

Background: The role of CD14 on TLR4 signaling in epithelial cells is not well understood.
Results: Blockade of CD14 inhibits TLR4 internalization and IRF3 signaling in human epithelial cells.
Conclusion: CD14 regulates TLR4 internalization and signaling in corneal epithelial cells and limits the severity of Pseudomonas aeruginosa corneal infection.
Significance: These data provide increased understanding of CD14 and TLR4 signaling in epithelial cells.
Keywords: Cornea, Epithelial Cell, Inflammation, Lipopolysaccharide (LPS), Neutrophil, Pseudomonas aeruginosa, Toll-like Receptors (TLRs)
Abstract
In the current study, we examined the role of CD14 in regulating LPS activation of corneal epithelial cells and Pseudomonas aeruginosa corneal infection. Our findings demonstrate that LPS induces Toll-like receptor 4 (TLR4) internalization in corneal epithelial cells and that blocking with anti-CD14 selectively inhibits TLR4 endocytosis, spleen tyrosine kinase (Syk) and IRF3 phosphorylation, and production of CCL5/RANTES and IFN-β, but not IL-8. Using a murine model of P. aeruginosa corneal infection, we show that although infected CD14−/− corneas produce less CCL5, they exhibit significantly increased CXC chemokine production, neutrophil recruitment to the corneal stroma, and bacterial clearance than C57BL/6 mice. We conclude that CD14 has a critical role in mediating TLR4 signaling through IRF3 in resident corneal epithelial cells and macrophages and thereby modulates TLR4 cell surface activation of the MyD88/NF-κB/AP-1 pathway and production of CXC chemokines and neutrophil infiltration to infected tissues.
Introduction
LPS activation of the TLR43/MD-2 receptor complex results in two distinct pathways based on activation of TIR containing adaptor molecules: the rapid TIRAP/MyD88 pathway from cell surface TLR4/MD-2 that activates NF-κB and AP-1, resulting in expression of CXC chemokines that recruits neutrophils; and the TRAM/TRIF pathway following internalization of TLR4/MD-2 to endosomes and activation of IRF1 and expression of type I IFN and CCL5/RANTES (1, 2).
CD14 was the first LPS-binding protein identified and was thought to be the LPS receptor prior to the discovery of TLR4 (1, 3). CD14 is a glycosylphosphatidylinositol-anchored protein and therefore cannot directly induce cell signaling; however, CD14 and lipid A-binding protein are accessory molecules to the TLR4/MD-2 receptor, and CD14 binds LPS and transfers it to MD-2 to initiate TLR4 dimerization and signaling (4, 5). In the absence of CD14, LPS-induced MyD88/TIRAP signaling is only partially reduced, whereas TRAM/TRIF signaling is completely impaired (6, 7). TLR4 internalization leads to activation of the TRIF/TRAM pathway (8), and this selective activity on macrophages and dendritic cells was recently shown to be mediated by CD14 through a spleen tyrosine kinase (Syk) and phospholipase Cγ-dependent pathway (7).
Although macrophages and dendritic cells have an important role in the host response to LPS and infection with Gram-negative bacteria, epithelial cells are generally the first cells that are exposed to LPS from the environment. Under homeostatic conditions, epithelial cells lining the gut, lungs, and ocular surface down-regulate LPS responsiveness; however, epithelial cells can respond directly to LPS following infectious and inflammatory conditions. We recently demonstrated that IFN-γ induces JAK2/STAT1 activation in corneal epithelial cells, which in turn leads to binding of STAT1 to GAS sites on the MD-2 promoter and induces transcription and cell surface localization of MD-2 and confers LPS responsiveness (9). Similar findings were reported for gut, lung, and conjunctival epithelial cells (10, 11), indicating that this pathway is common among epithelial cells. We also showed that corneal epithelial cells utilize the TRIF pathway when activated by TLR3 (12).
In the present study, we demonstrate that CD14 is expressed on corneal epithelial cells and is required for TLR4 internalization. We also show that blockade of CD14 inhibits IRF3, but not NF-κB phosphorylation, and is dependent on Syk activation. Using a murine model of Pseudomonas aeruginosa corneal infection, we found significantly elevated neutrophil infiltration and corneal opacification in the absence of CD14, which we attribute to impaired TLR4 internalization and consequently increased CXC chemokine production in the infected corneas. CD14-mediated TLR4 internalization and activation of the TRIF/IRF3 pathway therefore have an important role in bacterial infections by modulating MyD88/NF-κB/AP-1-mediated production of CXC chemokines and neutrophil-mediated inflammation and resulting tissue damage.
MATERIALS AND METHODS
Source of Reagents
Keratinocyte serum-free medium, trypsin, Hanks' balanced salt solution, and gentamycin were purchased from Invitrogen. The Ultrapure LPS was purchased from Invivogen. Anti-CD4 antibodies were from ELISA detection kits for IL-6, CXCL8/IL-8, CXCL1, IL-1β, and IFN-β, and recombinant human IFN-γ was purchased from R&D Systems. Peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were obtained from Santa Cruz Biotechnology. Blocking antibody to CD14 and all of the antibodies used for flow cytometry analysis were from eBiosciences (San Diego, CA). Piceatannol was purchased from Sigma.
Animals
C57BL/6 and CD14−/− mice (4–6 weeks old) were purchased from the Jackson Laboratory. All animals were housed in specific pathogen-free conditions in microisolator cages. All protocols were approved by Case Western Reserve University IACUC.
Mouse Models of Corneal Infection
Mice were anesthetized, and corneal epithelium was abraded with three parallel 1-mm scratches using a 26-gauge needle and infected with 2.5 μl of PBS containing 1 × 105 colony-forming units (cfu) of P. aeruginosa strain PAO1 as described (13). Mice were examined under a stereomicroscope for corneal opacification, ulceration, and perforation at 24 or 48 h after infection.
Imaging Corneal Opacity
Mice were euthanized, and corneal opacification (bright field) was visualized in the intact eye using MZFLIII microscope (Leica Microsystems) and Spot RT Slider KE camera (Diagnostics Instruments). All images were captured using SpotCam software (RT Slider KE).
Colony-forming Unit Quantification
At 24 and 48 h after infection, mice were euthanized, and the whole eye was homogenized in 1 ml of PBS using the Mixer Mill MM300 (Retsch, Newtown, PA) at 33 Hz for 4 min. Serial log dilutions of the eye homogenate were plated onto brain heart infusion agar plates. After overnight incubation at 37 °C, cfu were determined by direct counting.
Histology
Eyes were enucleated and placed in 10% formalin/PBS, 5-mm corneal sections were prepared, and H&E staining was performed as described (13). In brief, H&E staining was performed using Gill's hematoxylin for 3–5 min following deparaffinization. The slides were dipped in glacial acetic acid, stained with Shandon Bluing solution (Thermo Shandon, Pittsburgh, PA), and counterstained with alcohol eosin Y solution (Thermo Shandon). Slides were then dehydrated and mounted under Permount medium (Vector Laboratories).
Corneal Epithelial Cell Lines
The SV40-immortalized human corneal epithelial cell line 10.014 pRSV-T (HCEC) was obtained from the ATCC (Manassas, VA) and maintained by culturing in keratinocyte serum-free medium with bovine pituitary extract and recombinant human epidermal growth factor (Invitrogen) at 37 °C and 5% CO2. The hTCEpi cell lines are telomerase-immortalized human corneal epithelial cells (HCET) and were obtained from Dr. James Jester, University of California, Irvine, who generated them (14).
Primary Human Corneal Epithelial Cells
Primary cells were isolated from donor corneas obtained from Midwest Eye Banks (Chicago, IL). Tissue procurement was approved by the Case Western Reserve University (Cleveland, OH)/University Hospitals of Cleveland Institutional Review Board. Bulbar conjunctival tissue was removed, and cornea was excised and placed in sterile Hanks' balanced salt solution containing 10 mg/ml dispase and 5 mg/ml gentamycin for 4 h at 4 °C. The corneal epithelium was then collected by gentle scraping and incubation with 5 ml of 0.25% trypsin, 5 min at 37 °C. The epithelial cell suspension was transferred to DMEM containing 10% FCS to block further enzyme activity. Epithelial cells from each cornea were then collected by centrifugation and resuspended in keratinocyte serum-free medium containing epithelial growth factor and bovine pituitary extract with antibiotics. After passage 1, antibiotics were omitted from the culture medium, and cells were harvested and transferred to a 50-cm2 flask. The medium was changed every 4 days, and cells from passages 2–5 were used for experiments when cells were 70% confluent (15).
Isolation of Bone Marrow-derived Macrophages (BMDMs)
Bone marrow cells were derived from C5BL/6 and CD14−/− mice as described (13). In brief, following euthanasia, femurs and tibia were removed, cleaned, and centrifuged at 10,000 rpm for 30 s. The pellet was resuspended in RBC lysis buffer for 3 min and recovered by centrifugation at 1200 rpm for 10 min. Cells were incubated for 10 days in DMEM containing 5% FBS and 30% L929 medium during which they differentiated into macrophages (12). Macrophages were then incubated with live or antibiotic-killed PAO1 at a 10:1 ratio of bacteria to macrophages and processed for Western blotting and ELISA.
Cytokine ELISA
Infected and PBS-treated corneas were excised and homogenized in 150 μl of PBS using a Mixer Mill MM300 (Retsch). BMDMs were grown overnight and incubated with antibiotic-killed PAO1 for 4 h, and the supernatants were used directly. Human corneal cells were grown overnight in 12-well plates (4 × 105 cells/well), treated with LPS, and supernatants were collected. Cytokine were determined by sandwich ELISA according to the manufacturer's protocol (R&D Systems). Absorption was measured at 450 nm on a Versa Max microplate reader using SoftMaxPro software 5.2 (Molecular Devices).
Western Blot Analysis
BMDM from C57BL/6 and CD14−/− mice were incubated with PAO1 at a 10:1 ratio of bacteria to macrophages for indicated times. The cells were washed with ice-cold PBS and lysed using cold 1× lysis buffer (Cell Signaling Technology). Total protein was quantified using standard BCA technique (Thermo Scientific), and 20 μg of protein was separated by 12% SDS-polyacrylamide gel, transferred to nitrocellulose membrane, and probed with p-IκBα, total IκBα, p-IRF3, total IRF3, p-ERK, total ERK, p-JNK, total JNK, p-p38, total p38, and, β-actin (Cell Signaling Technology). All of the primary antibodies were used in 1:1000 dilutions. Proteins were detected using HRP-conjugated secondary antibodies and developed with Supersignal West Femto Maximum Sensitivity Substrate (Pierce).
p65 Nuclear Translocation
BMDMs were cultured as 5 × 104 cells/well in 12-well plates on sterile coverslips and incubated with P. aeruginosa at a 10:1 ratio of bacteria to epithelial cells for 60 min. Translocation of the NF-κB p65 subunit was determined as described (13, 16). Briefly, cells were fixed with 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.1% Triton X-100, and incubated with rabbit anti-mouse p65 (1:100; eBioscience). p65 binding was detected using Alexa Fluor 488-labeled goat anti-rabbit IgG antibody (Molecular Probes).
Flow Cytometry
Corneas dissected 24 and 48 h after infection were processed for flow cytometry as described (13). Briefly, after digestion in collagenase, Fc receptors were blocked for 20 min followed by incubation with Alexa Fluor 488-NIMP-R14 for 45 min to detect neutrophils. The cells were then washed and fixed with 1% paraformaldehyde. For staining of epithelial cells, HCEC, HCET, and primary human corneal epithelial cells were incubated with anti-CD14 antibody for 2 h followed by incubation with LPS for the indicated time periods. The cells were then incubated with human IgG (20 μg/ml) for 15 min followed by incubation with anti-TLR4 (eBioscience) or isotype control antibody for 45 min at 4 °C. Cells were fixed with 1% paraformaldehyde and analyzed using an Accuri c6 flow cytometry.
Immunocytochemistry
Human corneal epithelial cells were incubated for 2 h with IFN-γ (250 ng/ml) and then stimulated for 1 h with LPS (1 μg/ml). For extracellular staining, cells were blocked for 1 h in 10% goat serum and incubated for 1 h with mouse TLR4 antibody (Abcam) followed by incubation with Alexa Fluor 488 goat anti-mouse IgG for 40 min. After three washes with PBS, cells were counterstained with DAPI and visualized by fluorescence microscopy. For intracellular staining, cells were first fixed in 4% paraformaldehyde followed by permeabilization using 0.1% Triton X-100 and then stained as described above. Cells were examined using a Leica DM6000B inverted microscope.
Statistics
Statistical analysis was performed using an unpaired t test (Prism; GraphPad Software). p values < 0.05 were considered significant.
RESULTS
CD14 Regulates LPS-induced TLR4 Endocytosis in Human Corneal Epithelial Cells
Our previous studies showed that although TLR4 is constitutively expressed by corneal epithelial cells, LPS responsiveness is dependent on IFN-γ induction of MD-2 (9, 16), which forms the functional lipid A receptor. Therefore, in the experiments described below, corneal epithelial cells were stimulated with LPS in the presence of recombinant IFN-γ.
Fig. 1 shows TLR4 staining by confocal microscopy. Unstimulated HCET cells exhibit a cell surface TLR4 staining pattern consistent with the known presence of TLR4 on lipid rafts (17). In contrast, after a 1-h stimulation with LPS and IFN-γ, TLR4 staining is primarily intracellular.
FIGURE 1.
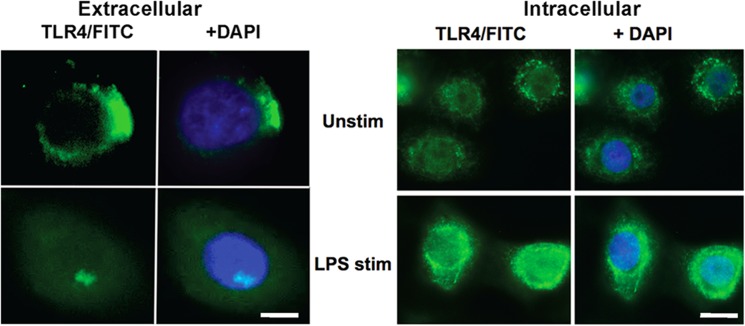
TLR4 is internalized in corneal epithelial cells following LPS activation. Extracellular and intracellular TLR4 expression on unstimulated (Unstim) human corneal epithelial cells and following incubation with LPS and IFN-γ. DAPI stain shows cell nucleus. Scale bars on left panels, 5 μm; on right panels, 10 μm.
To determine whether LPS induces CD14 expression on corneal epithelial cells, HCEC and HCET cell lines were incubated with LPS, and CD14 expression was detected by flow cytometry. As shown in Fig. 2A, CD14 is expressed on unstimulated HCEC and HCET cells and is not further elevated after stimulation with LPS, demonstrating that the CD14 is constitutively expressed on the cell surface of human corneal epithelial cells.
FIGURE 2.
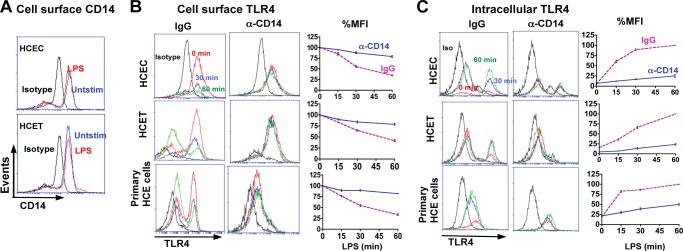
Role of CD14 in TLR4 internalization in corneal epithelial cells. A, constitutive expression of CD14 on the surface of HCEC and HCET human corneal epithelial cell lines analyzed by flow cytometry after 4-h incubation with LPS and IFN-γ. B, reduction in cell surface TLR4 after 15-min, 30-min, and 1-h incubation with LPS together with either control IgG or blocking anti-CD14. Left panels show representative profiles, and right panels show time course as a percentage of initial mean fluorescence intensity (MFI). C, intracellular TLR4 under the same conditions. These experiments were repeated three times with similar results.
As CD14 regulates TLR4 endocytosis in murine macrophages and dendritic cells (7), we examined whether CD14 has a similar role in epithelial cells. HCEC, HCET, and primary human corneal epithelial cells were incubated with LPS in the presence or absence of anti-CD14 blocking antibody, and cell surface and intracellular TLR4 expression were examined by flow cytometry. Results are shown as representative profiles after 30 and 60 min of incubation with blocking CD14 Ab or isotype control (IgG), and the percentage reduction of surface TLR4 expression was compared with unstimulated (no LPS) cells. Fig. 2B shows decreased cell surface TLR4 after a 30- or 60-min incubation with LPS, whereas in the presence of blocking CD14 Ab, TLR4 cell surface expression was not reduced. Quantification of mean fluorescence intensity shows 50% of the initial cell surface expression after a 30-min LPS incubation and 25–35% expression after 60 min compared with 80–90% in the presence of blocking anti-CD14 (Fig. 2B).
Conversely, flow cytometry showed increasing intracellular TLR4 following LPS stimulation (Fig. 2C), which is consistent with confocal images shown in Fig. 1. However, in the presence of anti-CD14, intracellular TLR4 was not increased. Taken together with Fig. 1, these findings demonstrate that in corneal epithelial cells, LPS induces relocalization of TLR4 from the cell surface to an intracellular location, which is mediated by CD14.
CD14 Mediates LPS-induced IRF3 but Not IκB Phosphorylation by Corneal Epithelial Cells
In macrophages and dendritic cells, cell surface TLR4 signals through the MyD88 pathway and activates NF-κB, whereas endosomal TLR4 signals through TRIF to activate IRF3-dependent cytokines (8). As we showed that TRIF is active in corneal epithelial cells (12), we anticipated that LPS would selectively activate the TRIF pathway and that this would be inhibited by blocking CD14.
HCEC and HCET cells were incubated with LPS and anti-CD14, and p-IRF3 and p-IκB were examined by Western blotting. As shown in Fig. 3A, p-IRF3 increased after a 30- and 60-min incubation with LPS; however, phosphorylation was inhibited 60–70% in the presence of anti-CD14 as shown by densitometry. In contrast, p-IκB was not inhibited by anti-CD14 in HCEC cells and only partially in HCET cells (Fig. 3B).
FIGURE 3.

Role of CD14 in LPS/TLR4 signaling in corneal epithelial cells. A and B, p-IRF3 (A) and p-IκB (B) expression in LPS/IFN-γ-activated HCEC and HCET cells in the presence of anti-CD14 antibody. C, IL-8 and CCL5/RANTES production by cells stimulated for 6 h with LPS and IFN-γ in the presence of anti-CD14 or control IgG. A and B, representative Western blots; C, Mean ± S.D. of two replicate experiments.
Consistent with these findings, production of CCL5/RANTES and IFN-β, which are activated by TRIF/IRF3 signaling (5, 18), was lower in the presence of anti-CD14 in both cell lines and in primary corneal epithelial cells (Fig. 3C). IL-8 was also lower in anti-CD14-treated cells (Fig. 3C), which is consistent with TRIF activation of NF-κB (5). These findings indicate that in addition to mediating TLR4 internalization, CD14 also regulates IRF3 signaling in human corneal epithelial cells.
Production of CCL5/RANTES Is Dependent on Syk Phosphorylation
In macrophages, Syk activation by LPS is CD14-dependent, but TLR4-independent (7). To determine the role of Syk in LPS activation, HCEC and HCET were stimulated with LPS in the presence of both anti-CD14 and the Syk inhibitor piceatannol.
As shown in Fig. 4A, p-Syk was detected in both cell lines after a 30- and 60-min incubation with LPS, and Syk phosphorylation was inhibited in the presence of anti-CD14. Consistent with this finding, LPS-induced CCL5 production was completely absent in the presence of piceatannol (Fig. 4B), whereas there was no difference in IL-8 production between LPS and LPS/piceatannol-treated HCEC and HCET cells.
FIGURE 4.
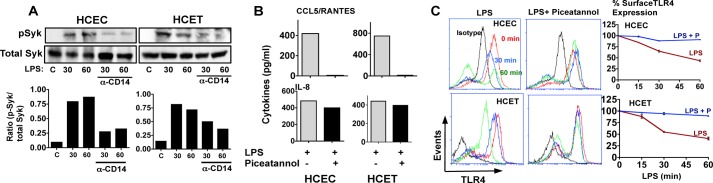
Role of Syk in CD14-mediated TLR4 internalization. A, total and phosphorylated Syk in HCEC and HCET cell lines after incubation with LPS/IFN-γ and either control IgG or CD14 blocking antibody. B, IL-8 and CCL5/RANTES production by LPS-stimulated HCEC and HCET cells in the presence of piceatannol. C, cell surface TLR4 on HCEC and HCET cell lines stimulated for 15 min, 30 min, or 1 h with LPS in the presence of the Syk inhibitor piceatannol. A, representative Western blots; B, mean ± S.D. of two replicate experiments. These experiments were repeated twice with similar results.
To examine whether p-Syk regulates LPS-induced TLR4 internalization, cells were incubated with LPS and piceatannol, and TLR4 cell surface expression was examined by flow cytometry. We found that in contrast to LPS stimulation alone where TLR4 cell surface expression decreased >50%, cell surface TLR4 remained unchanged over 60 min in LPS/piceatannol-treated cells (Fig. 4C). These data indicate that Syk phosphorylation mediates TLR4 internalization and selective activation of the TRIF pathway in human corneal epithelial cells.
CD14 Regulates Cytokine Production, Neutrophil Infiltration, and Bacterial Clearance in P. aeruginosa-infected Corneas
As CD14 regulates TLR4 signaling and cytokine production in vitro, we used gene knock-out mice to examine the role of CD14 in a murine model of P. aeruginosa corneal infection, which is dependent on TLR4 (13, 38).
Corneas of C57BL/6 and CD14−/− mice were abraded and infected topically with 1 × 105 virulent P. aeruginosa as described (13). After 24 h, corneas were homogenized, and cytokines were measured by ELISA. Fig. 5A shows that as with corneal epithelial cells, CCL5/RANTES production was significantly lower in infected corneas from CD14−/− compared with C57BL/6 mice. Conversely, CD14−/− mice had significantly elevated IL-6, CXCL1, and CXCL2 compared with C57BL/6 mice.
FIGURE 5.

Role of CD14 in P. aeruginosa corneal infection. A, cytokine production in corneas of C57BL/6 and CD14−/− mice 24 h after infection with 1 × 105 P. aeruginosa strain PAO1. Data are mean ± S.D. (error bars) of five mice per group. B, total neutrophils and macrophages in infected corneas 24 and 48 h postinfection after quantification by flow cytometry. C, P. aeruginosa cfu derived 2 h (indicating the inoculum), 24 h, and 48 h after infection. p values compare C57BL/6 and CD14−/− mice at the 24- and 48-h time points. D, representative corneas of C57BL/6 and CD14−/− mice 24 and 48 h after infection. E, representative histological sections from C57BL/6 and CD14−/− corneas 48 h after infection. Scale bar, 100 μm. B and C, data points represent individual corneas. Data are from a single representative experiment, which was repeated twice.
To determine the effect of CD14 deficiency on cellular infiltration, total cells in the cornea were quantified by flow cytometry using the Ly6G monoclonal Ab NIMPR-14 and the F4/80 macrophage Ab. As shown in Fig. 5B, there were significantly more neutrophils in the corneas of CD14−/− mice compared with C57BL/6 mice, whereas there was no significant difference in the number of macrophages. Consistent with the increased neutrophil numbers, total cfu in infected corneas of CD14−/− mice were significantly lower than C57BL/6 mice (Fig. 5D).
Normal mammalian corneas are transparent, and although there are resident fibroblasts and macrophages, neutrophils migrate into the corneal stroma from peripheral limbal vessels following infection. As shown in Fig. 5D, both groups of mice developed corneal opacification 24 h after infection; however, at 48 h, CD14−/− mice had more pronounced corneal opacification. This was consistent with increased cellular infiltration to the corneal stroma as shown in representative histological sections (Fig. 5E).
These findings are consistent with epithelial cell data shown above, and indicate that in vivo CD14 is required for CCL5 production associated with the TRIF pathway, whereas in the absence of CD14, production of CXC chemokines associated with the MyD88 pathway is elevated. This increased CXC chemokine production mediates neutrophil recruitment to the corneal stroma, more rapid bacterial killing, and exacerbated corneal disease (13).
CD14 Regulates P. aeruginosa-induced Signaling in Macrophages
Our previous studies using murine models of P. aeruginosa keratitis and LPS-induced inflammation showed that resident corneal macrophages are derived from the bone marrow and have a role in neutrophil recruitment by producing proinflammatory and chemotactic cytokines (13, 19).
To examine the role of CD14 in P. aeruginosa macrophage activation, BMDMs from C57BL/6 and CD14−/− mice were incubated with antibiotic killed P. aeruginosa strain PAO1, and cytokine production was examined after 3 h. Fig. 6A shows that as with corneal epithelial cells and in vivo, P. aeruginosa-stimulated CCL5/RANTES production was lower in CD14−/− compared with C57BL/6 macrophages, whereas IL-6 and CXCL2 were elevated. Further, p-IRF3 was elevated within 15 min in C57BL/6 macrophages, but was not detected in CD14−/− macrophages (Fig. 6B). Phospho-IκB was elevated in macrophages from both mouse strains; although expression was slightly higher in CD14−/− macrophages after 1 h, there was no difference in p65 translocation (Fig. 6, B and C).
FIGURE 6.

Role of CD14 in P. aeruginosa-activated macrophages. A, RANTES, CCL2/MIP2, and IL-6 production by C57BL/6 and CD14−/− macrophages after 6-h incubation with antibiotic-killed P. aeruginosa. Data are mean ± S.D. of two replicate experiments. B, total and phosphorylated IκB and IRF3 after incubation with antibiotic-killed P. aeruginosa. C, representative C57BL/6 and CD14−/− BMDMs showing translocation of p65 subunit of NF-κB into the nucleus 1 h after incubation with antibiotic-killed P. aeruginosa. D, total and phosphorylated p38, ERK, and JNK after incubation with antibiotic-killed P. aeruginosa. Experiments were repeated twice with similar results. Scale bar, 20 μm.
To examine the role of CD14 in the activation of the MAPK pathway, C57BL/6 and CD14−/− macrophages were incubated with P. aeruginosa, and p-p38, p-ERK, and p-JNK were examined. As shown in Fig. 6D, p-ERK and p-JNK were detected within 15 min activation of C57BL/6 macrophages, whereas p-p38 was not found until 1 h after stimulation. In contrast, CD14−/− macrophages generated p-p38 within 15 min but had decreased p-ERK compared with C57BL/6 macrophages. There was no significant difference in JNK activation between C57BL/6 and CD14−/− macrophages. Together, these data show that CD14 selectively regulates MAPK and IRF3 phosphorylation and CCL5 production by P. aeruginosa-stimulated macrophages.
DISCUSSION
CD14 mediates TLR4 signaling through its role as an ancillary LPS-binding protein that transfers LPS to the TLR4/MD-2 receptor complex (5). The selective role for CD14 in mediating the TLR4/TRIF pathway was first indicated in a report showing that CD14−/− macrophages have impaired IP-10 (CXCL10), but not TNF-α or IL-1β production (20). This role was confirmed in a study using a forward genetics approach that showed that CD14 was only required for MyD88-independent responses (6). However, the underlying mechanism for the selective role of CD14 was shown only recently to mediate Syk-dependent transport of TLR4 from the cell surface to endosomes to activate the TRIF signaling pathway (7). The role for CD14 in MyD88-dependent responses can be overcome by increasing LPS concentration, whereas its role as a TLR4/MD-2 chaperone to the endosome cannot be compensated by increasing LPS (7, 20).
In the current study, we demonstrate that CD14 has a similar role in epithelial cells, as blockade of CD14 inhibits TLR4 internalization, IRF3 phosphorylation and CCL5 production by LPS-stimulated corneal epithelial cells. We also show that as with dendritic cells and macrophages (7), this process is dependent on phosphorylation of Syk.
We also examined the role of CD14 in vivo using a well established murine model of P. aeruginosa corneal infection (13, 21, 22). Neutrophils are essential for bacterial killing and cause tissue damage, which manifests as corneal opacification. In the current study, we show that infected CD14−/− mice have a more pronounced inflammatory response than C57BL/6 mice, with significantly higher neutrophils infiltrating resulting in more severe corneal opacity. Increased neutrophils also contribute to the more rapid bacterial clearance found in these mice.
Given that CD14 has an important role as an LPS co-receptor (5, 6, 23), our in vivo findings showing an inhibitory role for CD14 appear somewhat counterintuitive. However, other studies support our observations by showing exacerbated inflammation in CD14−/− mice. For example, in a model of Escherichia coli peritonitis, CD14−/− mice exhibit increased neutrophil infiltration associated with bacterial clearance (24, 25). Similarly, CD14−/− mice infected intranasally with Streptococcus pneumoniae or Burkholderia pseudomallei exhibited accelerated bacterial killing compared with control mice (26, 27). Further, brains of S. pneumoniae-infected CD14−/− mice produced elevated CXCL2/MIP-2, resulting in increased neutrophil infiltration and more severe disease (28), which is similar to our findings with P. aeruginosa-infected corneas. Also, CD14−/− mice infected with Borrelia burgdorferi had more severe inflammation than control mice, which was attributed to increased production of regulatory proteins including SOCS1 and SOCS3 (29). Although B. burgdorferi activates TLR2 rather than TLR4, these authors also showed reduced p38 phosphorylation, which is important for SOCS3 expression (30). In the current study we found elevated phospho-p38 in P. aeruginosa stimulated CD14−/− macrophages, suggesting that this pathway is not involved. However, we also found decreased p-ERK, which is in agreement with an earlier report showing impaired MAPK phosphorylation in LPS-stimulated CD14−/− macrophages (31).
In the E. coli peritonitis model, the increased number of neutrophils infiltrating CD14−/− corneas was attributed to increased CXC chemokines in the peritoneal cavity compared with the blood (32), whereas in the S. pneumoniae meningitis model, the increased disease related to elevated CXCR2 expression in CD14−/− neutrophils (28). We also demonstrated increased CXC chemokine production by CD14−/− macrophages and showed that CD14 blocks LPS activation in human corneal epithelial cells; however, all of these reports show correlations and do not address the apparent anomaly of an inhibitory role for CD14 in vivo and accessory role in vitro.
The role of CD14 in infection with Gram-positive bacteria and spirochetes is likely due to its reported ability to bind bacterial components other than LPS, including peptidoglycan, and lipoteichoic acid (1, 33). Additional complicating factors include a role for CD14 in opsonization of P. aeruginosa and the possible role for CD14-independent activation of TLR4 by ExoS (34–36). The role of CD14 in mediating TLR4 signaling also depends on the carbohydrate side chains of LPS, as CD14 is required for activation by smooth, but not rough LPS (6).
Taken together, our findings demonstrate that CD14 mediates TLR4 internalization and IRF3 signaling in corneal epithelial cells, resulting in increased CCL5 and to a lesser extent IFN-β production. As intestinal and conjunctival epithelial cells regulate LPS responsiveness through IFN-γ/MD-2 expression using similar mechanisms as corneal epithelial cells (10, 11), it is very likely that CD14 has a similar role on epithelial cells from other tissues. As reported for macrophages and dendritic cells (7), our data show that in the absence of CD14-mediated endocytosis, the TLR4/MD-2 receptor complex remains on the epithelial cell membrane, leading to increased CXC chemokine and proinflammatory cytokine production and increased neutrophil recruitment. As we also show that CD14−/− macrophages produce elevated proinflammatory cytokines, it is likely that it is the combined role of CD14 on these cells that mediate the enhanced neutrophil infiltration in infected CD14−/− corneas. The increased numbers of neutrophil then contribute to disease pathogenesis by increasing total corneal thickness and by degranulation and release of cytotoxic mediators including reactive oxygen and nitrogen species that kill resident keratocytes and by production of serine proteases and matrix metalloproteinases that degrade the collagen fibrils in the cornea (21, 37).
In conclusion, CD14 has a critical role in regulating TLR4 responses not only by mediating the TLR4/TRIF/IRF3 signaling pathway in macrophages and corneal epithelial cells, but is also required to modulate production of TLR4/MyD88/NF-κB-dependent proinflammatory cytokines that have the potential to induce exacerbated responses that can result in increased disease manifestations.
Acknowledgments
We thank Scott Howell, Dawn Smith, and Catherine Doller of the Visual Sciences Core facility for expert technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants R01EY14362 and P30EY11373 (to E. P.). This work was also supported by the Research to Prevent Blindness Foundation and the Ohio Lions Eye Research Foundation.
- TLR4
- Toll-like receptor 4
- BMDM
- bone marrow-derived macrophage
- HCEC
- SV40-immortalized human corneal epithelial cell line 10.014 pRSV-T
- HCET
- telomerase-immortalized human corneal epithelial cell line
- IRF3
- interferon regulatory transcription factor
- Syk
- spleen tyrosine kinase
- TIRAP
- TIR (Toll/IL-1R intracellular domain)-associated protein
- TRIF
- TIR domain-containing adaptor inducing IFN-β
- TRAM
- TRIF-related adaptor molecule
- CCL
- CXCL, chemokine ligand
- RANTES
- regulated on activation, normal T cell expressed and secreted.
REFERENCES
- 1. Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS-binding protein. Science 249, 1431–1433 [DOI] [PubMed] [Google Scholar]
- 2. Visintin A., Latz E., Monks B. G., Espevik T., Golenbock D. T. (2003) Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J. Biol. Chem. 278, 48313–48320 [DOI] [PubMed] [Google Scholar]
- 3. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 4. Kawai T., Akira S. (2011) Toll-like receptors and their cross-talk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 5. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 6. Jiang Z., Georgel P., Du X., Shamel L., Sovath S., Mudd S., Huber M., Kalis C., Keck S., Galanos C., Freudenberg M., Beutler B. (2005) CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570 [DOI] [PubMed] [Google Scholar]
- 7. Zanoni I., Ostuni R., Marek L. R., Barresi S., Barbalat R., Barton G. M., Granucci F., Kagan J. C. (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kagan J. C., Medzhitov R. (2006) Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 9. Roy S., Sun Y., Pearlman E. (2011) Interferon-γ-induced MD-2 protein expression and lipopolysaccharide (LPS) responsiveness in corneal epithelial cells is mediated by Janus tyrosine kinase-2 activation and direct binding of STAT1 protein to the MD-2 promoter. J. Biol. Chem. 286, 23753–23762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Talreja J., Dileepan K., Puri S., Kabir M. H., Segal D. M., Stechschulte D. J., Dileepan K. N. (2005) Human conjunctival epithelial cells lack lipopolysaccharide responsiveness due to deficient expression of MD2 but respond after interferon-γ priming or soluble MD2 supplementation. Inflammation 29, 170–181 [DOI] [PubMed] [Google Scholar]
- 11. Abreu M. T., Arnold E. T., Thomas L. S., Gonsky R., Zhou Y., Hu B., Arditi M. (2002) TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J. Biol. Chem. 277, 20431–20437 [DOI] [PubMed] [Google Scholar]
- 12. Johnson A. C., Li X., Pearlman E. (2008) MyD88 functions as a negative regulator of TLR3/TRIF-induced corneal inflammation by inhibiting activation of c-Jun N-terminal kinase. J. Biol. Chem. 283, 3988–3996 [DOI] [PubMed] [Google Scholar]
- 13. Sun Y., Karmakar M., Roy S., Ramadan R. T., Williams S. R., Howell S., Shive C. L., Han Y., Stopford C. M., Rietsch A., Pearlman E. (2010) TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J. Immunol. 185, 4272–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robertson D. M., Li L., Fisher S., Pearce V. P., Shay J. W., Wright W. E., Cavanagh H. D., Jester J. V. (2005) Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest. Ophthalmol. Vis. Sci. 46, 470–478 [DOI] [PubMed] [Google Scholar]
- 15. Adhikary G., Sun Y., Pearlman E. (2008) C-Jun NH2-terminal kinase (JNK) is an essential mediator of Toll-like receptor 2-induced corneal inflammation. J. Leukoc. Biol. 83, 991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roy S., Bonfield T., Tartakoff A. M. (2013) Non-apoptotic toxicity of Pseudomonas aeruginosa toward murine cells. PLoS One 8, e54245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Motshwene P. G., Moncrieffe M. C., Grossmann J. G., Kao C., Ayaluru M., Sandercock A. M., Robinson C. V., Latz E., Gay N. J. (2009) An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 284, 25404–25411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chinnery H. R., Carlson E. C., Sun Y., Lin M., Burnett S. H., Perez V. L., McMenamin P. G., Pearlman E. (2009) Bone marrow chimeras and c-fms conditional ablation (Mafia) mice reveal an essential role for resident myeloid cells in lipopolysaccharide/TLR4-induced corneal inflammation. J. Immunol. 182, 2738–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perera P. Y., Vogel S. N., Detore G. R., Haziot A., Goyert S. M. (1997) CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or Taxol. J. Immunol. 158, 4422–4429 [PubMed] [Google Scholar]
- 21. Pearlman E., Sun Y., Roy S., Karmakar M., Hise A. G., Szczotka-Flynn L., Ghannoum M., Chinnery H. R., McMenamin P. G., Rietsch A. (2013) Host defense at the ocular surface. Int. Rev. Immunol. 32, 4–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun Y., Karmakar M., Taylor P. R., Rietsch A., Pearlman E. (2012) ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J. Immunol. 188, 1884–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haziot A., Ferrero E., Kontgen F., Hijiya N., Yamamoto S., Silver J., Stewart C. L., Goyert S. M. (1996) Resistance to endotoxin shock and reduced dissemination of Gram-negative bacteria in CD14-deficient mice. Immunity 4, 407–414 [DOI] [PubMed] [Google Scholar]
- 24. Metkar S., Awasthi S., Denamur E., Kim K. S., Gangloff S. C., Teichberg S., Haziot A., Silver J., Goyert S. M. (2007) Role of CD14 in responses to clinical isolates of Escherichia coli: effects of K1 capsule expression. Infect. Immun. 75, 5415–5424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haziot A., Hijiya N., Gangloff S. C., Silver J., Goyert S. M. (2001) Induction of a novel mechanism of accelerated bacterial clearance by lipopolysaccharide in CD14-deficient and Toll-like receptor 4-deficient mice. J. Immunol. 166, 1075–1078 [DOI] [PubMed] [Google Scholar]
- 26. Wiersinga W. J., de Vos A. F., Wieland C. W., Leendertse M., Roelofs J. J., van der Poll T. (2008) CD14 impairs host defense against Gram-negative sepsis caused by Burkholderia pseudomallei in mice. J. Infect. Dis. 198, 1388–1397 [DOI] [PubMed] [Google Scholar]
- 27. Dessing M. C., Knapp S., Florquin S., de Vos A. F., van der Poll T. (2007) CD14 facilitates invasive respiratory tract infection by Streptococcus pneumoniae. Am. J. Respir. Crit. Care Med. 175, 604–611 [DOI] [PubMed] [Google Scholar]
- 28. Echchannaoui H., Frei K., Letiembre M., Strieter R. M., Adachi Y., Landmann R. (2005) CD14 deficiency leads to increased MIP-2 production, CXCR2 expression, neutrophil transmigration, and early death in pneumococcal infection. J. Leukoc. Biol. 78, 705–715 [DOI] [PubMed] [Google Scholar]
- 29. Benhnia M. R., Wroblewski D., Akhtar M. N., Patel R. A., Lavezzi W., Gangloff S. C., Goyert S. M., Caimano M. J., Radolf J. D., Sellati T. J. (2005) Signaling through CD14 attenuates the inflammatory response to Borrelia burgdorferi, the agent of Lyme disease. J. Immunol. 174, 1539–1548 [DOI] [PubMed] [Google Scholar]
- 30. Bode J. G., Ludwig S., Freitas C. A., Schaper F., Ruhl M., Melmed S., Heinrich P. C., Häussinger D. (2001) The MKK6/p38 mitogen-activated protein kinase pathway is capable of inducing SOCS3 gene expression and inhibits IL-6-induced transcription. Biol. Chem. 382, 1447–1453 [DOI] [PubMed] [Google Scholar]
- 31. Perera P. Y., Mayadas T. N., Takeuchi O., Akira S., Zaks-Zilberman M., Goyert S. M., Vogel S. N. (2001) CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and Taxol-inducible gene expression. J. Immunol. 166, 574–581 [DOI] [PubMed] [Google Scholar]
- 32. Metkar S., Kim K. S., Silver J., Goyert S. M. (2012) Differential expression of CD14-dependent and -independent pathways for chemokine induction regulates neutrophil trafficking in infection. J. Leukoc. Biol. 92, 389–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kusunoki T., Hailman E., Juan T. S., Lichenstein H. S., Wright S. D. (1995) Molecules from Staphylococcus aureus that bind CD14 and stimulate innate immune responses. J. Exp. Med. 182, 1673–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berenger B. M., Hamill J., Stack D., Montgomery E., Huston S. M., Timm-McCann M., Epelman S., Mody C. H. (2011) Membrane CD14, but not soluble CD14, is used by exoenzyme S from P. aeruginosa to signal proinflammatory cytokine production. J. Leukoc. Biol. 90, 189–198 [DOI] [PubMed] [Google Scholar]
- 35. Epelman S., Stack D., Bell C., Wong E., Neely G. G., Krutzik S., Miyake K., Kubes P., Zbytnuik L. D., Ma L. L., Xie X., Woods D. E., Mody C. H. (2004) Different domains of Pseudomonas aeruginosa exoenzyme S activate distinct TLRs. J. Immunol. 173, 2031–2040 [DOI] [PubMed] [Google Scholar]
- 36. Wilkinson T. S., Dhaliwal K., Hamilton T. W., Lipka A. F., Farrell L., Davidson D. J., Duffin R., Morris A. C., Haslett C., Govan J. R., Gregory C. D., Sallenave J. M., Simpson A. J. (2009) Trappin-2 promotes early clearance of Pseudomonas aeruginosa through CD14-dependent macrophage activation and neutrophil recruitment. Am. J. Pathol. 174, 1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trinkaus-Randall V., Leibowitz H. M., Ryan W. J., Kupferman A. (1991) Quantification of stromal destruction in the inflamed cornea. Invest. Ophthalmol. Vis. Sci. 32, 603–609 [PubMed] [Google Scholar]
- 38. Huang X., Du W., McClellan S. A., Barrett R. P., Hazlett L. D. (2006) TLR4 is required for host resistance in Pseudomonas aeruginosa keratitis. Invest. Ophthalmol. Vis. Sci. 47, 4910–4916 [DOI] [PubMed] [Google Scholar]