreated with 100 μm Bay 11-7082 or l-cysteine for 30 min at 4 °C before adding 1 μm biotin-HMNS.
Measurement of NLRP3 ATPase Activity
Recombinant NLRP3 was incubated with the indicated concentrations of MNS inhibitor in reaction buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 1 mm PMSF, and 1× proteinase inhibitor) for 15 min. DMSO was used as a vehicle control. The reaction mixtures were further incubated for 1 h at 37 °C after the addition of 250 μm ATP. The hydrolysis of ATP by NLRP3 was measured by Pi ColorLock Gold phosphate detection system according to the manual.
Cytotoxicity Assay
The percentage of cell death was determined using the lactate dehydrogenase release assay (Promega). The absorbance at 490 nm was measured, and the percentage of cytotoxicity was calculated relative to the 100% release value obtained by lysis of cells with a solution of 0.1% Triton X-100.
Statistical Analysis
Student's t test was used to determine statistically significant difference between two groups. One-way analysis of variance was used to analyze differences among multiple groups. A p value <0.05 was considered significant.
RESULTS
Identification of MNS as a Potent Inhibitor for ATP-induced NLRP3 Inflammasome Activation
Given the universal role of kinases in signal transduction, we sought to investigate the signaling mechanism of NLRP3 inflammasome activation by screening a kinase inhibitory library containing 160 selected kinase inhibitors. As Toll-like receptor ligands or cytokines are required to induce the expression of Nlrp3 and pro-IL-1β through NF-κB signaling in mouse bone marrow macrophages (21), we primed macrophages with LPS before the treatment with inhibitors to exclude their potential inhibitory effects on the NF-κB signaling pathway. Macrophages were treated with each kinase inhibitor (10 μm) for 15 min before stimulation with the NLRP3 activator ATP. We used ELISA to measure the level of IL-1β in the culture supernatant as a readout for NLRP3 inflammasome activation. The compound Bay 11-7082 inhibited Nlrp3 inflammasome activation in accordance with a previous report (22). We identified an additional chemical compound, MNS, that consistently abolished IL-1β secretion in our screening experiments (Fig. 1A).
FIGURE 1.

Identification of MNS as a potent inhibitor for ATP-induced NLRP3 inflammasome activation. A, chemical structure of MNS is shown. B and C, BMDMs were primed with LPS (100 ng/ml) for 4 h and then treated with indicated doses of MNS for 15 min before ATP stimulation, or left untreated. The culture supernatants were analyzed for IL-1β (B) or TNF-α (C) by ELISA. D, BMDMs were stimulated by ATP in the absence or presence of MNS (0.5, 1, 2.5, 5, 10 μm). Culture supernatants (Sup.) and cell extracts (Lys.) were immunoblotted for caspase-1, IL-1β, IL-18, Asc, or Nlrp3. Actin was a loading control. E, LPS-primed BMDMs were left unstimulated or stimulated with 5 mm ATP for 30 min in the presence of indicated concentrations of inhibitors. Culture supernatants and cell extracts were immunoblotted for caspase-1. F, immortalized Nlrp3 KO macrophages with reconstitution of Nlrp3 were stimulated with 5 mm ATP for 30 min in the absence or presence of different concentrations of MNS (1.25, 2.5, 5, 10 μm). After stimulation, culture supernatant and cell lysate were immunoblotted for caspase-1 and Nlrp3. Actin was a loading control. Data are representative of three independent experiments. Bar graphs shown are the mean ± S.D. (error bars) of triplicate wells.
To further investigate the inhibitory activity of MNS, we examined ATP-induced IL-1β secretion by LPS-primed mouse macrophages in the presence of different concentrations of the compound. As shown in Fig. 1B, MNS inhibited IL-1β secretion with an approximate IC50 of 2 μm. In contrast, the levels of TNF-α secretion between untreated and MNS-treated macrophages were comparable (Fig. 1C). Consistent with the ELISA results, MNS inhibited the production of mature IL-1β in the cell supernatant as shown by immunoblotting (Fig. 1D). Furthermore, IL-18 secretion and caspase-1 cleavage were also inhibited in the presence of MNS. Notably, MNS did not affect the protein levels of Nlrp3, Asc, or caspase-1 in mouse macrophages (Fig. 1D). As a comparison, MNS was slightly more potent than Bay 11-7082 in inhibiting NLRP3-mediated caspase-1 cleavage (Fig. 1E). In addition, treatment with MNS also inhibited ATP-induced NLRP3 inflammasome activation in Nlrp3-null immortalized macrophages reconstituted with Nlrp3, which do not need priming for Nlrp3 induction (Fig. 1F). These results indicate that chemical compound MNS potently inhibits ATP-induced NLRP3 inflammasome activation.
MNS Specifically Inhibits NLRP3 Inflammasome Activation
ATP activates the NLRP3 inflammasome through P2X7 receptor signaling (23, 24). To determine whether MNS specifically targets P2X7 receptor signaling or a common signaling component(s) for NLRP3 inflammasome activation, we tested the inhibitory activity of MNS on inflammasome activation induced by two additional NLRP3 activators, nigericin and silica, which act independently of the P2X7 receptor. As it was observed with ATP, treatment with MNS inhibited IL-1β secretion induced by both NLRP3 activators in a dose-dependent manner (Fig. 2, A and B). Consistent with reduced secretion of IL-1β, immunoblotting of culture supernatant revealed that the cleavage of caspase-1 was abolished in the presence of MNS (Fig. 2C). In contrast, MNS treatment did not affect caspase-1 activation induced by Salmonella, a pathogen known to trigger NLRC4 inflammasome activation (Fig. 2D) (25, 26). Transfection of synthetic poly(dA·dT) induces AIM2 inflammasome activation in macrophages (27, 28). Likewise, MNS did not affect AIM2 inflammasome activation by poly(dA·dT) (Fig. 2E). Caspase-1 activation triggers pyropotosis in macrophages, which can be revealed by measuring the release of lactate dehydrogenase (29). Consistently, MNS prevented ATP- and nigericin-induced lactate dehydrogenase release, but not that induced by Salmonella and poly(dA·dT) (Fig. 2F). These data indicate that MNS is a specific inhibitor of the NLRP3 inflammasome.
FIGURE 2.

MNS specifically inhibits the NLRP3 inflammasome. A and B, LPS-primed BMDMs were treated with indicated doses of MNS for 15 min and then stimulated by 10 μm nigericin for 1 h (A) or 500 μg/ml silica for 4 h (B). The culture supernatants were analyzed for IL-1β. C, BMDMs were primed with LPS and then stimulated with nigericin (1 h) or silica (4 h) in the absence or presence of 5 μm MNS. DMSO was used as a vehicle control. Culture supernatants (Sup.) and cell extracts (Lys.) were immunoblotted for caspase-1. Actin was a loading control. UT, untreated. D, primed BMDMs were infected with S. enterica sv. typhimurium (S. typhi) (m.o.i. = 10, 1 h) or stimulated by ATP (5 mm, 30 min) in the absence or presence of 5 μm MNS. Culture supernatants and cell extracts were immunoblotted for caspase-1. Actin was a loading control. E, primed BMDMs were transfected with 2 μg/ml poly(dA·dT) (4 h) or stimulated with 5 mm ATP (30 min) in the absence or presence of 5 μm MNS. Culture supernatants and cell extracts were immunoblotted for caspase-1. Actin was a loading control. F, cytotoxicity (lactate dehydrogenase (LDH release)) was measured from cells stimulated with ATP (5 mm, 30 min) or nigericin (10 μm, 1 h), infected with S. typhi (m.o.i. = 10, 1h) or transfected with poly(dA·dT) (2 μg/ml, 4 h) in the absence or presence of 5 μm MNS. Data are representative of three independent experiments. Bar graphs shown represent the mean ± S.D. (error bars) of triplicate wells. *, p < 0.05.
Syk Kinase Contributes to Pro-IL-1β Production but Is Dispensable for NLRP3 Inflammasome Activation in Macrophages
MNS is categorized as a Syk kinase inhibitor in our screening kinase inhibitor library. A previous study has shown that Syk was not required for NLRP3 inflammasome activation by nigericin but was essential for Candida albicans-induced NLRP3 inflammasome activation in dendritic cells (30). To examine further the role of Syk in NLRP3 inflammasome activation, we generated Syk-deficient BMDMs from chimeric mice. Syk depletion was confirmed in Syk−/− BMDMs by immunoblotting (Fig. 3A). There was minimal or no impairment in caspase-1 cleavage in response to the NLRP3 activators ATP, nigericin and silica in Syk−/− macrophages compared with wild-type macrophages (Fig. 3B). However, IL-1β secretion was reduced in Syk−/− macrophages, consistent with the reduced levels of pro-IL-1β in Syk−/− macrophages (Fig. 3, B and C). In contrast, IL-18 secretion was slightly elevated in Syk−/− macrophages as well as the intracellular levels of pro-IL-18 (Fig. 3B). This elevated secretion of IL-18 in Syk−/− macrophages was also confirmed by ELISA (Fig. 3D). The secretion of cytokine TNF-α was not affected by Syk deficiency (Fig. 3E). These data suggest that the Syk kinase contributes to pro-IL-1β induction by LPS but is largely dispensable for MNS-mediated inhibition of the NLRP3 inflammasome.
FIGURE 3.

Syk kinase is dispensable for NLRP3 inflammasome activation. A, BMDMs from WT or Syk−/− mice were primed with 100 ng/ml LPS for 4 h and then stimulated with 5 mm ATP (30 min), 10 μm nigericin (1 h), or 500 μg/ml silica (4 h). B, culture supernatants (Sup.) and cell extracts (Lys.) were immunoblotted for caspase-1, IL-1β, IL-18, or Syk. Actin was a loading control. C–E, the culture supernatants were also analyzed for IL-1β (C), IL-18 (D), or TNF-α (E) by ELISA. Data are representative of three independent experiments. Bar graphs shown represent the mean ± S.D. (error bars) of triplicate wells. *, p < 0.05.
MNS Prevents NLRP3 Agonist-induced ASC Speck Formation without Blocking Potassium Efflux
Potassium efflux is required for NLRP3 inflammasome activation in response to ATP, bacterial toxins, and particulate matter (20, 23, 24, 31). To test whether MNS inhibits NLRP3 inflammasome activation by preventing potassium efflux, we compared the levels of intracellular potassium after ATP stimulation in the presence or absence of MNS. ATP stimulation induced the drop of intracellular potassium in DMSO-treated macrophages, and this was unaffected by MNS pretreatment, suggesting that MNS acts downstream of potassium efflux to inhibit NLRP3 activation (Fig. 4A). A common event associated with inflammasome activation is the formation of large intracellular ASC aggregates called ASC specks that can be observed by microscopy or detected as oligomers after chemical cross-linking by immunoblotting (29). MNS prevented ASC speck formation induced by NLRP3 agonists, but did not affect ASC speck formation induced by stimuli that activate NLRC4 or AIM2 (Fig. 4, B and C). Biochemically, the presence of MNS abolished ASC oligomerization induced by activation of NLRP3, but not of the NLRC4 inflammasome (Fig. 4D). In contrast, YVAD, an inhibitor of caspase-1, blocked caspase-1 cleavage but did not affect ASC oligomerization (Fig. 4D). These results indicate that MNS specifically inhibits NLRP3-mediated ASC speckle formation.
FIGURE 4.

MNS inhibits NLRP3-mediated ASC speck formation without blocking potassium efflux. A, LPS-primed BMDMs were treated with DMSO or MNS (10 μm) for 15 min before ATP stimulation. The intracellular potassium was measured and expressed as the percentage of intracellular potassium in cells without ATP stimulation. B, LPS-primed BMDMs were stimulated with ATP (5 mm, 30 min) or nigericin (10 μm, 1 h), infected with S. enterica sv. typhimurium (S. typhi.) (m.o.i. = 10, 1 h), or transfected with poly(dA·dT) (2 μg/ml, 4h) in the absence or presence of 5 μm MNS. Cells were fixed, permeabilized, and stained for Asc (green). C, the percentage of cells containing ASC speck was quantified from three different view fields. D, LPS-primed BMDMs were untreated or pretreated with 5 μm MNS or 50 μm YVAD for 15 min. Cells were then stimulated with ATP (5 mm, 30 min) or nigericin (10 μm, 1 h), or infected with S. typhi. (m.o.i. = 10, 1 h). Macrophages were extracted with PBS buffer containing 0.5% Triton X-100, and the Triton-insoluble pellets were cross-linked with disuccinimidyl suberate. The cell lysates were immunoblotted for caspase-1. The cross-linked pellets were immunoblotted for Asc. Data are representative of three independent experiments. Bar graphs shown represent the mean ± S.D. (error bars) of triplicate wells. *, p < 0.05.
Structure-Activity Assay of MNS Analogues
To explore the structure-activity relationship of MNS and the NLRP3 inflammasome, we examined the inhibitory activity of MNS analogues on NLRP3 inflammasome activation. We selected five analogues for our experiments: 3,4-(methylenedioxy)cinnamic acid (C1), 1,2-methylenedioxy-4-propenylbenzene (C2), trans-β-nitrostyrene (C3), benzoylnitromethane (C4), and trans-4-hydroxy-3-methoxy-β-nitrostyrene (C5), which have modifications on the nitrovinyl group or dioxole group (Fig. 5A). LPS-primed macrophages were pretreated with different doses of MNS analogues before ATP stimulation. MNS analogues lacking the nitrovinyl side chain (3,4-(methylenedioxy)cinnamic acid, 1,2-methylenedioxy-4-propenylbenzene, and benzoylnitromethane) did not inhibit NLRP3-mediated caspase-1 cleavage and IL-1β secretion. In contrast, trans-β-nitrostyrene and trans-4-hydroxy-3-methoxy-β-nitrostyrene retained inhibitory activity (Fig. 5, B and C). These results suggest that the nitrovinyl side chain is essential for MNS-mediated inhibition of the NLRP3 inflammasome.
FIGURE 5.
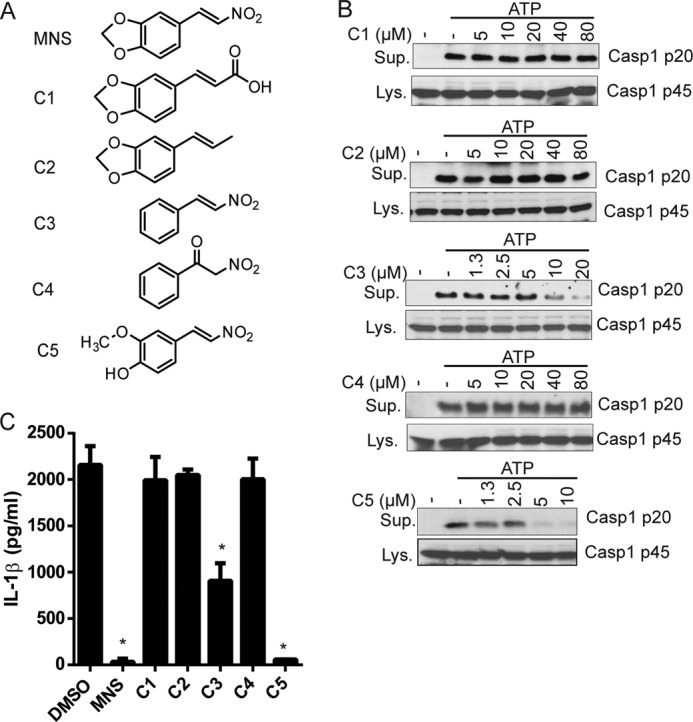
Structure-activity relationship of MNS analogues. A, chemical structures of MNS analogues. B, inhibitory activities of MNS analogues on NLRP3 inflammasome activation. LPS-primed BMDMs were pretreated with the indicated doses of chemical compounds for 15 min before ATP stimulation. The culture supernatants (Sup.) and cell lysates (Lys.) were collected and immunoblotted for caspase-1. C, LPS-primed BMDMs pretreated with DMSO or MNS derivatives (10 μm) for 15 min before ATP stimulation. IL-1β from culture supernatant was analyzed by ELISA. Data are representative of three independent experiments. Bar graphs shown represent the mean ± S.D. (error bars) of triplicate wells. *, p < 0.05.
MNS Binds Directly to NLRP3 and Inhibits Its ATPase Activity
As MNS was previously reported to inhibit tyrosine kinases (32), we examined the pattern of tyrosine phosphorylation in BMDMs stimulated by ATP or nigericin in the absence or presence of inhibitors. Cells treated with MNS and C4 had comparable patterns of tyrosine phosphorylation as detected by immunoblotting with anti-Tyr antibody (data not shown). To determine the mechanism by which MNS inhibits NLRP3 inflammasome activation, we examined MNS-targeted proteins in mouse macrophages. The compound HMNS was biotinylated through hydrogen oxide group (biotin-HMNS) (Fig. 6A). Biotin-HMNS retained inhibitory activity on NLRP3 inflammasome, albeit at a higher concentration (Fig. 6B). Macrophage proteins pulled down by biotin-HMNS were fractionated by SDS-PAGE and analyzed by mass spectrometry (Fig. 6C and supplemental Table). Notably, NLRP3 protein was one of the most abundant proteins pulled down by biotin-HMNS. The association between biotin-HMNS and NLRP3 protein was further confirmed by immunoprecipitation followed by immunoblotting (Fig. 6D). Consistent with the finding that MNS does not inhibit NLRC4 inflammasome, we could not detect biotin-HMNS associated with the NLRC4 protein by mass spectrometry or immunoprecipitation assay (Fig. 6D and supplemental Table). Furthermore, biotin-HMNS could pull down recombinant NLRP3, and this was abolished in the presence of excess HMNS (Fig. 6E). These data suggest that NLRP3 is directly targeted by MNS. To map the targeting domains of NLRP3 by MNS, we treated HEK293 cells expressing full-length wild-type NLRP3 (FL) or mutants lacking the pyrin domain (ΔPyrin) or the leucine-rich repeat domain (ΔLRR) with biotin-HMNS. Biotin-HMNS could pull down WT NLRP3 and NLRP3 mutants with deletion of either the pyrin or LRR domain (Fig. 6F). Moreover, biotin-HMNS could pull down the NOD and the LRR domain, but not the pyrin domain (Fig. 6G). Furthermore, MNS inhibited the ATPase activity of NLRP3 in a concentration-dependent manner, as measured by the release of free phosphate (Fig. 6H). Bay 11-7082 has been reported to inhibit the ATPase activity of NLRP3 possibly through the mechanism of cysteine modification (22). We examined whether Bay 11-7082 and l-cysteine could compete off the pulldown of NLRP3 by biotin-HMNS. Addition of excess amounts of Bay 11-7082 and l-cysteine abolished the pulldown of NLRP3 by biotin-HMNS (Fig. 6I). Taken together, these results suggest that MNS directly binds to NLRP3 and inhibit its ATPase activity.
FIGURE 6.

MNS binds to NLRP3 and inhibits its ATPase activity. A, chemical structure of biotin-HMNS is shown. B, LPS-primed BMDMs were stimulated with 5 mm ATP for 30 min in the absence or presence of the indicated concentrations of biotin-HMNS. Culture supernatant (Sup.) and cell lysate (Lys.) were immunoblotted for caspase-1. C, NLRP3 was pulled down by biotin-HMNS from cell lysates. Lysates from LPS-primed BMDMs were incubated with biotin (1 μm) or biotin-HMNS (1 μm) at 4 °C for 1 h. Bound proteins were pulled down by streptavidin beads and fractioned on a SDS-polyacrylamide gel. The identity of bound proteins was determined by mass spectrometry. D, biotin-HMNS bound to NLRP3 in cells. BMDMs were pretreated (IP) with or without biotin-HMNS (10 μm) for 30 min and lysed in 0.5% Nonidet P-40 TBS buffer. The bound proteins in the lysate were pulled down by streptavidin beads. The cell lysates and bound proteins were immunoblotted for NLRP3 and NLRC4. GAPDH was used as a loading control. E, recombinant human NLRP3 was bound by biotin-HMNS. Recombinant human NLRP3 was incubated with biotin (1 μm) or biotin-HMNS (1 μm) in the presence or absence of free compound HMNS, 100 μm) at 4 °C for 1 h. The bound NLRP3 was pulled down by streptavidin beads and detected by Western blotting. F and G, HEK293T cells were transfected with FLAG-tagged full-length NLRP3 (FL), pyrin deletion mutant (ΔPyrin), LRR deletion mutant (ΔLRR), pyrin domain, NOD domain, or LRR domain for 16 h. Cells were treated with 10 μm biotin-HMNS for 30 min before lysis. The bound proteins were pulled down by streptavidin beads. The cell lysates and bound proteins were immunoblotted (IB) by using anti-FLAG antibody. Actin was a loading control. H, MNS inhibited NLRP3 ATPase activity. The release of NLRP3-hydrolyzed inorganic phosphate in the presence or absence of MNS was determined using a Pi ColorLock Gold phosphate detection system. The data were subtracted from the results of experiments without addition of NLRP3 and expressed as the percentage of control. I, lysates from LPS-primed BMDMs were incubated with biotin-HMNS (1 μm) in the presence of Bay 11-7082 (100 μm) or l-cysteine (100 μm) at 4 °C for 1 h. Bound proteins were pulled down by streptavidin beads and immunoblotted for NLRP3. The lysate blot was shown as an input control. Data are representative of two or three independent experiments.
CONCLUSION
Inappropriate activation of the NLRP3 inflammasome has been implicated in the pathogenesis of inflammation-associated diseases, such as gouty arthritis, silicosis, type 2 diabetes, Alzheimer disease, and cryopyrin-associated periodic fever syndromes (2, 33). Thus, inhibition of NLRP3 inflammasome activation could have therapeutic potential for treating these diverse inflammatory diseases. Based on research in mouse macrophages, a two-step mechanism has been proposed for NLRP3 inflammasome activation. In the first step, known as priming, NLRP3 is transcriptionally up-regulated by TLR ligands or cytokines through the NF-κB signaling pathway (21). However, NLRP3 induction is not sufficient for activation of the NLRP3 inflammasome in that a second step triggered by diverse stimuli such as ATP, nigericin, or particulate matter, is necessary for NLRP3 activation (7, 34). Although kinases such as IκK and TAK1 are required for the priming step of NLRP3 induction, it is not clear whether kinases are also involved in the activating step. A previous study showed that the kinase PKR was required for the activation of all known inflammasomes including the NLRP3 inflammasome (35). However, these results could not be confirmed by a different study (36). Furthermore, PKCδ was reported to regulate NLRC4 inflammasome activation by phosphorylating NLRC4 (37). In this study, we sought to explore the role of kinases in NLRP3 inflammasome activation by screening a kinase inhibitor library. We identified a Syk kinase inhibitor, MNS, as an inhibitor of NLRP3 inflammasome activation. MNS did not affect the activation of the NLRC4 and AIM2 inflammasomes at concentrations that abrogated NLRP3 inflammasome activation. Surprisingly, deletion of the Syk kinase in macrophages did not affect NLRP3 inflammasome activation induced by ATP, nigericin and silica, indicating that Syk is dispensable for NLRP3 inflammasome activation. These results are consistent with a previous study that Syk was required for caspase-1 activation induced by the fungus C. albicans but was not necessary for nigericin-induced NLRP3 inflammasome activation in dendritic cells (30). MNS was reported to inhibit other tyrosine kinases such as Src and FAK at higher concentrations (IC50 > 27.3 μm) (32). Thus, it is possible that those tyrosine kinase(s) may play a redundant role in NLRP3 inflammasome activation in macrophages. However, we found that MNS treatment had patterns of tyrosine phosphorylation comparable with those of C4 treatment after ATP or nigericin stimulation. Alternatively, MNS may inhibit NLRP3 inflammasome activation through a novel kinase-independent fashion (see below). Further studies are needed to understand how MNS inhibits the NLRP3 inflammasome.
The blockade of IL-1β function by anakira is currently used in the clinic to treat cryopyrin-associated periodic fever syndrome patients (38). In addition to IL-1β maturation, NLRP3 inflammasome activation induces other cell responses such as cell death and IL-18 maturation, suggesting that therapeutic strategies that directly target NLRP3 inflammasome activation would be more effective than blocking IL-1β alone. Previous studies have identified several inhibitors of NLRP3 inflammasome activation, including glyburide, parthenolide, Bay 11-7082, bromoxone, CRID3, and deubiquitinase inhibitors (22, 39–44). These inhibitors have been suggested to target the NLRP3 ATPase activity, ASC oligomerization, or upstream signaling events required for NLRP3 activation. Here, we have identified MNS as a novel inhibitor of NLRP3 inflammasome activation. Previously MNS was reported to inhibit platelet aggregation, tumor cell growth, and apoptosis (45–47). Effects of nitrostyrene on tyrosine kinases, protein phosphatases, and telomerase activity have been suggested to account for those inhibitions. Structurally, the nitrovinyl group was essential for MNS inhibitory activity as tested compounds with this group retained inhibitory activity for NLRP3 inflammasome activation. In contrast, the dioxole group was dispensable for NLRP3 inhibitory activity. One possibility is that the first carbon of nitrovinyl group is attacked by the thiol group of cysteines in target proteins. This is further supported by the data that the excess amounts of l-cysteine prevent the pulldown of NLRP3 by MNS. Furthermore, our studies suggest that MNS binds to both the NOD and LRR domains of NLRP3, providing a direct target for inflammasome inhibition. Consistent with the latter notion, nitrostyrene inhibited the ATPase activity of NLRP3, suggesting that cysteine(s) of NLRP3 may be a direct target of this compound. Further biochemical studies on the mechanism of action of nitrostyrene on NLRP3 may provide additional insight for the development of new therapeutic strategies for blocking abnormal NLRP3 inflammasome activation.
Acknowledgments
We thank Dr. Hollis Showalter for biotinylating trans-4-hydroxy-3-methoxy-β-nitrostyrene, Dr. Venkatesha Basrur for mass spectrometry, and Dr. Tyler Nygaard for a critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI063331 and R01DK091191.

This article contains a supplemental table.
- NOD
- nucleotide binding oligomerization domain
- ASC
- apoptosis-associated speck-like protein containing a CARD
- AIM2
- absent in melanoma 2
- BMDM
- bone marrow-derived macrophage
- DMSO
- dimethyl sulfoxide
- HMNS
- trans-4-hydroxy-3-methoxy-β-nitrostyrene
- LRR
- leucine-rich repeat
- MNS
- 3,4-methylenedioxy-β-nitrostyrene
- m.o.i.
- multiplicity of infection
- NLRC4
- NLR family CARD domain-containing protein 4
- NLRP3
- NACHT, LRR and PYD domains-containing protein 3.
REFERENCES
- 1. Dinarello C. A. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 [DOI] [PubMed] [Google Scholar]
- 2. Martinon F., Mayor A., Tschopp J. (2009) The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 [DOI] [PubMed] [Google Scholar]
- 3. Franchi L., Muñoz-Planillo R., Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 5. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Halle A., Hornung V., Petzold G. C., Stewart C. R., Monks B. G., Reinheckel T., Fitzgerald K. A., Latz E., Moore K. J., Golenbock D. T. (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 [DOI] [PubMed] [Google Scholar]
- 8. Cassel S. L., Eisenbarth S. C., Iyer S. S., Sadler J. J., Colegio O. R., Tephly L. A., Carter A. B., Rothman P. B., Flavell R. A., Sutterwala F. S. (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masters S. L., Dunne A., Subramanian S. L., Hull R. L., Tannahill G. M., Sharp F. A., Becker C., Franchi L., Yoshihara E., Chen Z., Mullooly N., Mielke L. A., Harris J., Coll R. C., Mills K. H., Mok K. H., Newsholme P., Nuñez G., Yodoi J., Kahn S. E., Lavelle E. C., O'Neill L. A. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T. C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D. T. (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ting J. P., Kastner D. L., Hoffman H. M. (2006) CATERPILLERs, pyrin and hereditary immunological disorders. Nat. Rev. Immunol. 6, 183–195 [DOI] [PubMed] [Google Scholar]
- 13. Hoffman H. M., Mueller J. L., Broide D. H., Wanderer A. A., Kolodner R. D. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feldmann J., Prieur A. M., Quartier P., Berquin P., Certain S., Cortis E., Teillac-Hamel D., Fischer A., de Saint Basile G. (2002) Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 71, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rathinam V. A., Vanaja S. K., Fitzgerald K. A. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He Y., Franchi L., Núñez G. (2013) TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J. Immunol. 190, 334–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franchi L., Kamada N., Nakamura Y., Burberry A., Kuffa P., Suzuki S., Shaw M. H., Kim Y. G., Núñez G. (2012) NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 13, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giubellino A., Shi Z. D., Jenkins L. M., Worthy K. M., Bindu L. K., Athauda G., Peruzzi B., Fisher R. J., Appella E., Burke T. R., Bottaro D. P. (2008) Selectivity and mechanism of action of a growth factor receptor-bound protein 2 SRC homology 2 domain binding antagonist. J. Med. Chem. 51, 7459–7468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Franchi L., Eigenbrod T., Núñez G. (2009) Cutting edge: TNF-α mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 183, 792–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., Núñez G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B. G., Fitzgerald K. A., Hornung V., Latz E. (2009) Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Juliana C., Fernandes-Alnemri T., Wu J., Datta P., Solorzano L., Yu J. W., Meng R., Quong A. A., Latz E., Scott C. P., Alnemri E. S. (2010) Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franchi L., Kanneganti T. D., Dubyak G. R., Núñez G. (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282, 18810–18818 [DOI] [PubMed] [Google Scholar]
- 24. Kahlenberg J. M., Dubyak G. R. (2004) Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 286, C1100–1108 [DOI] [PubMed] [Google Scholar]
- 25. Miao E. A., Alpuche-Aranda C. M., Dors M., Clark A. E., Bader M. W., Miller S. I., Aderem A. (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 7, 569–575 [DOI] [PubMed] [Google Scholar]
- 26. Franchi L., Amer A., Body-Malapel M., Kanneganti T. D., Ozören N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A., Grant E. P., Núñez G. (2006) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in Salmonella-infected macrophages. Nat. Immunol. 7, 576–582 [DOI] [PubMed] [Google Scholar]
- 27. Fernandes-Alnemri T., Yu J. W., Datta P., Wu J., Alnemri E. S. (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., Fitzgerald K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E. S. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gross O., Poeck H., Bscheider M., Dostert C., Hannesschläger N., Endres S., Hartmann G., Tardivel A., Schweighoffer E., Tybulewicz V., Mocsai A., Tschopp J., Ruland J. (2009) Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436 [DOI] [PubMed] [Google Scholar]
- 31. Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., Tschopp J. (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 [DOI] [PubMed] [Google Scholar]
- 32. Wang W. Y., Hsieh P. W., Wu Y. C., Wu C. C. (2007) Synthesis and pharmacological evaluation of novel β-nitrostyrene derivatives as tyrosine kinase inhibitors with potent antiplatelet activity. Biochem. Pharmacol. 74, 601–611 [DOI] [PubMed] [Google Scholar]
- 33. Davis B. K., Wen H., Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mariathasan S., Weiss D. S., Newton K., McBride J., O'Rourke K., Roose-Girma M., Lee W. P., Weinrauch Y., Monack D. M., Dixit V. M. (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 [DOI] [PubMed] [Google Scholar]
- 35. Lu B., Nakamura T., Inouye K., Li J., Tang Y., Lundbäck P., Valdes-Ferrer S. I., Olofsson P. S., Kalb T., Roth J., Zou Y., Erlandsson-Harris H., Yang H., Ting J. P., Wang H., Andersson U., Antoine D. J., Chavan S. S., Hotamisligil G. S., Tracey K. J. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He Y., Franchi L., Núñez G. (2013) The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 43, 1147–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qu Y., Misaghi S., Izrael-Tomasevic A., Newton K., Gilmour L. L., Lamkanfi M., Louie S., Kayagaki N., Liu J., Kömüves L., Cupp J. E., Arnott D., Monack D., Dixit V. M. (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 [DOI] [PubMed] [Google Scholar]
- 38. López-Castejón G., Pelegrín P. (2012) Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin. Investig. Drugs 21, 995–1007 [DOI] [PubMed] [Google Scholar]
- 39. Lamkanfi M., Mueller J. L., Vitari A. C., Misaghi S., Fedorova A., Deshayes K., Lee W. P., Hoffman H. M., Dixit V. M. (2009) Glyburide inhibits the cryopyrin/NALP3 inflammasome. J. Cell Biol. 187, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gong Y. N., Wang X., Wang J., Yang Z., Li S., Yang J., Liu L., Lei X., Shao F. (2010) Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 20, 1289–1305 [DOI] [PubMed] [Google Scholar]
- 41. Py B. F., Kim M. S., Vakifahmetoglu-Norberg H., Yuan J. (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 [DOI] [PubMed] [Google Scholar]
- 42. Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E. S. (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 287, 36617–36622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lopez-Castejon G., Luheshi N. M., Compan V., High S., Whitehead R. C., Flitsch S., Kirov A., Prudovsky I., Swanton E., Brough D. (2013) Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 288, 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coll R. C., O'Neill L. A. (2011) The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS One 6, e29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang W. Y., Wu Y. C., Wu C. C. (2006) Prevention of platelet glycoprotein IIb/IIIa activation by 3,4-methylenedioxy-β-nitrostyrene, a novel tyrosine kinase inhibitor. Mol. Pharmacol. 70, 1380–1389 [DOI] [PubMed] [Google Scholar]
- 46. Kim J. H., Lee G. E., Lee J. E., Chung I. K. (2003) Potent inhibition of human telomerase by nitrostyrene derivatives. Mol. Pharmacol. 63, 1117–1124 [DOI] [PubMed] [Google Scholar]
- 47. Kaap S., Quentin I., Tamiru D., Shaheen M., Eger K., Steinfelder H. J. (2003) Structure activity analysis of the pro-apoptotic, antitumor effect of nitrostyrene adducts and related compounds. Biochem. Pharmacol. 65, 603–610 [DOI] [PubMed] [Google Scholar]