

Background: TSC1 is a negative regulator for mTORC1 that is implicated in cancer.
Results: mTORC1 activation by TSC1 deletion promotes mammary tumor growth through increasing autophagy and Akt activation of tumor cells.
Conclusion: mTORC1 activation in mammary tumor cells is important in promoting breast cancer progression in vivo.
Significance: New insights into the mechanisms of breast cancer progression may offer potential targets in breast cancer therapy.
Keywords: Autophagy, Breast Cancer, Mouse, mTOR Complex (mTORC), Signaling
Abstract
The mammalian target of rapamycin complex 1 (mTORC1) is a master regulator of cell growth and proliferation. Recent studies have suggested that constitutive activation of mTORC1 in normal cells could lead to malignant tumor development in several tissues. However, the mechanisms of mTORC1 hyperactivation to promote the growth and metastasis of breast or other cancers are still not well characterized. Here, using a new inducible deletion system, we show that deletion of Tsc1 in mouse primary mammary tumor cells, either before or after their transplantation, significantly increased their growth in vivo. The increase in tumor growth was completely rescued by rapamycin treatment, suggesting a major contribution from mTORC1 hyperactivation. Interestingly, glucose starvation-induced autophagy, but not amino acid starvation-induced autophagy, was increased significantly in Tsc1-null tumor cells. Further analysis of these cells also showed an increased Akt activation but no significant changes in Erk signaling. Together, these results provide insights into the mechanism by which hyperactivation of mTORC1 promotes breast cancer progression through increasing autophagy and Akt activation in vivo.
Introduction
Tuberous sclerosis complex (Tsc)3 is an autosomal dominant disease that is caused by mutations in either the Tsc1 or Tsc2 gene and is characterized by the formation of benign tumors in multiple systems such as skin, brain, lung, liver, heart, and kidney (1–3). Tsc1 and Tsc2 proteins form a complex that negatively regulates cell growth/proliferation through the inhibition of mammalian target of rapamycin (mTOR). mTOR is a large protein kinase associated with different protein partners to form two independently regulated hetero-oligomeric complexes, the rapamycin-sensitive and rapamycin-insensitive mTOR complexes (mTORC) 1 and 2, respectively (4, 5). mTORC1 contains the catalytic subunit mTOR and its binding partners mLST8/GβL, PRAS40, and Raptor, which have been well documented to promote the growth and proliferation of a variety of cells through the phosphorylation of two main regulators of mRNA translation and ribosome biogenesis, ribosomal S6 kinase (S6K) and eukaryotic initiation factor 4E binding protein 1 (4EBP1) (4, 6), although a multitude of other targets have also been suggested in recent studies (7).
mTORC1 has been shown to play a major role in the regulation of autophagy by phosphorylating components of the autophagy induction machinery (8). Autophagy is an evolutionarily conserved process involved in the degradation of bulk cytoplasmic materials via sequestering them in the double-membraned structures called autophagosomes, followed by delivery to lysosomes for degradation (9, 10). In addition to its essential role in a variety of physiological processes, autophagy dysfunction has been linked to many diseases, including cancer, although the underlying molecular mechanisms are not very clear at present (9, 11–13).
Although abnormalities in Tsc/mTOR signaling are best illustrated in the development of benign tumors in many organs, recent studies have also suggested potential functions of this key pathway in the development and progression of several malignant cancers. For example, knockout of raptor inhibited mTORC1 activation and leukemia propagation (14). Liver-specific knockout of Tsc1 led to increased mTORC1 signaling and development of hepatocellular carcinoma in mice (15). Mice with conditional knockout of Tsc1 in prostate epithelial cells developed prostate cancer at an old age (16). However, a potential role and mechanisms of Tsc/mTOR signaling have not been examined directly in breast cancer in vivo, although a previous study showed that overexpression of Tsc1 in a murine breast cancer cell line, 4T1, reduced their growth in nude mice by inhibiting mTORC1-mediated VEGF expression and tumor angiogenesis (17).
In this work, we created a novel system that allows one to delete Tsc1 in primary mammary tumor cells both in vitro and in vivo in an inducible manner and demonstrated directly that deletion of Tsc1 and consequent activation of mTORC1 promoted mammary tumor growth and metastasis. Moreover, we showed that Tsc1 deletion increased glucose starvation-induced autophagy as well as Akt activation, which could promote tumor cell survival and contribute to the increased tumor growth in vivo.
EXPERIMENTAL PROCEDURES
Analysis of Human Breast Cancer Gene Expression Datasets
Oncomine was used to examine Tsc1 expression in several human cancer datasets, including Radvanyi Breast, GSE1477 (18); Richardson Breast 2, GSE3744 (19); The Cancer Genome Atlas, http://tcga-data.nci.nih.gov/tcga/; and Curtis (20).
Mice and Genotyping
MMTV-PyMT transgenic mice in the FVB/n background were as described previously (21). Tsc1f/f mice (C57/B6 background) were provided by Dr. David Kwiatkowski (22) and were used to cross with MMTV-PyMT mice to produce Tsc1f/f;MMTV-PyMT mice with mixed FVB and C57/B6 backgrounds. Genotyping for the Tsc1 and PyMT alleles was performed as described previously (21, 22). Mice were housed and handled by following the local, state, and federal regulations. The guidelines of the Institutional Animal Care and Use Committee at the University of Michigan were used in all experiments with mice.
Generation of Primary Mammary Tumor Cells Capable of Inducible Deletion of Tsc1
Primary mammary tumor cells were isolated from female Tsc1f/f;MMTV-PyMT mice as described previously (21). The primary Tsc1f/f,PyMT tumor cells were then transduced with supernatants containing recombinant MSCV-CreERT2-puro retroviruses (Addgene plasmid no. 22776, generated by the laboratory of Tyler Jacks at the Massachusetts Institute of Technology). The cells were incubated in medium with puromycin, and the pool of infected cells resistant to puromycin selection were obtained as primary Tsc1f/f,PyMT,CreER tumor cells whose Tsc1 can be deleted by activation of Cre recombinase either in vitro or following transplantation in vivo.
Primary Tsc1f/f,PyMT,CreER tumor cells were incubated with 4-hydroxytamoxifen to activate Cre recombinase to delete Tsc1 gene or mock media to generate Tsc1 KO and Ctrl cells, respectively, which were used in most experiments. In some experiments, these cells were transplanted into the mammary fat pads of nude mice as described below. After the appearance of mammary tumors (about 2 mm in diameter), tamoxifen was injected into the recipient mice to delete Tsc1 in the tumor cells. Lastly, primary Tsc1f/f,PyMT tumor cells were transplanted, and the recipient mice were treated by tamoxifen in a similar manner to serve as a control for the above experiments.
Cell Multiplication, Proliferation, Apoptosis, Migration, and Invasion Assays
Primary tumor cells were seeded in 6-well plates in DMEM containing 10% FBS. The cells were harvested by trypsinization at regular intervals and counted to determine cell multiplication. For measuring cell proliferation, cultured primary tumor cells or tumor cell sections were subjected to immunohistochemical staining using antibody against Ki67 (M3060, Spring Bioscience) as described previously (21). For detection of apoptosis, tumor sections were stained using antibody against cleaved caspase 3 (catalog no. 9661S, Cell Signaling Technology) or subjected to a TUNEL assay as described previously (21). Cell migration assays were performed using a 48-well Boyden chamber as described previously (23). For invasion assays, the upper chamber of a Millicell-PCF culture insert (8 μm, catalog no. P18P01250, Millipore) was coated with 100 μl Matrigel at 1 mg/ml and dried for 6 h at 37 °C. Tumor cells in DMEM (100 μl at 106 cells/ml) were plated in the upper chamber, and the chamber was inserted in 1.5 ml of DMEM with 10% FBS and 10 μg/ml fibronectin (Sigma, catalog no. F1141) as chemoattractants in a well of 6-well plates. After incubation for 24 h at 37 °C, the non-invasive cells in the upper chamber were wiped off with a cotton swab. The invasive cells on the underside of the membrane were fixed in pure methanol for 10 min at room temperature, immersed in double-distilled H2O, air dried, and Giemsa-stained (Sigma, catalog no. GS500) (1:10 dilution in double-distilled H2O) for 1 h at room temperature. Finally, the membrane was immersed in double-distilled H2O for 10 s, put on a glass slide, covered with a cover slide, and then cells were counted under a microscope.
Tumor Cell Transplantation, Tumor Monitoring, and Tail Vein Injection
Transplantation of primary tumor cells was performed as described previously (24). Briefly, cells were harvested, washed twice in DMEM, and then injected (106 cells in DMEM with 5 μg/ml Matrigel) into the no. 4 inguinal mammary fat pads of 8-week-old female athymic nude mice (The Jackson Laboratory). After tumors appeared, tumor size was monitored as described previously (21, 24). Tail vein injection was performed to monitor experimental metastasis to the lungs of recipient nude mice as described previously (25). The number of nodules on the surface of the lungs was counted as described previously (21).
Amino Acid and Glucose Starvation of Tumor Cells
For amino acid starvation, tumor cells were washed twice with Earle's starvation buffer (HyClone) and then incubated in Earle's starvation buffer for various times before harvesting for analysis. For glucose starvation experiments, tumor cells were washed in glucose-free DMEM (catalog no. D5030, Sigma) and then incubated in this medium for various times prior to harvesting for analyses.
Gene Knockdown by shRNAs
Lentivirus vectors encoding various shRNAs and packaging vectors (pSPAX2 and pMD2.G) were purchased from the University of Michigan Biochemical Research Core. The shRNA vectors used in this study included shAkt#1 (V3LMM_497479) and shAkt#2 (V3LMM_497482) (both for Akt1). The shRNA vectors and the packaging plasmids were cotransfected into HEK293T cells. Two days after transfection, supernatants were harvested and passed through a sterile 0.45-μm filter to remove cell debris. Primary tumor cells were infected with the supernatants containing recombinant lentiviruses and Polybrene (8 μg/ml) three times every other day and then incubated in medium containing puromycin to select for pools of stable cells expressing the shRNAs.
Immunohistochemistry and Western Blot Analysis
Immunohistochemical staining using antibody against Phospho-S6 (P-S6, Ser-240/Ser-244) (catalog no. 2215, Cell Signaling Technology) was performed as described previously (24). For Western blot analysis using various antibodies, protein lysates were prepared from tumor cells or frozen tumor tissues that had been ground with a ceramic mortar and pestle on dry ice using Nonidet P-40 lysis buffer (5 ml of Nonidet P-40, 10 ml of 1 m Tris-HCl (pH 8), 4 g of NaCl, 50 ml of glycerol, 500 ml of 1 m Na3VO4, and 435 ml of double-distilled water) in the presence of protease inhibitor mixture (Sigma, catalog no. P2714) by sonication. SDS (0.2% w/v) was added to extract insoluble proteins such as p62. Then, protein lysates were loaded on an SDS-PAGE gel to separate different sizes of proteins by electrophoresis. Western blot analyses were quantified by densitometry using the ImageJ program. The following antibodies were used in this study: LC3 (catalog no. 0231-100/LC3-5F10) from NanoTools; β-actin (catalog no. A5441) from Sigma; p62 (catalog no. BML-PW9860) from Enzo Life Sciences; Erk (catalog no. Sc-94) and p70 S6 kinase (S6K) (catalog no. Sc-230) from Santa Cruz Biotechnology; and Tsc1 (catalog no. 4906S), phospho-p70 S6 kinase (P-S6K) (Thr-389) (catalog no. 9205S), phospho-Erk (P-Erk) (Thr-202/Tyr-204) (catalog no. 9101S), phospho-Akt (P-Akt) (Ser-473) (catalog no. 4060S), Akt (catalog no. 9272S), and cleaved caspase 3 (catalog no. 9661S) from Cell Signaling Technology.
Statistics
Student's t test was applied to analyze statistical significance. A value of p < 0.05 is indicative of statistical significance.
RESULTS
Tsc1 Deletion Increases Mammary Tumorigenesis and Metastasis
Tsc1 and Tsc2 are well known tumor suppressors whose loss-of-function mutations lead to the formation of benign tumors in different organs (1–3). However, the alterations of Tsc1 or Tsc2 and their potential roles and mechanisms in the development and progression of breast and other tissue malignancies are not well documented. Bioinformatics analysis of several datasets in the Oncomine databases (26) showed significantly decreased expression levels of Tsc1 mRNA in human breast cancer samples compared with that of normal breast tissues (Fig. 1A). This correlation suggests that low or diminished Tsc1 protein expression may have a role in breast cancer development and progression.
FIGURE 1.

Tsc1 is down-regulated in human breast cancers, and Tsc1 deletion increases multiplication, proliferation, migration, and invasion of primary mouse mammary tumor cells. A, microarray data of the relative expression levels of Tsc1 in human invasive breast ductal carcinoma versus normal breast tissues were extracted from Oncomine (26). The analyzed datasets include Radvanyi Breast, Richardson Breast 2, The Cancer Genome Atlas Breast, and Curtis Breast (see “Experimental Procedures” for information on the datasets). B, schematics for the generation of Ctrl and Tsc1 KO cells as described under “Experimental Procedures.” 4-OHT, 4-hydroxytamoxifen. C, genomic DNA was prepared from Ctrl and Tsc1 KO cells and then analyzed by PCR to detect Tsc1 floxed and deleted alleles. D, lysates of Ctrl and Tsc1 KO cells were prepared and analyzed by immunoblotting using antibodies against various proteins, as indicated. E–H, Ctrl and Tsc1 KO cells were analyzed for multiplication in culture (E), proliferation by Ki67 staining (F), migration using Boyden chambers (G), and invasion through Matrigel (H), as described under “Experimental Procedures.” ***, p < 0.001 when compared with the value of Ctrl cells.
To study the role of Tsc1 and mTOR signaling in breast cancer cells directly, we generated primary mammary tumor cells whose Tsc1 gene can be deleted in an inducible manner. The floxed Tsc1 mice (22) were crossed with the MMTV-PyMT transgenic mice model of human breast cancer (27) to generate Tsc1f/f;MMTV-PyMT mice, which developed metastatic breast cancer induced by the PyMT oncoprotein in a similar manner as the parental MMTV-PyMT mice (data not shown). Primary mammary tumor cells were isolated from these mice and then infected by recombinant retroviruses encoding CreER. As expected, treatment of these tumor cells with 4-hydroxytamoxifen-activated Cre recombinase, leading to the conversion of the floxed Tsc1 allele to a deleted allele (designated as Tsc1 KO cells), whereas mock treatment did not affect the floxed Tsc1 allele (designated as Ctrl cells) (Fig. 1, B and C). Consistent with the genotyping results, Western blotting of tumor cell lysates showed a significantly diminished Tsc1 expression in Tsc1 KO cells compared with Ctrl cells (Fig. 1D). Moreover, phosphorylation of p70 S6 kinase (P-S6K) was increased in Tsc1 KO cells relative to those in Ctrl cells, indicating the activation of mTORC1 upon the loss of the Tsc1-Tsc2 complex as a result of Tsc1 deletion in these cells as expected.
To determine the effect of Tsc1 deletion and consequent activation of mTORC1 on mammary tumorigenesis and metastasis, we first evaluated various cellular activities of Tsc1 KO and Ctrl cells in vitro. We found that Tsc1 KO cells showed a high rate of proliferation, whereas Ctrl cells multiplied quite slowly under the same culture conditions in vitro (Fig. 1E). Staining of the cells with Ki67 (a marker for proliferation) confirmed the significantly increased proliferation of Tsc1 KO cells compared with Ctrl cells (Fig. 1F). Interestingly, we also found increased migratory and invasive activity for Tsc1 cells relative to Ctrl cells (Fig. 1, G and H). We next examined the effect of Tsc1 deletion on tumor growth by transplanting Tsc1 KO and Ctrl cells into the mammary fat pads of recipient nude mice. Consistent with the increased proliferation in vitro (Fig. 1, E and F), mammary tumors derived from Tsc1 KO cells grew at a significantly higher rate compared with those from Ctrl cells in the recipient mice (Fig. 2A). Moreover, immunohistochemical staining of tumor sections showed an increased fraction of Ki67+ cells in tumors from Tsc1 KO cells compared with those from Ctrl cells (Fig. 2, B and C), suggesting that increased proliferation may be responsible for the higher growth rate of Tsc1-null mammary tumors.
FIGURE 2.
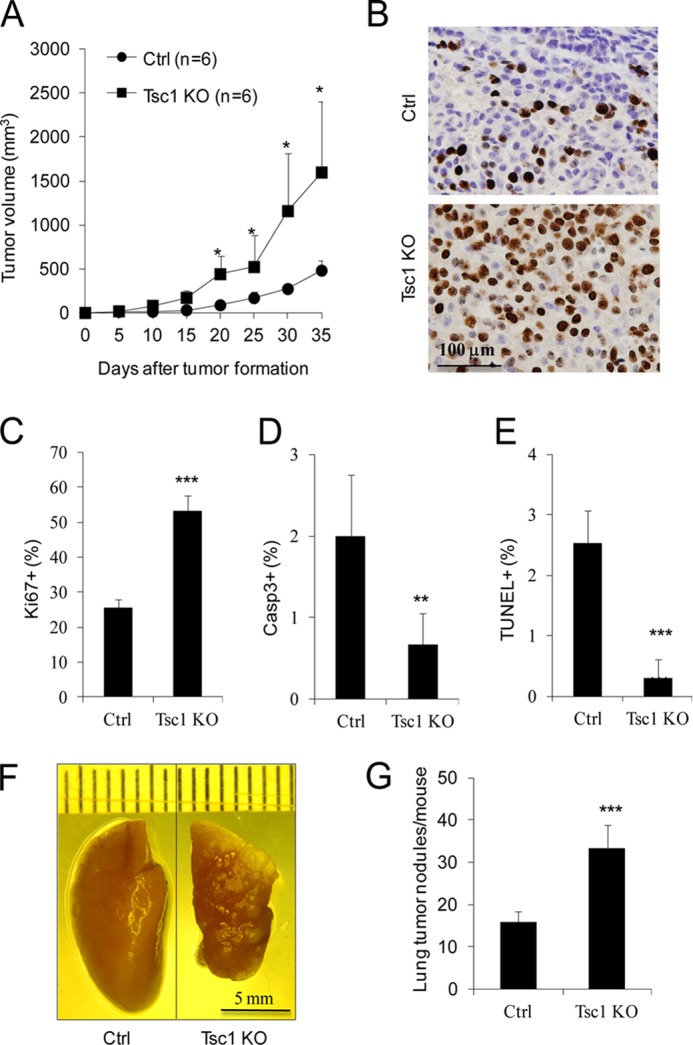
Tsc1 KO cells have increased mammary tumor growth and lung metastasis in recipient mice. Ctrl and Tsc1 KO cells were injected into the mammary fat pads of 8-week-old athymic nude mice. A, mean cumulative mammary tumor volume per mouse at different times after the initial primary tumor appearance. B and C, sections from primary tumors in the recipient mice were analyzed by immunohistochemistry using anti-Ki67. Representative images are shown in B. The means ± S.E. of the percentage of Ki67+ cells in the sections are shown in C (n = 4). D and E, sections from primary tumors in the recipient mice were analyzed by immunohistochemistry using anti-cleaved caspase 3 or TUNEL. The means ± S.E. of the percentage of Casp3+ (D) or TUNEL+ (E) cells in the sections are shown (n = 10). F and G, lungs were harvested from the recipient mice at 7 weeks after injection. Representative images are shown in F. The means ± S.E. of the number of metastatic nodules on the surfaces of lungs per mouse are shown in G (n = 7). *, p < 0.05; **, p < 0.01; ***, p < 0.001 when compared with the value of Ctrl cells.
We next examined the apoptosis index in the mammary tumors. Immunohistochemical staining of tumor sections showed a significantly reduced fraction of cells positive for cleaved caspase 3 labeling in Tsc1 KO tumors compared with that in Ctrl tumors (Fig. 2D). Likewise, a lower fraction of apoptotic cells was also found in sections of Tsc1 KO tumors compared with that in Ctrl tumors by TUNEL staining (Fig. 2E), indicating that increased cell survival also plays an important role in the elevated mammary tumor growth of Tsc1 KO cells. Lastly, we used tail vein injection of nude mice to determine the experimental metastatic potential of Tsc1 KO and Ctrl cells. As shown in Fig. 2, F and G, a significantly increased number of metastatic nodules was found in the lungs of recipient mice injected with Tsc1 KO cells compared with those injected with Ctrl cells. Taken together, these results suggest that the deletion of Tsc1 and the consequent increase in mTORC1 signaling promotes mammary tumor development and metastasis.
Sustained mTORC1 Hyperactivation Is Required for the Increased Tumor Growth of Tsc1 KO Cells in Vivo
To exclude a potential adaptive response of Tsc1 KO cells derived from in vitro deletion of the Tsc1 flox allele in culture, we also examined the effect of deletion of Tsc1 in vivo after transplantation (Fig. 3A). To this end, tumor cells from Tsc1f/f;MMTV-PyMT mice infected with recombinant retroviruses encoding CreER were injected to the no.4 inguinal mammary fat pads of nude mice. After tumors reached a size of about 2 mm in diameter, tamoxifen (Tam) was administrated for three consecutive days to the mice to induce deletion of the Tsc1 flox allele in vivo. We found that Tam treatment of the recipient mice increased the tumor growth rate compared with vehicle treatment (Fig. 3B), which is consistent with the observation made when Tsc1 was deleted before transplantation (see Fig. 2A). Similarly, Ki67 staining of tumor sections showed a significantly increased proliferation of tumor cells following Tam treatment compared with vehicle treatment (Fig. 3, C and D). We also observed an increased level of phosphorylated S6 (P-S6) in the tumors of Tam-treated recipient mice (Figs. 4, A and B), which is consistent with the activation of mTORC1 signaling in the tumors. Western blot analysis of tumor lysates showed an increased phosphorylated Akt (P-Akt) in the tumors following Tam treatment (Fig. 4, C and D). Lastly, mammary tumors derived from the Tam treatment group also showed reduced apoptosis compared with that from the vehicle group (Fig. 4, E and F). In contrast with cells with CreER expression, treatment of recipient mice that had been transplanted with Tsc1f/f;MMTV-PyMT tumor cells (but not infected by recombinant retroviruses encoding CreER) with Tam did not alter mammary tumor growth in these mice (data not shown).
FIGURE 3.

Deletion of Tsc1 in mammary tumor cells in vivo increases tumor growth in an mTORC1-dependent manner. A, schematics for the deletion of Tsc1 induced by tamoxifen (100 mg/kg) after transplantation of primary tumor cells followed by rapamycin (Rap) (1 mg/kg) treatment, as described under “Experimental Procedures.” B, mean cumulative mammary tumor volume per mouse at different times after the initial primary tumor appearance. The arrow marks the beginning of rapamycin injection every other day, starting at 20 days after the initial tumor appearance and tamoxifen injection. C and D, sections from primary tumors in the recipient mice were analyzed by immunohistochemistry using anti-Ki67. Representative images are shown in C. The means ± S.E. of the percentage of Ki67+ cells in the sections are shown in D (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001 when Tam-treated mice were compared with vehicle-treated mice or when Tam + Rap-treated mice were compared with Tam-treated mice.
FIGURE 4.
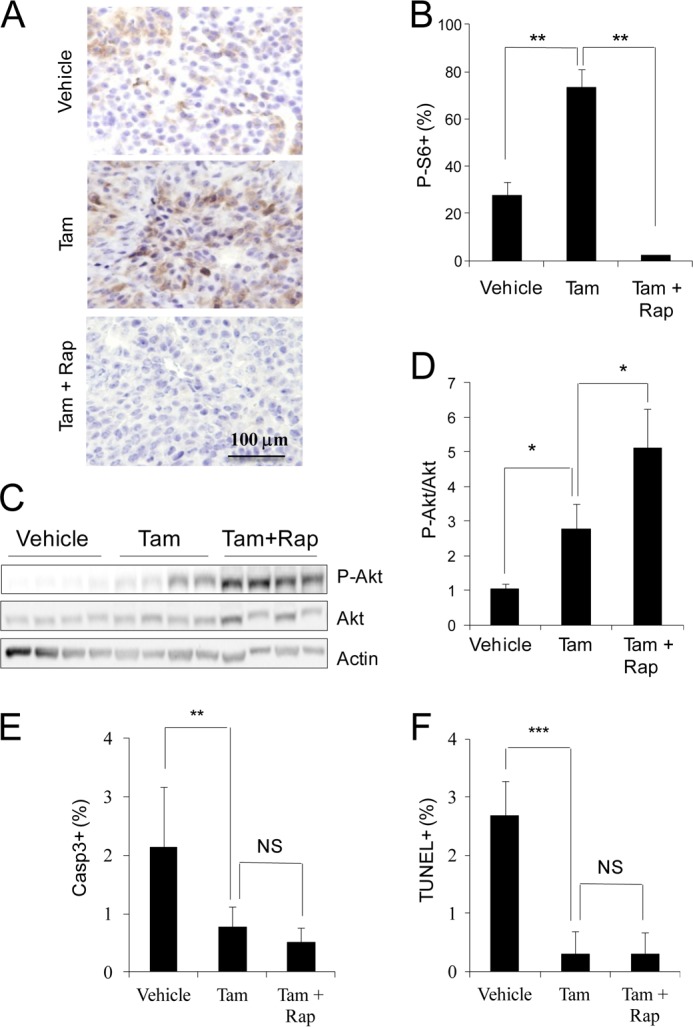
Analysis of tumor cell signaling and apoptosis following Tsc1 deletion in vivo. Mice were treated as outlined in Fig. 3A. A and B, sections from primary tumors in the recipient mice were analyzed by immunohistochemistry using anti-P-S6. Representative images are shown in A. The means ± S.E. of the percentage of P-S6+ cells in the sections are shown in B (n = 5). Rap, rapamycin. C and D, lysates were prepared from mammary tumors of recipient mice and then analyzed by immunoblotting using antibodies against various proteins, as indicated. Representative results are shown in C. The intensities of the P-Akt and Akt bands were determined from four independent experiments by densitometry. The means ± S.E. of the relative ratio of P-Akt/Akt (normalized to vehicle-treated mice) are shown in D. E and F, Sections from primary tumors in the recipient mice were analyzed by immunohistochemistry using anti-cleaved caspase 3 or TUNEL. The means ± S.E. of the percentage of Casp3+ (E) or TUNEL+ (F) cells in the sections are shown (n = 10). NS, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001 when Tam-treated mice were compared with Vehicle-treated mice or when Tam + Rap-treated mice were compared with Tam-treated mice.
To further validate whether the increased mammary tumor cell proliferation and growth are caused by hyperactivation of mTORC1, we employed rapamycin (a widely used mTORC1 inhibitor) to treat recipient mice with in vivo Tsc1 deletion and examined the effects on mammary tumor growth. As shown in Fig. 3B, rapamycin treatment significantly reduced mammary tumor growth in the recipient mice with Tam-induced Tsc1 deletion to a similar level as those in the control mice treated with vehicle alone (i.e. without Tsc1 deletion). Ki67 staining of the tumor sections also showed corresponding decreases in tumor cell proliferation by rapamycin treatment (Fig. 3, C and D). As expected, the elevated P-S6 level was abolished by rapamycin treatment (Fig. 4, A and B). Interestingly, rapamycin treatment led to a further increase in the level of P-Akt (Fig. 4, C and D), which was likely due to the feedback activation of mTORC2 upon inhibition of mTORC1 by rapamycin. However, the lower level of apoptosis of mammary tumors from Tsc1 KO cells was not affected by rapamycin treatment (Fig. 4, E and F). Together, these results suggest that deletion of Tsc1 after tumor development in vivo promoted their growth and that sustained mTORC1 activation is required for maintaining the increased mammary tumor growth in vivo.
Glucose Starvation-induced Autophagy Flux Is Increased in Tsc1 KO Cells
Recent studies have suggested a role of autophagy in promoting survival and proliferation of a number of cancer cells (21, 28–30). Interestingly, mTORC1 has been documented to play a major role in the regulation of autophagy (8, 31). Thus, we first examined autophagy in Tsc1 KO cells to evaluate the potential contribution of dysregulated autophagy to the increased tumor growth in our breast cancer mouse model. In growing cells, an elevated LC3-II was found both in the absence and presence of NH4Cl, a lysosomal inhibitor to block LC3-II degradation, in Tsc1 KO cells compared with those in Ctrl cells (Fig. 5A), indicating increased basal autophagy upon Tsc1 deletion in these cells. Moreover, the level of the autophagy substrate p62 was decreased in growing Tsc1 KO cells compared with Ctrl cells. P62 accumulated to a similarly higher level in both Tsc1 KO and Ctrl cells in the presence of NH4Cl, suggesting that the reduced p62 amount was caused by the increased basal autophagy in Tsc1 KO cells.
FIGURE 5.
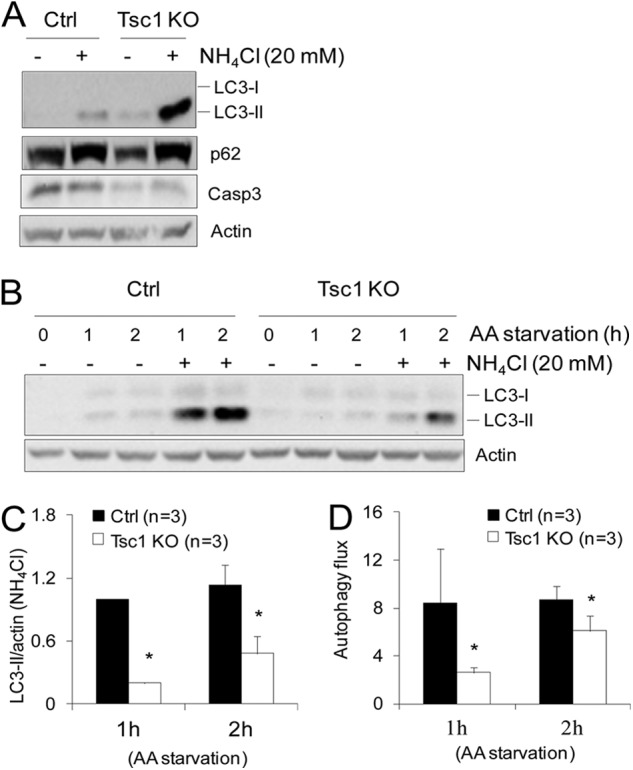
Analysis of autophagy of Tsc1 KO cells in growing and amino acid starvation conditions. A, Ctrl and Tsc1 KO cells were incubated in fresh growth medium in the absence or presence of 20 mm NH4Cl for 16 h. Lysates were then prepared and analyzed by immunoblotting using antibodies against various proteins as indicated. B–D, Ctrl and Tsc1 KO cells were subjected to amino acid (AA) starvation (i.e. incubation in Earle's starvation buffer) for various times in the absence or presence of 20 mm lysosomal inhibitor (NH4Cl), as indicated, as described under “Experimental Procedures.” Lysates were then prepared and analyzed by immunoblotting using antibodies against various proteins as indicated. Representative results are shown in B. The intensity of the LC3-II band (normalized to that of actin) was determined from three independent experiments by densitometry. The means ± S.E. of the relative autophagy activity (normalized to Ctrl cells at 1 h after starvation) are shown in C. The means ± S.E. of the autophagy flux (calculated as LC3-II/actin with NH4Cl divided by LC3-II/actin without NH4Cl) are shown in D. *, p < 0.05, when compared with the value of Ctrl cells.
To further explore the underlying mechanisms of increased autophagy in Tsc1 KO cells, we analyzed LC3-II levels as well as the calculated autophagy flux (defined as the value of LC3-II/actin in the presence of lysosomal inhibitor divided by LC3-II/actin in the absence of the inhibitor) (32) in these cells in response to amino acid or glucose starvation. We found that amino acid starvation-induced autophagy was decreased in Tsc1 KO cells compared with that in Ctrl cells, as measured by the accumulation of LC3-II in the presence of NH4Cl (Fig. 5, B and C) as well as by calculated autophagy flux (Fig. 5D), which is consistent with previous observations of negative regulation of autophagy by mTORC1 under nutrient depletion conditions (31). Interestingly, however, the autophagy activity as well as the autophagy flux induced by glucose starvation was increased in Tsc1 KO cells compared with that in Ctrl cells (Figs. 6, A–C). Moreover, we found that although amino acid starvation caused comparable levels of apoptosis in Ctrl and Tsc1 KO cells, glucose starvation-induced apoptosis was suppressed significantly in Tsc1 KO cells compared with that in Ctrl cells (Fig. 6D). Similarly reduced apoptosis was also found in growing Tsc1 KO cells compared with Ctrl cells (Fig. 5A), which is also consistent with the reduced apoptosis in tumors derived from Tsc1 KO cells (see Fig. 2, D and E). Together, these results suggest that, under growing conditions, Tsc1 deletion and consequent mTORC1 activation in mammary tumor cells resulted in increased autophagy, which could contribute to the elevated tumor cell survival and growth by decreasing cellular stress-induced apoptosis.
FIGURE 6.
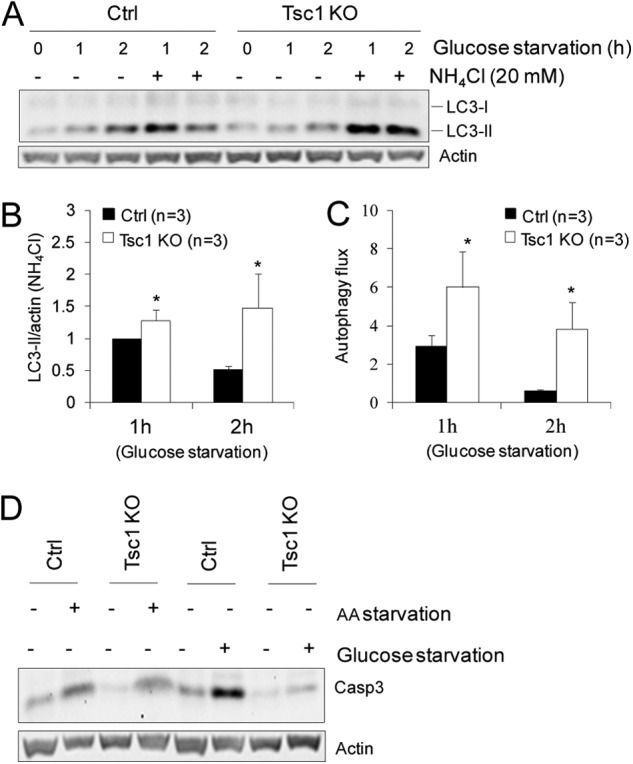
Tsc1 deletion increases autophagy and survival of mammary tumor cells in response to glucose starvation. A–C, Ctrl and Tsc1 KO cells were subjected to glucose starvation (i.e. incubation in glucose-free DMEM) for various times in the absence or presence of 20 mm lysosomal inhibitor (NH4Cl), as indicated, as described under “Experimental Procedures.” Lysates were then prepared and analyzed by immunoblotting using antibodies against various proteins, as indicated. Representative results are shown in A. Intensity of the LC3-II band (normalized to that of actin) was determined from three independent experiments by densitometry. The means ± S.E. of the relative autophagy activity (normalized to Ctrl cells at 1 h after starvation) are shown in B. The means ± S.E. of the autophagy flux (calculated as LC3-II/actin with NH4Cl divided by LC3-II/actin without NH4Cl) are shown in C. *, p < 0.05 when compared with the value of Ctrl cells. D, Ctrl and Tsc1 KO cells were incubated in normal medium (- lanes) or subjected to amino acid (AA) or glucose starvation (+ lanes) for 16 h. Lysates were then prepared and analyzed by immunoblotting using antibodies against various proteins, as indicated.
Role of Akt Activation in the Increased Mammary Tumor Growth upon Tsc1 Deletion
To examine other potential downstream signaling pathways that may play an important role in the increased tumor growth of Tsc1 KO cells, we analyzed the activation status of Erk and Akt in mammary tumors derived from Tsc1 KO and Ctrl cells in the recipient mice. Analysis of tumor lysates showed slightly (except one with a much higher level), but statistically not significant, increased Erk activation in tumors from Tsc1 KO cells compared with those from Ctrl cells without Tsc1 deletion (Fig. 7A). In contrast, significantly increased Akt activation was found in Tsc1 KO tumors compared with that in Ctrl tumors. Consistent with results obtained from tumor samples, we also observed increased Akt, but not Erk, activation in primary Tsc1 KO cells compared with Ctrl cells (Fig. 7B). The increased Akt activation in Tsc1 KO cells was also observed under amino acid- and glucose-starved conditions (Fig. 7, C and D), although the elevated P-S6K in these cells was only found under growing and glucose-starvation conditions but not in amino acid starvation condition (Figs. 7, C and E, and 1D). We next examined the potential role of the elevated Akt signaling in the increased survival by employing lentiviral shRNA to deplete Akt in Tsc1 KO cells. As shown in Fig. 8A, infection of Tsc1 KO cells with lentiviral vectors encoding shRNA targeting two different Akt sequences both reduced the expression and phosphorylation of Akt in these cells. Inhibition of Akt signaling by knockdown significantly increased apoptosis of Tsc1 KO cells (Fig. 8A, bottom two panels) and also decreased the elevated multiplication of these cells (B). Together, these results suggest that increased Akt signaling and tumor cell survival also contribute to the promotion of mammary tumor growth by Tsc1 deletion and consequent mTORC1 activation in vivo.
FIGURE 7.
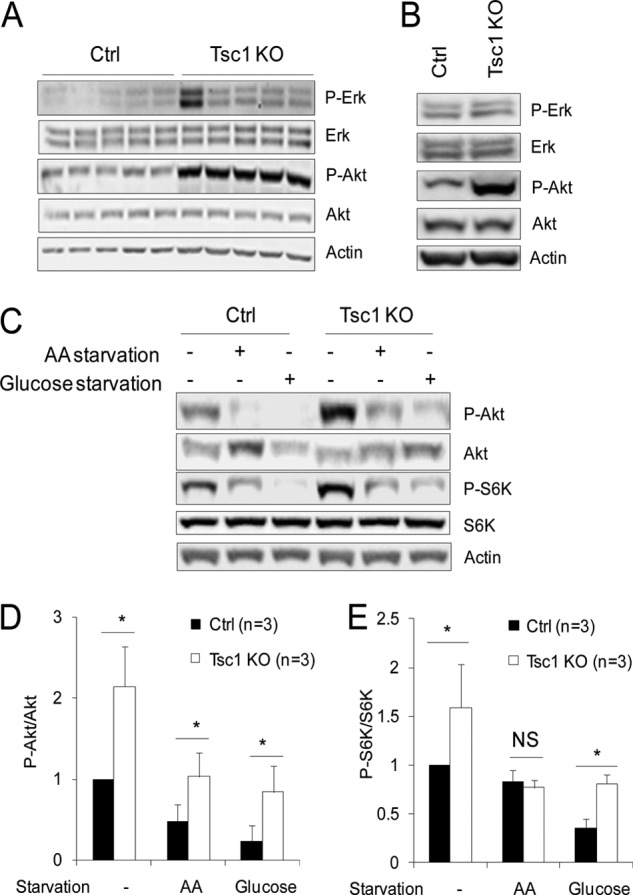
Increased Akt activation in Tsc1 KO cells. A and B, lysates were prepared from mammary tumors of recipient mice developed from transplanted Ctrl and Tsc1 KO cells (A) or Ctrl and Tsc1 KO cells directly (B). They were then analyzed by immunoblotting using antibodies against various proteins, as indicated. C–E, Ctrl and Tsc1 KO cells were subjected to amino acid (AA) (i.e. incubation in Earle's starvation buffer) or glucose (i.e. incubation in glucose-free DMEM) starvation for 24 h, as indicated, as described under “Experimental Procedures.” Lysates were then prepared and analyzed by immunoblotting using antibodies against various proteins, as indicated. Representative results are shown in C. The intensities of the P-Akt, Akt, P-S6K, and S6K bands were determined from three independent experiments by densitometry. The means ± S.E. of the relative ratio of P-Akt/Akt and P-S6K/S6K (normalized to Ctrl cells without starvation) are shown in D and E, respectively. NS, not significant; *, p < 0.05 when compared with the value of Ctrl cells.
FIGURE 8.

Role of Akt activation in the increased survival and multiplication of Tsc1 KO cells. Tsc1 KO cells were infected with recombinant lentiviruses encoding Akt1 shRNA or a control shRNA. Lysates from aliquots of the infected cells were analyzed by immunoblotting using antibodies against various proteins as indicated (A, top three panels). The cells were also analyzed for apoptosis after 4 days in culture by immunoblotting using antibodies against cleaved caspase 3 (A, bottom two panels) and multiplication in culture (B). **, p < 0.01, when compared with the value of Tsc1 KO cells infected by control shRNA.
DISCUSSION
Loss-of-function mutations in Tsc1 or Tsc2 and consequent activation of mTORC1 signaling has been implicated in a variety of diseases, including the development of benign tumors in different organs (1–3). Recent studies also showed that mTORC1 is frequently hyperactivated by overexpression of its upstream positive regulator Rheb in human cancers (33), suggesting that activation of mTORC1 might play a key role in the rapid growth of human cancers. Interestingly, our bioinformatics analyses showed that, in human breast carcinoma, Tsc1 expression is significantly lower compared with that in normal breast tissues, raising the possibility that Tsc1 down-regulation also contributes to hyperactivation of mTORC1 in breast cancers. By generating and analyzing a unique model with inducible deletion of Tsc1 in mouse primary mammary tumor cells, we show here a novel mechanism showing that deletion of Tsc1 and consequent activation of mTORC1 promotes mammary tumor growth through increased autophagy under glucose starvation conditions as well as increased Akt activation.
Recent studies suggested that both the mTORC1 and mTORC2 complexes regulate a variety of downstream pathways and cellular activities (34), which could conceivably promote mammary tumor growth as observed in our studies. However, as rapamycin could fully reverse the increased growth of Tsc1-null tumors, hyperactivation of mTORC1 likely play a major role in the increased mammary tumor growth and metastasis in the recipient mice. It is well known that mTORC1-mediated phosphorylation of S6K and 4EBP1 promotes protein translation and cell growth, which are important in proliferation of many normal and cancer cells (6, 35, 36). This study suggest the additional interesting mechanism that elevated activation of Akt signaling and increased survival of Tsc1-null mammary tumor cells is also important for the increased progression and metastasis of these cells. These results are consistent with previous observations that Akt1 deficiency inhibited breast cancer proliferation and migration (37) and that Tsc1 and Tsc2 were down-regulated, whereas Akt1 was up-regulated, in human oral squamous cell carcinoma (38).
Although the precise mechanisms are not well understood at present, our results also suggest a role for the altered autophagy in mediating cell survival under glucose starvation conditions in Tsc1-null tumor cells, which likely contributes to their increased survival in vitro and tumorigenicity and metastasis in vivo. Autophagy has been shown to play dual roles in either suppressing or promoting tumorigenesis and progression in previous studies (21, 28, 29, 39, 40), which are likely dependent on the context of the stage of cancer progression, types, or other factors. Consistent with previous studies (31), we found that Tsc1 deletion and consequent mTORC1 hyperactivation reduced amino acid starvation-induced autophagy in Tsc1-null tumor cells. Surprisingly, however, we found that glucose starvation-induced autophagy was elevated in these cells. More importantly, Tsc1-null tumor cells exhibited increased autophagy under growing conditions, likely caused by the rapid uptake of glucose during the growth of cancer cells that might lead to “glucose starvation,” even in growth medium, because of the addiction of cancer cells to glucose uptake and aerobic glycolysis (i.e. the Warburg effect) (41, 42). These results are consistent with previous findings that autophagy increased tumor cell growth (21, 28, 29, 43). Future studies will be required to investigate the interesting possibility that Tsc1 deletion in mammary tumor cells may promote Akt phosphorylation by a novel mechanism through the increased autophagy under glucose starvation and/or other stress conditions in these cells.
Previous studies using breast cancer cell lines demonstrated that loss-of-function Tsc1 mutation stimulated cancer progression by up-regulation of VEGF production to induce tumor angiogenesis (17). Although our studies focused on an intrinsic role of mTORC1 hyperactivation in the promotion of mammary tumor cell proliferation in Tsc1-null tumor cells, potentially increased tumor angiogenesis in the recipient mice could also contribute to increased breast cancer growth and metastasis in vivo. Although future studies will be necessary to fully understand the mechanisms, these results provide further support for targeting Tsc/mTOR signaling in cancer therapy because they may be very effective by blocking both tumor cell growth and tumor angiogenesis through intrinsic and autocrine functions of hyperactivated mTORC1.
Acknowledgments
We thank Dr. David J. Kwiatkowski (Harvard Medical School) for Tsc1f/f mice and Dr. Xiaokui Mo (Ohio State University) for help with statistics. We also thank Drs. Huaping Fan, Ming Luo, Shaogang Sun, and Chenran Wang for help with various experimental procedures and members of our laboratory for discussions, critical reading of the manuscript, and comments.
This research was supported, in whole or in part, by National Institutes of Health Grants CA150926 and CA163493 (to J. L. G.).
- Tsc
- tuberous sclerosis complex
- mTOR
- mammalian target of rapamycin
- mTORC
- mammalian target of rapamycin complex
- Ctrl
- control
- Tam
- tamoxifen
- P-Akt
- phospho-Akt.
REFERENCES
- 1. Kwiatkowski D. J. (2003) Tuberous sclerosis. From tubers to mTOR. Ann. Hum. Genet. 67, 87–96 [DOI] [PubMed] [Google Scholar]
- 2. Inoki K., Guan K. L. (2009) Tuberous sclerosis complex. Implication from a rare genetic disease to common cancer treatment. Hum. Mol. Genet. 18, R94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orlova K. A., Crino P. B. (2010) The tuberous sclerosis complex. Ann. N.Y. Acad. Sci. 1184, 87–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 5. Alessi D. R., Pearce L. R., García-Martínez J. M. (2009) New insights into mTOR signaling. mTORC2 and beyond. Sci. Signal 2, pe27. [DOI] [PubMed] [Google Scholar]
- 6. Hay N., Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 [DOI] [PubMed] [Google Scholar]
- 7. Yu Y., Yoon S. O., Poulogiannis G., Yang Q., Ma X. M., Villén J., Kubica N., Hoffman G. R., Cantley L. C., Gygi S. P., Blenis J. (2011) Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 332, 1322–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan E. Y. (2009) mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal 2, pe51. [DOI] [PubMed] [Google Scholar]
- 9. Mizushima N., Komatsu M. (2011) Autophagy. Renovation of cells and tissues. Cell 147, 728–741 [DOI] [PubMed] [Google Scholar]
- 10. Chen Y., Klionsky D. J. (2011) The regulation of autophagy. Unanswered questions. J. Cell Sci. 124, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Choi A. M., Ryter S. W., Levine B. (2013) Autophagy in human health and disease. N. Engl. J. Med. 368, 651–662 [DOI] [PubMed] [Google Scholar]
- 12. White E. (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rubinsztein D. C., Mariño G., Kroemer G. (2011) Autophagy and aging. Cell 146, 682–695 [DOI] [PubMed] [Google Scholar]
- 14. Hoshii T., Tadokoro Y., Naka K., Ooshio T., Muraguchi T., Sugiyama N., Soga T., Araki K., Yamamura K., Hirao A. (2012) mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Invest. 122, 2114–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Menon S., Yecies J. L., Zhang H. H., Howell J. J., Nicholatos J., Harputlugil E., Bronson R. T., Kwiatkowski D. J., Manning B. D. (2012) Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci. Signal 5, ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kladney R. D., Cardiff R. D., Kwiatkowski D. J., Chiang G. G., Weber J. D., Arbeit J. M., Lu Z. H. (2010) Tuberous sclerosis complex 1. An epithelial tumor suppressor essential to prevent spontaneous prostate cancer in aged mice. Cancer Res. 70, 8937–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee D. F., Kuo H. P., Chen C. T., Hsu J. M., Chou C. K., Wei Y., Sun H. L., Li L. Y., Ping B., Huang W. C., He X., Hung J. Y., Lai C. C., Ding Q., Su J. L., Yang J. Y., Sahin A. A., Hortobagyi G. N., Tsai F. J., Tsai C. H., Hung M. C. (2007) IKK β suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455 [DOI] [PubMed] [Google Scholar]
- 18. Radvanyi L., Singh-Sandhu D., Gallichan S., Lovitt C., Pedyczak A., Mallo G., Gish K., Kwok K., Hanna W., Zubovits J., Armes J., Venter D., Hakimi J., Shortreed J., Donovan M., Parrington M., Dunn P., Oomen R., Tartaglia J., Berinstein N. L. (2005) The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 102, 11005–11010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richardson A. L., Wang Z. C., De Nicolo A., Lu X., Brown M., Miron A., Liao X., Iglehart J. D., Livingston D. M., Ganesan S. (2006) X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 9, 121–132 [DOI] [PubMed] [Google Scholar]
- 20. Curtis C., Shah S. P., Chin S. F., Turashvili G., Rueda O. M., Dunning M. J., Speed D., Lynch A. G., Samarajiwa S., Yuan Y., Gräf S., Ha G., Haffari G., Bashashati A., Russell R., McKinney S., METABRIC Group, Langerød A., Green A., Provenzano E., Wishart G., Pinder S., Watson P., Markowetz F., Murphy L., Ellis I., Purushotham A., Børresen-Dale A. L., Brenton J. D., Tavaré S., Caldas C., Aparicio S. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei H., Wei S., Gan B., Peng X., Zou W., Guan J. L. (2011) Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 25, 1510–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uhlmann E. J., Wong M., Baldwin R. L., Bajenaru M. L., Onda H., Kwiatkowski D. J., Yamada K., Gutmann D. H. (2002) Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 52, 285–296 [DOI] [PubMed] [Google Scholar]
- 23. Shen T. L., Park A. Y., Alcaraz A., Peng X., Jang I., Koni P., Flavell R. A., Gu H., Guan J. L. (2005) Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell Biol. 169, 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo M., Fan H., Nagy T., Wei H., Wang C., Liu S., Wicha M. S., Guan J. L. (2009) Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 69, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan H., Guan J. L. (2011) Compensatory function of Pyk2 protein in the promotion of focal adhesion kinase (FAK)-null mammary cancer stem cell tumorigenicity and metastatic activity. J. Biol. Chem. 286, 18573–18582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abbas H. A., Maccio D. R., Coskun S., Jackson J. G., Hazen A. L., Sills T. M., You M. J., Hirschi K. K., Lozano G. (2010) Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7, 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guy C. T., Cardiff R. D., Muller W. J. (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene. A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 12, 954–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J. M., Dell'antonio G., Mautner J., Tonon G., Haigis M., Shirihai O. S., Doglioni C., Bardeesy N., Kimmelman A. C. (2011) Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lock R., Roy S., Kenific C. M., Su J. S., Salas E., Ronen S. M., Debnath J. (2011) Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kon M., Kiffin R., Koga H., Chapochnick J., Macian F., Varticovski L., Cuervo A. M. (2011) Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 3, 109ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Castets P., Lin S., Rion N., Di Fulvio S., Romanino K., Guridi M., Frank S., Tintignac L. A., Sinnreich M., Ruegg M. A. (2013) Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744 [DOI] [PubMed] [Google Scholar]
- 32. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., Al-Younes H. M., Al-Zeer M. A., Albert M. L., Albin R. L., Alegre-Abarrategui J., Aleo M. F., Alirezaei M., Almasan A., Almonte-Becerril M., Amano A., Amaravadi R., Amarnath S., Amer A.O., Andrieu-Abadie N., Anantharam V., Ann D. K., Anoopkumar-Dukie S., Aoki H., Apostolova N., Arancia G., Aris J. P., Asanuma K., Asare N. Y., Ashida H., Askanas V., Askew D. S., Auberger P., Baba M., Backues S. K., Baehrecke E. H., Bahr B. A., Bai X. Y., Bailly Y., Baiocchi R., Baldini G., Balduini W., et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu Z. H., Shvartsman M. B., Lee A. Y., Shao J. M., Murray M. M., Kladney R. D., Fan D., Krajewski S., Chiang G. G., Mills G. B., Arbeit J. M. (2010) Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 70, 3287–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR. From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dowling R. J., Topisirovic I., Alain T., Bidinosti M., Fonseca B. D., Petroulakis E., Wang X., Larsson O., Selvaraj A., Liu Y., Kozma S. C., Thomas G., Sonenberg N. (2010) mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guertin D. A., Sabatini D. M. (2007) Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 [DOI] [PubMed] [Google Scholar]
- 37. Ju X., Katiyar S., Wang C., Liu M., Jiao X., Li S., Zhou J., Turner J., Lisanti M. P., Russell R. G., Mueller S. C., Ojeifo J., Chen W. S., Hay N., Pestell R. G. (2007) Akt1 governs breast cancer progression in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 7438–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chakraborty S., Mohiyuddin S. M., Gopinath K. S., Kumar A. (2008) Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer 8, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathew R., Karp C. M., Beaudoin B., Vuong N., Chen G., Chen H. Y., Bray K., Reddy A., Bhanot G., Gelinas C., Dipaola R. S., Karantza-Wadsworth V., White E. (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takamura A., Komatsu M., Hara T., Sakamoto A., Kishi C., Waguri S., Eishi Y., Hino O., Tanaka K., Mizushima N. (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 25, 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Annibaldi A., Widmann C. (2010) Glucose metabolism in cancer cells. Curr. Opin. Clin. Nutr. Metab. Care 13, 466–470 [DOI] [PubMed] [Google Scholar]
- 42. Shaw R. J. (2006) Glucose metabolism and cancer. Curr. Opin. Cell Biol. 18, 598–608 [DOI] [PubMed] [Google Scholar]
- 43. Amaravadi R. K. (2011) Cancer. Autophagy in tumor immunity. Science 334, 1501–1502 [DOI] [PubMed] [Google Scholar]