

Background: c-FLIPL is a regulator of caspase-8 activity in T lymphocytes.
Results: Caspase-8 activity is lost upon deletion of c-FLIPL. p43FLIP rescues caspase-8 activity through Raf1, TRAF2, and RIPK1 association, augmenting ERK and NF-κB pathways.
Conclusion: The FLIPL cleavage product p43FLIP promotes activation of pathways involved with T cell growth.
Significance: This study provides new insight into the regulation of caspase-8 activity by c-FLIP.
Keywords: Apoptosis, Caspase, Cell Proliferation, Molecular Cell Biology, T Cell, FLIP
Abstract
Caspase-8 is now appreciated to govern both apoptosis following death receptor ligation and cell survival and growth via inhibition of the Ripoptosome. Cells must therefore carefully regulate the high level of caspase-8 activity during apoptosis versus the modest levels observed during cell growth. The caspase-8 paralogue c-FLIP is a good candidate for a molecular rheostat of caspase-8 activity. c-FLIP can inhibit death receptor-mediated apoptosis by competing with caspase-8 for recruitment to FADD. However, full-length c-FLIPL can also heterodimerize with caspase-8 independent of death receptor ligation and activate caspase-8 via an activation loop in the C terminus of c-FLIPL. This triggers cleavage of c-FLIPL at Asp-376 by caspase-8 to produce p43FLIP. The continued function of p43FLIP has, however, not been determined. We demonstrate that acute deletion of endogenous c-FLIP in murine effector T cells results in loss of caspase-8 activity and cell death. The lethality and caspase-8 activity can both be rescued by the transgenic expression of p43FLIP. Furthermore, p43FLIP associates with Raf1, TRAF2, and RIPK1, which augments ERK and NF-κB activation, IL-2 production, and T cell proliferation. Thus, not only is c-FLIP the initiator of caspase-8 activity during T cell activation, it is also an initial caspase-8 substrate, with cleaved p43FLIP serving to both stabilize caspase-8 activity and promote activation of pathways involved with T cell growth.
Introduction
The functions of caspase-8 have expanded from its original description as an upstream initiator caspase in cell death to a mediator in a variety of other signal pathways, including signaling of certain Toll-like receptors, T cell survival and growth, and the RIG-I helicase pathway (1–4). In nearly all instances in which it was examined, the enzymatic activity of caspase-8 was required, most likely to cleave RIPK1 (receptor (TNFRSF)-interacting serine/threonine protein kinase 1) and prevent formation of the Ripoptosome (5–9). This raised the question of how the level of caspase-8 activity is regulated to mediate such divergent functions as cell growth versus death.
c-FLIP (cellular FLICE-like inhibitory protein; CFLAR, Casper) is an enzymatically inactive paralogue of caspase-8, arising most likely as a gene duplication of caspase-8, as both genes are located in proximity on human chromosome 2 (10–12). c-FLIPL (full-length c-FLIP-Long) differs in the C-terminal enzymatic region of caspase-8 (10), and as a consequence, c-FLIPL has no intrinsic caspase activity of its own. The original descriptions of c-FLIP differed in their claims to function, with some investigators reporting that c-FLIP inhibited caspase-8 activation by death receptors such as Fas (CD95) and inhibited cell death (10), whereas others reported that it enhanced caspase activity and promoted cell death (13). With the subsequent realization that caspase-8 functions not only in promoting cell death but also in cell survival and growth (14, 15), interest increased in c-FLIP as a potential regulator of caspase-8 activity in these opposing processes. Further suggestion of this model came from structural studies showing that c-FLIPL could heterodimerize with caspase-8 (16). The structural findings revealed that c-FLIPL contains in its C terminus a loop that actually activates the enzymatic pocket of caspase-8 and stabilizes its activity in the full-length form (16). Hence, in addition to its first described function as an inhibitor of caspase-8 activation by competitively binding to FADD following death receptor ligation, c-FLIPL has now emerged also as an activator of caspase-8. This provides an explanation for the earlier opposite functions described for c-FLIP.
This dual function of c-FLIP raised a further issue regarding how c-FLIP could maintain caspase-8 activity at a moderate level for cell survival and growth because it contained an activation domain for caspase-8. Indeed, overexpression systems for c-FLIPL often revealed that it induced cell death by virtue of intensely activating caspase-8 (13, 17). A potentially simple and elegant solution came with the realization that the activation loop in the C terminus of c-FLIPL contains a caspase-8 cleavage site at Asp-376, which would yield a predicted p43FLIP cleavage product (16). It was thus possible that, temporally, c-FLIPL could initiate caspase-8 activation, and then caspase-8 would cleave c-FLIPL to produce truncated p43FLIP.
c-FLIPL thus emerged as both the initiator of caspase-8 activity and potentially its initial substrate. This model left unclear whether c-FLIPL was still required to maintain caspase-8 activity once caspase-8 was activated. Conceivably, in the absence of c-FLIPL, caspase-8 would spontaneously form homodimers, resulting in apoptosis. Conversely, the persistence of c-FLIPL in the caspase complex might also continue to augment caspase-8 activation to the point of cell death. This led us to consider two possibilities for p43FLIP function. First, p43FLIP might be a critical substrate product of caspase-8 and supplant the need for continued caspase-8 activity. Second, p43FLIP might be ideally suited for the function of maintaining caspase-8 activity once initiated by c-FLIPL and supplant the continued need for c-FLIPL. We therefore sought to test the function of p43FLIP specifically following T cell activation, once caspase-8 activity is established. We examined this question by transgenically expressing p43FLIP in T lymphocytes in situations in which either caspase-8 or endogenous c-FLIP could be deleted following T cell activation and the initiation of caspase-8 activity. Our findings reveal that following T cell activation in wild-type T cells, c-FLIPL is cleaved to form p43FLIP, which is in complex with active caspase-8, and can recruit the adaptor proteins Raf1, RIPK1, and TRAF2 (TNF receptor-associated factor 2), which link to the ERK and NF-κB pathways. Furthermore, the acute loss of c-FLIP in effector T cells leads to reduced caspase-8 activity and impairment of cell growth, whereas p43FLIP can rescue T cells from the acute loss of c-FLIP by maintaining caspase-8 in an active form.
EXPERIMENTAL PROCEDURES
Production of p43FLIP Transgenic Mice and Stably p43FLIP-transfected Jurkat T Cells
Murine p43FLIP was expressed transgenically in the T cell compartment. Briefly, FLAG-tagged murine p43FLIP cDNA was inserted into a target vector containing the lck proximal promoter and a downstream human growth hormone locus (18). Transgenic mice were produced using C57BL/6 fertilized eggs. Mice were screened by PCR of ear DNA for the human growth hormone sequence contained within the p43FLIP construct using the following primers: 5′-primer, 5′-GGAGCCAGGGCTGGGCATAAAA-3′; and 3′-primer, 5′-GACTCACCCTGAAGTTCTCAGGATCC-3′.
Immunoblotting using anti-FLIP monoclonal antibody (Dave-2, Millipore, Billerica, MA) further confirmed expression of the transgene. Three p43FLIP transgenic mouse lines that gave similar phenotypes were obtained. One line (line 34) was selected for the studies reported here.
The same p43FLIP was cloned into the pEF-BOS vector (19) and transfected into Jurkat T cells. Stable transfectants were obtained under puromycin selection and then cloned by limiting dilution. Five separate clones that yielded similar characteristics of signaling were derived.
Additional Mouse Lines
Mice containing loxP sites surrounding either the caspase-8 or c-FLIP locus have been described previously (15, 20). ER-Cre mice (CAG-Cre/Esr1) were obtained from The Jackson Laboratory (Bar Harbor, ME). These transgenic mice have a tamoxifen-inducible Cre-mediated recombination system driven by the chicken β-actin promoter/enhancer coupled with the CMV immediate-early enhancer. The transgene insert contains a fusion product involving Cre recombinase and a mutant form of the mouse estrogen receptor ligand-binding domain that does not bind natural ligand estrogen at physiological concentrations but will bind the synthetic ligand 4-hydroxytamoxifen. Restricted to the cytoplasm, the Cre/ESR1 protein can gain access to the nuclear compartment after only exposure to tamoxifen. When crossed with a mouse strain containing a loxP site-flanked sequence of interest, the offspring are useful for generating tamoxifen-induced, Cre-mediated targeted deletions.
T Cell Subset Purification, Culture, and Growth
Spleen cells after hemolysis with Gey's solution were combined with lymph node cells, followed by negative selection to enrich for T cells. To purify cell subsets, samples were incubated with antibodies to MHC class II (3F12), B220 (RA3-6B2), NK1.1 (PK136), and CD11b (Tib-128) for 30 min to bind B cells, NK cells, and macrophages. Samples were washed and then incubated with goat anti-rat/mouse IgG-labeled magnetic beads (Qiagen, Valencia, CA) for 45 min, followed by magnetic field separation. Purity was confirmed by flow cytometry and was routinely >90%.
T cells were activated in RPMI 1640 medium (Mediatech, Herndon, VA) supplemented with 100 units/ml penicillin/streptomycin (Invitrogen), 25 mm HEPES (Sigma), 50 μm 2-mercaptoethanol (Sigma), 2.5 mg/ml glucose (Sigma), 110 μg/ml pyruvate (Invitrogen), 292.3 μg/ml glutamine (Invitrogen), 10 μg/ml folate (Invitrogen), and 5% FBS (HyClone, Logan, UT) using plate-bound anti-CD3 antibody (10 μg/ml; clone 145-2C11), soluble anti-CD28 ascites antibody (1:1000), and recombinant human IL-2 (50 units/ml; Cetus, Emeryville, CA) for 2 days. Cells were then removed from anti-CD3 antibody-coated wells and fed with fresh culture medium containing IL-2. Cell proliferation was measured using [3H]thymidine incorporation. Cells were plated at 1.0 × 105/well in wells coated with anti-CD3 antibody (10 μg/ml) and soluble anti-CD28 antibody (1:100). On day 3, cells were pulsed with [3H]thymidine (1 μCi/well) for 16 h and then harvested and counted.
Antibodies and Flow Cytometry
Anti-murine CD8α monoclonal antibody conjugated to phycoerythrin-Texas Red® was purchased from Invitrogen. Anti-murine CD4 monoclonal antibodies conjugated to phycoerythrin-Cy5.5 or phycoerythrin were purchased from Invitrogen. For flow cytometry, 750,000 cells were incubated in 0.1 ml of PBS containing 1.0% BSA Fraction V (Sigma), 0.001% (w/v) sodium azide (PBS/azide), and the appropriate antibodies at 4 °C for 30 min. After washing with PBS/azide, cells were fixed in 1% methanol-free formaldehyde (Ted Pella, Inc., Reading, CA) in PBS/azide. Samples were stored at 4 °C until they were analyzed with an BD LSR II flow cytometer (BD Biosciences).
For TUNEL assay of apoptosis, surface staining was first completed as described above, with the exception that cells were fixed on ice for 15 min using 10% formaldehyde in PBS. Cells were then washed twice with PBS, fixed in 70% ice-cold ethanol for 15 min, and washed twice with PBS. Nicked DNA was labeled by incubating cells with terminal deoxynucleotidyltransferase buffer (2.5 mm CoCl2, 1 unit of terminal deoxynucleotidyltransferase, and 0.5 nmol of FITC-dUTP (Roche Diagnostics)) in a total volume of 50 μl at 37 °C for 1 h. Samples were then washed twice with 1% BSA in PBS and fixed using 1% formaldehyde in PBS. Cytokine levels were assessed using Bio-Plex (Bio-Rad) in supernatants from purified T cells (106/ml) stimulated with anti-CD3/anti-CD28 antibody for 24 h.
Immunoblot Analysis
Cells were washed once with ice-cold PBS and solubilized in lysis buffer (0.2% Nonidet P-40, 20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm sodium orthovanadate, 10% glycerol, and cOmplete protease inhibitor (Roche Diagnostics)). Post-nuclear lysate proteins (20 μg/lane) were separated by 12.5% SDS-PAGE. Proteins were transferred to PVDF membranes (Bio-Rad), and blots were blocked and probed with the indicated antibodies in 4% nonfat milk in TBS and 0.1% Tween 20. Immunoreactive proteins were visualized using HRP-labeled conjugates (Jackson ImmunoResearch Laboratories, West Grove, PA) and developed using LumiGLO (KPL Laboratories, Gaithersburg, MD). The antibodies used were specific for caspase-8 (a kind gift of Dr. Andreas Strasser, Walter and Eliza Hall Institute); c-FLIP (ProSci, Poway, CA); caspase-3 (a kind gift of Dr. Y. Lazebnik, Cold Spring Harbor Laboratory); and RIPK1, RIPK3, TRAF2, and Raf1 (BD Biosciences).
T Cell Activation and Analysis of ERK and NK-κB Activity
Purified T cells were activated using biotinylated anti-CD3 and anti-CD28 antibodies (each 5 μg/ml) and avidin (Sigma) in the absence or presence of the proteasome blocker MG132 (Sigma). Reactions were stopped after the times indicated using cold PBS. Cells were then lysed in the appropriate buffer for analysis using Bio-Plex and by immunoblotting using antibodies to phospho-MEK1, total MEK1, phospho-ERK, total ERK, phospho-IκBα, and total IκBα.
Biotinylated VAD-fluoromethyl Ketone Active Caspase and Anti-FLAG Precipitation Assays
Viable day 4 T cell lymphoblasts were disrupted using lysis buffer containing 20 μm biotinylated Val-Ala-Asp(OMe)-fluoromethyl ketone (biotin-VAD; MP Biomedicals). 600 μg of protein lysate was then precleared by rocking with 40 μl of Sepharose 6B-agarose beads (Sigma) at 4 °C for 2 h. The supernatant was subsequently incubated overnight with 30 μl of streptavidin-Sepharose beads (Zymed Laboratories Inc., Inc., San Francisco, CA) on a rocker at 4 °C. Beads were washed five times with lysis buffer without cOmplete protease inhibitor and then boiled in loading buffer. Beads were removed by centrifugation, and immunoblot analysis was performed on the supernatants. Alternatively, FLAG-tagged p43FLIP was immunoprecipitated from 600 μg of protein lysate from the same cells using biotinylated anti-FLAG magnetic beads. Precipitates were prepared using a magnetic column, washed five times, and then eluted and immunoblotted for the indicated proteins.
Statistical Analyses
Student's t test or repeated measures analysis of variance (ANOVA)2 was conducted to assess differences between the indicated genotypes of mice.
RESULTS
p43FLIP Is Generated by Caspase-8 and Promotes Recruitment of Adaptor Proteins That Link to NF-κB and ERK
Both caspase-8 and c-FLIP are required to initiate T cell growth (14, 15, 21). However, it is not known whether the persistence of these molecules is required to sustain proliferation once initiated. We therefore used the inducible ER-Cre system to delete caspase-8 upon the addition of tamoxifen. T cells from ER-Cre-casp8loxP/loxP mice were activated with anti-CD3/anti-CD28 antibody plus IL-2 and cultured for 3 days, at which time tamoxifen (0.25–1.0 μm) was added for 24 h and then washed out, and cells were re-cultured in IL-2-containing medium. As shown in Fig. 1A, these doses of tamoxifen nearly completely eliminated the expression of caspase-8 but did not alter the levels of caspase-8 in casp8loxP/loxP T cells lacking ER-Cre. Of interest was that following the deletion of caspase-8 in effector T cells, growth was arrested (Fig. 1B). The loss of caspase-8 expression was also paralleled by a loss of cleaved p43FLIP in effector T cells, although total levels of c-FLIP were unchanged (Fig. 1C). This is consistent with a known caspase-8 cleavage site in c-FLIPL at Asp-376 (16).
FIGURE 1.
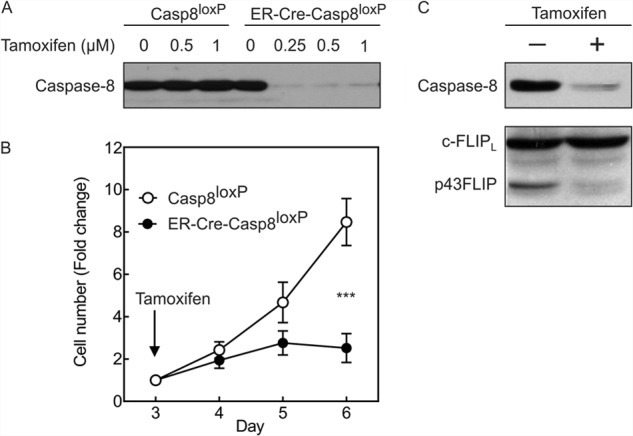
Deletion of caspase-8 reduces growth of effector T cells and prevents cleavage of c-FLIPL. Purified T cells from either casp8loxP/loxP mice (Casp8loxP) or ER-Cre-casp8loxP/loxP mice were stimulated with anti-CD3/anti-CD28 antibody plus IL-2. On day 3, tamoxifen was added at the indicated concentrations for 24 h and then washed out, and cells were re-cultured in medium containing IL-2. A, on day 5, cell lysates were made and immunoblotted (20 μg of protein/lane) for caspase-8. B, cell growth was monitored during days 3–6 following the addition of tamoxifen (0.35 μm) on day 3. Shown are the mean -fold changes in cell number ± S.D. of three experiments normalized to the cell number at the time of tamoxifen addition on day 3. ***, p < 0.0001 by repeated measures ANOVA. C, cell lysates from ER-Cre-casp8loxP/loxP T cells without or with tamoxifen treatment (48 h) were immunoblotted for caspase-8 and c-FLIP. These findings were consistent in four experiments.
We and others have previously observed using cell lines and overexpression systems that p43FLIP associated with RIPK1 and TRAF2 better than c-FLIPL (22, 23). This suggested that part of the function of caspase-8 in effector T cells could be to cleave c-FLIPL to p43FLIP to recruit these adaptor proteins. We thus developed a transgenic mouse expressing murine p43FLIP in T cells under the control of the lck proximal promoter. Three independent lines of mice with similar phenotypes were generated. There were no observed differences from wild-type mice in thymus cellularity or T cell development, and there were no differences in lymph node or spleen T cell numbers, CD4/CD8 ratio, or proportion of memory T cells based on CD44 expression (data not shown).
We examined whether p43FLIP would alter the activity of caspase-8 as well as associating molecules. Although p43FLIP did not alter the expression of total cellular caspase-8, there was an increase in the active fraction of caspase-8 in day 4 effector T cells, as determined by precipitation of active caspases using biotin-VAD and avidin-Sepharose (Fig. 2). This is consistent with the known ability of c-FLIP to stabilize active caspase-8 (16, 24, 25). Furthermore, p43FLIP was also in the active caspase complex. Of particular interest was that the active caspase complex in p43FLIP T cells also contained greater amounts of RIPK1, TRAF2, and Raf1 compared with wild-type T cells (Fig. 2). To further confirm the specificity of this association, we performed immunoprecipitation of the FLAG-tagged p43FLIP transgene and observed that it also associated with Raf1, TRAF2, and RIPK1 (Fig. 2B). The same observations were made in the p43FLIP-transfected Jurkat T cells (data not shown).
FIGURE 2.

p43FLIP augments active caspase-8 and recruits RIPK1, TRAF2, and Raf1 to the active caspase complex. Day 4 effector T cells from normal littermate control (NLC) mice or p43FLIP transgenic mice were lysed in biotin-VAD. Whole cell lysates (WCL; 20 μg/lane) and either biotin-VAD precipitates (A) or anti-FLAG precipitates (B) of FLAG-tagged p43FLIP (each precipitate from 600 μg of protein of whole cell lysates) were immunoblotted for the indicated molecules. The graphs represent densitometry of immunoblots of three experiments normalized to normal littermate control levels. Casp8, caspase-8; i.p., immunoprecipitate.
p43FLIP Augments Activation of NF-κB and ERK
Because Raf1, TRAF2, and RIPK1 are upstream regulators of the ERK MAPK and NF-κB pathways, we examined the activity of these pathways using Bio-Plex and by standard immunoblotting. Freshly purified T cells from wild-type or p43FLIP mice were activated with soluble biotinylated anti-CD3/anti-CD28 antibody and cross-linked with streptavidin for various time periods up to 30 min. Both assays detected a prominent increase in phosphorylation of ERK within 5 min in p43FLIP T cells compared with wild-type T cells (Fig. 3, A and C). However, both groups of T cells equivalently activated ERK with phorbol 12-myristate 13-acetate, indicating that p43FLIP was acting in proximity to T cell receptor signaling. MEK1, the upstream activator of ERK, also manifested greater phosphorylation and activation in p43FLIP T cells (Fig. 3C).
FIGURE 3.

Increased activation of ERK and NF-κB in p43FLIP T cells. Purified T cells from p43FLIP mice or normal littermate control (NLC) mice were stimulated with biotinylated anti-CD3/anti-CD28 antibody and cross-linked with streptavidin for the times indicated. The proteasome blocker MG132 was added to some samples to prevent degradation of phospho-IκBα. Lysates were then analyzed using Bio-Plex for ERK phosphorylation (A) and IκBα phosphorylation (B) as a ratio of the phosphorylated form of each molecule to the total amount of the same molecule. Shown are the means ± S.D. of three experiments expressed as -fold increase in the phospho-ERK/total ERK or phospho-IκBα/total IκBα ratios over unstimulated cells (time 0). Statistical analysis was done over the complete time period using repeated measures ANOVA (phospho-ERK, p < 0.0001; phospho-IκBα, p = 0.0038). Independently, lysates were also analyzed by immunoblotting for phospho-ERK, total ERK, phospho-MEK, and total MEK (C) and phospho-IκBα and total IκBα (D). The findings obtained by immunoblotting were consistent in two experiments using p43FLIP T cells, as well as in two experiments using p43FLIP Jurkat T cells (not shown). PMA, phorbol 12-myristate 13-acetate.
NF-κB activity was also increased in p43FLIP T cells. As shown in Fig. 3 (B and D), both assays revealed greater phosphorylation of IκBα in p43FLIP T cells, and this was considerably augmented in the presence of the proteasome blocker MG132. Similar results for the activation of NF-κB and ERK were seen in Jurkat T cells stably transfected with p43FLIP (data not shown).
As the ERK and NF-κB pathways contribute to IL-2 gene activation, we examined cytokine production in p43FLIP T cells. Compared with wild-type T cells, p43FLIP T cells produced approximately twice the amount of IL-2 within 24 h following stimulation with anti-CD3/anti-CD28 antibody (Fig. 4A). Bio-Plex analysis of cell supernatants demonstrated no differences in the production of IFNγ, IL-6, IL-10, IL-13, IL-17, or TNFα (data not shown). Proliferation of T cells was also moderately increased in p43FLIP T cells (Fig. 4B). This was likely due to the increased IL-2 production, as there were no differences in surface expression of CD25 (data not shown). Although IL-2 can also sensitize effector T cells to reactivation-induced death (26), we actually observed reduced death of day 4 effector T cells from c-FLIP mice following CD3 re-stimulation (Fig. 4C). As reactivation-induced T cell death in vitro is mediated largely by FasL (27), we directly examined cell death induced by FasL and found that this was also considerably reduced in p43FLIP T cells (Fig. 4D). This is likely due to the ability of c-FLIP to compete with recruitment of caspase-8 at the death-inducing signal complex (12).
FIGURE 4.
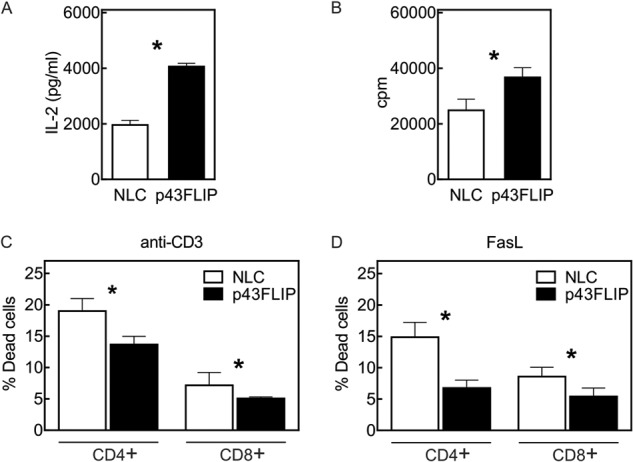
Increased IL-2 production, proliferation, and reduced cell death in p43FLIP T cells. Purified T cells from normal littermate control (NLC) mice or p43FLIP mice were stimulated with anti-CD3/anti-CD28 antibody. A, after 24 h, supernatants were analyzed for IL-2 production using Bio-Plex. B, after 72 h, cells were pulsed for 16 h with [3H]thymidine, and the amount of incorporated [3H]thymidine was measured. C and D, T cell lymphoblasts on day 4 were re-stimulated for 6 h with either cross-linked anti-CD3 antibody (C) or soluble FasL (D), and cell death was measured by TUNEL assay. Results are the means ± S.E. of triplicate cultures. Findings were consistent in four experiments for IL-2, five experiments for [3H]thymidine, and two experiments each for anti-CD3 antibody- and FasL-mediated cell death. *, p < 0.05.
c-FLIP Is Required to Maintain Caspase-8 Activity, and p43FLIP Rescues Loss of Endogenous c-FLIP
As c-FLIPL is both an activator of caspase-8 and an initial caspase-8 substrate, it was possible that p43FLIP might be able to rescue T cells from the loss of caspase-8. The ability of p43FLIP to associate with Raf1, RIPK1, and TRAF2 (28) suggested that once p43FLIP is formed, it might supplant the need for caspase-8 activity. We therefore crossed the p43FLIP mice with ER-Cre-casp8loxP/loxP mice. Deletion of caspase-8 did not alter the levels of total endogenous c-FLIP or transgenic p43FLIP (Fig. 5A). Conversely, as anticipated, p43FLIP did not alter the loss of caspase-8 following tamoxifen treatment. Of note is that p43FLIP also did not rescue T cells from death following the loss of caspase-8 (Fig. 5B). Thus, although p43FLIP can augment activation of caspase-8 as well as ERK and NF-κB, it cannot replace the need for caspase-8 in T cell survival. This is consistent with recent reports that identified a complex involving RIPK1, RIPK3, and FADD known as the Ripoptosome, which induces cell death known as necroptosis when RIPK1 and RIPK3 are not tonically cleaved by caspase-8 (9).
FIGURE 5.
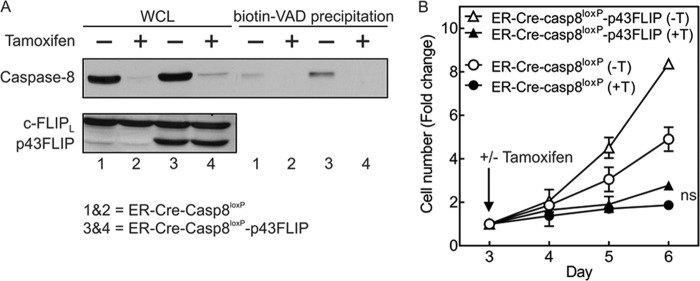
p43FLIP does not rescue T cells from loss of caspase-8. Purified T cells from ER-Cre-casp8loxP/loxP mice (lanes 1 and 2) or ER-Cre-casp8loxP/loxP-p43FLIP mice (lanes 3 and 4) were stimulated with anti-CD3/anti-CD28 antibody plus IL-2. On day 3, tamoxifen (0.35 μm) was either added (+) or not (−), and on day 5, cell lysates containing biotin-VAD were made. A, whole cell lysates (WCL) and biotin-VAD precipitates were immunoblotted for caspase-8 and c-FLIP. B, cell growth for the same cells following the addition of tamoxifen (+T) or vehicle control (−T) on day 3. Shown are the mean -fold changes in cell number ± S.D. of three experiments normalized to cell number at the time of tamoxifen addition on day 3. ns, no significant difference for ER-Cre-casp8loxP/loxP (+tamoxifen) versus ER-Cre-casp8loxP/loxP-p43FLIP (+tamoxifen) by repeated measures ANOVA.
Although loss of caspase-8 could not be supplanted by p43FLIP, it was not clear what the effect of loss of endogenous c-FLIP would be on maintaining caspase-8 activity in effector T cells. Conceivably, caspase-8 could maintain its own activity, independent of c-FLIP, through the formation of caspase-8 homodimers in effector T cells, as occurs following Fas ligation. However, we previously observed that neither Fas nor TNFR1 is required for the initiation or maintenance of caspase-8 activity in effector T cells (17). We thus considered whether the persistence of c-FLIP is required after T cell activation to maintain caspase-8 activity. To examine this, we developed ER-Cre-FLIPloxP/loxP mice. Treatment with tamoxifen greatly reduced c-FLIP expression within 48 h (Fig. 6A). Of particular interest was that caspase-8 activity became undetectable when levels of c-FLIP were diminished, and the association of RIPK1 and RIPK3 in the active caspase complex was also reduced. As a result, T cells devoid of c-FLIP rapidly stopped proliferating (Fig. 6B). Using mouse embryonic fibroblasts, we confirmed that the loss of c-FLIP also resulted in the loss of active caspase-8 while not altering the levels of total caspase-8 (data not shown).
FIGURE 6.

Deletion of c-FLIP reduces active caspase-8 but not active caspase-3. Purified T cells from ER-Cre-FLIPloxP/loxP mice were stimulated with anti-CD3/anti-CD28 antibody plus IL-2. A, on day 3, tamoxifen (0.35 μm) was either added (+) or not (−), and on day 5, cell lysates containing biotin-VAD were made. Shown are whole cell lysates (WCL) and biotin-VAD precipitates immunoblotted for c-FLIP, caspase-8, caspase-3, RIPK1, and RIPK3. The graphs represent densitometry of the displayed biotin-VAD immunoblots. Casp, caspase; Tmx, tamoxifen. B, cell growth for the same cells following the addition of tamoxifen on day 3. Shown are the mean -fold changes in cell number ± S.D. of three experiments normalized to cell number at the time of tamoxifen addition on day 3. **, p = 0.0058 by repeated measures ANOVA.
To further assess whether p43FLIP could rescue caspase-8 activity following the loss of endogenous c-FLIP, we developed ER-Cre-FLIPloxP/loxP-p43FLIP mice. The addition of tamoxifen to effector T cells from these mice still induced the loss of endogenous c-FLIP, but the presence of transgenic p43FLIP rescued the loss of caspase-8 activity (Fig. 7A). This was specific to caspase-8, as it did not alter the activity of caspase-3. Paralleling the rescue of active caspase-8, p43FLIP also enhanced the association of RIPK1 and RIPK3 in the active caspase complex. Finally, p43FLIP also rescued proliferation following deletion of c-FLIP (Fig. 7B).
FIGURE 7.

p43FLIP rescues loss of active caspase-8 and growth of FLIP-deficient T cells. Lymph node T cells from the indicated strains of mice were activated with anti-CD3/anti-CD28 antibody plus IL-2 for 3 days, and tamoxifen (0.35 μm) added for an additional 2 days. A, whole cell lysates (WCL) and biotin-VAD precipitates were immunoblotted for c-FLIP, caspase-8, caspase-3, RIPK1, and RIPK3. The graphs represent densitometry of the displayed immunoblots. Casp, caspase. B, cell growth for the same cells following the addition of tamoxifen (+T) or vehicle control (−T) on day 3. Shown are the mean -fold changes in cell number ± S.D. of three experiments normalized to cell number at the time of tamoxifen addition on day 3. ***, p < 0.0001 for ER-Cre-FLIPloxP/loxP (+tamoxifen) versus ER-Cre-FLIPloxP/loxP-p43FLIP (+tamoxifen) by repeated measures ANOVA.
DISCUSSION
The current findings underscore the intricate interactions of c-FLIP and caspase-8 (Fig. 8). c-FLIPL is both an activator and an initial substrate of caspase-8 in effector T cells. c-FLIPL contains an activation domain in its C terminus that can activate caspase-8 upon their heterodimerization (16). Active caspase-8 then rapidly cleaves c-FLIPL at Asp-376 to form p43FLIP, which serves to limit further activation of caspase-8 but maintains its activity even with the loss of c-FLIPL. p43FLIP can also recruit adaptor proteins that link to the ERK and NF-κB pathways. In T cells, this manifests as increased IL-2 production and cell proliferation and survival. Thus, p43FLIP emerges as an important caspase-8 cleavage product that is critical for maintaining caspase-8 activity at a precise intermediate level for cell survival and growth while also promoting signal pathways for cell growth.
FIGURE 8.
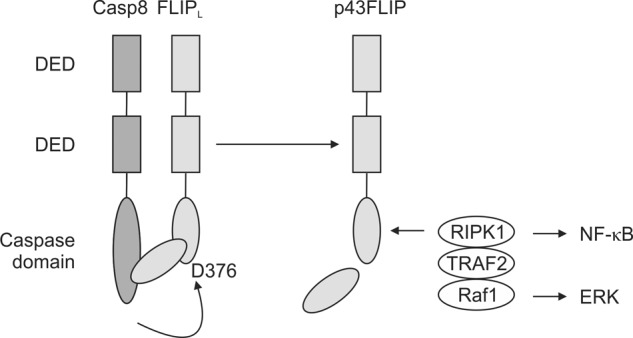
Model of c-FLIP regulation of caspase-8 activity. Caspase-8 (Casp8) and c-FLIPL heterodimerize by their mutual death effector domains (DED). c-FLIPL activates caspase-8 in its full-length form via an activation loop in the C terminus of c-FLIPL, and active caspase-8 then cleaves c-FLIPL at Asp-376. Cleaved p43FLIP maintains caspase-8 activity while associating with the adaptor proteins RIPK1 and TRAF2 to promote activation of NF-κB and with Raf1 to promote activation of ERK.
In our previous studies (17, 29), we noted that expression of either c-FLIPL or c-FLIPS in T cells results paradoxically in increased cell death. In the case of c-FLIPL, this was the result of greatly augmented caspase-8 activation, whereas c-FLIPS provoked considerably reduced caspase-8 activation compared with wild-type T cells. This underscores the relatively narrow window of caspase-8 activity that is required for T cell survival and growth. These observations are consistent with mathematical modeling studies that predicted that increased c-FLIPL in complex with caspase-8 would lead to excessive activation of caspase-8 (30), as we saw in T cells from c-FLIPL mice (17). These findings are also consistent with studies showing that non-cleavable c-FLIPL excessively activates caspase-8, presumably because caspase-8 cannot cleave the C-terminal activation domain of non-cleavable c-FLIPL (30). In the case of c-FLIPS, the reduction in caspase-8 activity is consistent with the lack of the C-terminal caspase-8 activation domain in c-FLIPS, and thus, this may lead to activation of the Ripoptosome and cell death. In addition, in T cells from both c-FLIPL and c-FLIPS transgenic mice, NF-κB activity was reduced, which may have further contributed to loss of cell viability. Because p43FLIP lacks the activation domain but can remain associated with caspase-8, p43FLIP is able to maintain moderate levels of active caspase-8 without its further activation. Thus, in contrast c-FLIPL and c-FLIPS, cleaved p43FLIP is the only c-FLIP form that enhanced cell survival, in conjunction with its ability to maintain moderate levels of caspase-8 activity and enhance activation of ERK and NF-κB. In particular, p43FLIP was sufficient to maintain active caspase-8 following the loss of endogenous c-FLIP. p43FLIP is thus an important caspase-8 cleavage product, which, unlike c-FLIPL or c-FLIPS, maintains caspase-8 activity at an optimal level to promote cell survival while also recruiting adaptor proteins for activation of ERK and NF-κB.
An unanticipated observation was that when c-FLIP was deleted from effector T cells, there was a concomitant loss of active caspase-8 rather than spontaneous homodimerization of caspase-8 and its complete cleavage and activation, as would occur with death receptor ligation. It is thus likely that different stoichiometric conditions and localization of caspase-8 activation govern caspase-8 homodimerization and activation at death receptors when forming a death-inducing signal complex for apoptosis versus caspase-8/c-FLIP heterodimers that are needed for cell survival and growth. However, there are several reports of Fas stimulating growth or activation of various cell types, including neurons, hepatocytes, dendritic cells, fibroblasts, and certain tumors (31–36). In some of these studies, analysis of signal pathways has shown up-regulation of c-FLIP and activation of ERK and/or NF-κB following Fas stimulation (33, 34). Thus, it is likely that with sufficient levels of c-FLIP, Fas signaling can form caspase-8/c-FLIP heterodimers that divert signals for cell survival and growth.
The inability of p43FLIP to rescue defective growth of effector T cells following their loss of caspase-8 is likely due to activation of the Ripoptosome. Recent studies have revealed that tonic caspase-8 activity is required to cleave RIPK1 to prevent formation of a complex that activates RIPK3 and MLKL to promote caspase-independent necroptosis (9, 37). Thus, although c-FLIPL is a critical caspase-8 substrate in T cell survival, the formation of p43FLIP cannot supplant the need for continued caspase-8 activity to cleave RIPK1. However, p43FLIP is required to maintain precise moderate amounts of caspase-8 activation and to promote recruitment of RIPK1 to the active caspase complex. Both caspase-8-deficient and c-FLIP-deficient mice are embryonic lethal (38, 39). In the case of caspase-8 deficiency, lethality can be rescued by the additional deletion of RIPK3 (5, 6). Of interest is that rescue of lethality from loss of c-FLIP requires the further loss of RIPK3 and FADD (40). This may allude to additional functions of c-FLIP beyond regulating caspase-8 activity.
Acknowledgment
We thank Dr. Takao Kataoka for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants GM103496, AI36333, and AI45666.
- ANOVA
- analysis of variance.
REFERENCES
- 1. Salmena L., Hakem R. (2005) Caspase-8 deficiency in T cells leads to a lethal lymphoinfiltrative immune disorder. J. Exp. Med. 202, 727–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beisner D. R., Ch'en I. L., Kolla R. V., Hoffmann A., Hedrick S. M. (2005) Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J. Immunol. 175, 3469–3473 [DOI] [PubMed] [Google Scholar]
- 3. Lemmers B., Salmena L., Bidère N., Su H., Matysiak-Zablocki E., Murakami K., Ohashi P. S., Jurisicova A., Lenardo M., Hakem R., Hakem A. (2007) Essential role for caspase-8 in Toll-like receptors and NFκB signaling. J. Biol. Chem. 282, 7416–7423 [DOI] [PubMed] [Google Scholar]
- 4. Rajput A., Kovalenko A., Bogdanov K., Yang S. H., Kang T. B., Kim J. C., Du J., Wallach D. (2011) RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351 [DOI] [PubMed] [Google Scholar]
- 5. Kaiser W. J., Upton J. W., Long A. B., Livingston-Rosanoff D., Daley-Bauer L. P., Hakem R., Caspary T., Mocarski E. S. (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oberst A., Dillon C. P., Weinlich R., McCormick L. L., Fitzgerald P., Pop C., Hakem R., Salvesen G. S., Green D. R. (2011) Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang H., Zhou X., McQuade T., Li J., Chan F. K., Zhang J. (2011) Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feoktistova M., Geserick P., Kellert B., Dimitrova D. P., Langlais C., Hupe M., Cain K., MacFarlane M., Häcker G., Leverkus M. (2011) cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F., Zachariou A., Lopez J., MacFarlane M., Cain K., Meier P. (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 [DOI] [PubMed] [Google Scholar]
- 10. Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J.-L., Schröter M., Burns K., Mattmann C., Rimoldi D., French L. E., Tschopp J. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 [DOI] [PubMed] [Google Scholar]
- 11. Thome M., Schneider P., Hofmann K., Fickenscher H., Meinl E., Neipel F., Mattmann C., Burns K., Bodmer J.-L., Schröter M., Schröter M., Scaffidi C., Krammer P. H., Peter M. E., Tschopp J. (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517–521 [DOI] [PubMed] [Google Scholar]
- 12. Tschopp J., Irmler M., Thome M. (1998) Inhibition of Fas death signals by FLIPs. Curr. Opin. Immunol. 10, 552–558 [DOI] [PubMed] [Google Scholar]
- 13. Shu H. B., Halpin D. R., Goeddel D. V. (1997) Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 6, 751–763 [DOI] [PubMed] [Google Scholar]
- 14. Chun H. J., Zheng L., Ahmad M., Wang J., Speirs C. K., Siegel R. M., Dale J. K., Puck J., Davis J., Hall C. G., Skoda-Smith S., Atkinson T. P., Straus S. E., Lenardo M. J. (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419, 395–399 [DOI] [PubMed] [Google Scholar]
- 15. Salmena L., Lemmers B., Hakem A., Matysiak-Zablocki E., Murakami K., Au P. Y., Berry D. M., Tamblyn L., Shehabeldin A., Migon E., Wakeham A., Bouchard D., Yeh W. C., McGlade J. C., Ohashi P. S., Hakem R. (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Micheau O., Thome M., Schneider P., Holler N., Tschopp J., Nicholson D. W., Briand C., Grütter M. G. (2002) The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 277, 45162–45171 [DOI] [PubMed] [Google Scholar]
- 17. Dohrman A., Russell J. Q., Cuenin S., Fortner K., Tschopp J., Budd R. C. (2005) Cellular FLIP long form augments caspase activity and death of T cells through heterodimerization with and activation of caspase-8. J. Immunol. 175, 311–318 [DOI] [PubMed] [Google Scholar]
- 18. Rincón M., Flavell R. A. (1997) Transcription mediated by NFAT is highly inducible in effector CD4+ T helper 2 (Th2) cells but not in Th1 cells. Mol. Cell. Biol. 17, 1522–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizushima S., Nagata S. (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang N., He Y.-W. (2005) An essential role for c-FLIP in the efficient development of mature T lymphocytes. J. Exp. Med. 202, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chau H., Wong V., Chen N. J., Huang H. L., Lin W. J., Mirtsos C., Elford A. R., Bonnard M., Wakeham A., You-Ten A. I., Lemmers B., Salmena L., Pellegrini M., Hakem R., Mak T. W., Ohashi P., Yeh W. C. (2005) Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J. Exp. Med. 202, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kataoka T., Tschopp J. (2004) N-terminal fragment of c-FLIPL processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-κB signaling pathway. Mol. Cell. Biol. 24, 2627–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dohrman A., Kataoka T., Cuenin S., Russell J. Q., Tschopp J., Budd R. C. (2005) Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-κB activation. J. Immunol. 174, 5270–5278 [DOI] [PubMed] [Google Scholar]
- 24. Boatright K. M., Deis C., Denault J. B., Sutherlin D. P., Salvesen G. S. (2004) Activation of caspases-8 and -10 by FLIPL. Biochem. J. 382, 651–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pop C., Oberst A., Drag M., Van Raam B. J., Riedl S. J., Green D. R., Salvesen G. S. (2011) FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 433, 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lenardo M. J. (1991) Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature 353, 858–861 [DOI] [PubMed] [Google Scholar]
- 27. Ju S.-T., Panka D. J., Cui H., Ettinger R., El-Khatib M., Sherr D. H., Stanger B. Z., Marshak-Rothstein A. (1995) Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 373, 444–448 [DOI] [PubMed] [Google Scholar]
- 28. Kataoka T., Budd R. C., Holler N., Thome M., Martinon F., Irmler M., Burns K., Hahne M., Kennedy N., Kovacsovics M., Tschopp J. (2000) The caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol. 10, 640–648 [DOI] [PubMed] [Google Scholar]
- 29. Hinshaw-Makepeace J., Huston G., Fortner K. A., Russell J. Q., Holoch D., Swain S., Budd R. C. (2008) c-FLIP-S reduces activation and caspase and NF-κB pathways and decreases T cell survival. Eur. J. Immunol. 38, 54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schleich K., Warnken U., Fricker N., Oztürk S., Richter P., Kammerer K., Schnölzer M., Krammer P. H., Lavrik I. N. (2012) Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol. Cell 47, 306–319 [DOI] [PubMed] [Google Scholar]
- 31. Aggarwal B. B., Singh S., LaPushin R., Totpal K. (1995) Fas antigen signals proliferation of normal human diploid fibroblast and its mechanism is different from tumor necrosis factor receptor. FEBS Lett. 364, 5–8 [DOI] [PubMed] [Google Scholar]
- 32. Desbarats J., Newell M. K. (2000) Fas engagement accelerates liver regeneration after partial hepatectomy. Nat. Med. 6, 920–923 [DOI] [PubMed] [Google Scholar]
- 33. Desbarats J., Birge R. B., Mimouni-Rongy M., Weinstein D. E., Palerme J. S., Newell M. K. (2003) Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat. Cell Biol. 5, 118–125 [DOI] [PubMed] [Google Scholar]
- 34. Collins C., Wolfe J., Roessner K., Shi C., Sigal L. H., Budd R. C. (2005) Lyme arthritis synovial γδ T cells instruct dendritic cells via Fas ligand. J. Immunol. 175, 5656–5665 [DOI] [PubMed] [Google Scholar]
- 35. Perlman H., Pagliari L. J., Liu H., Koch A. E., Haines G. K., 3rd, Pope R. M. (2001) Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum. 44, 21–30 [DOI] [PubMed] [Google Scholar]
- 36. Owen-Schaub L. B., Radinsky R., Kruzel E., Berry K., Yonehara S. (1994) Anti-Fas on nonhematopoietic tumors: levels of Fas/APO-1 and bcl-2 are not predictive of biological responsiveness. Cancer Res. 54, 1580–1586 [PubMed] [Google Scholar]
- 37. Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 [DOI] [PubMed] [Google Scholar]
- 38. Varfolomeev E. E., Schuchmann M., Luria V., Chiannilkulchai N., Beckmann J. S., Mett I. L., Rebrikov D., Brodianski V. M., Kemper O. C., Kollet O., Lapidot T., Soffer D., Sobe T., Avraham K. B., Goncharov T., Holtmann H., Lonai P., Wallach D. (1998) Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276 [DOI] [PubMed] [Google Scholar]
- 39. Yeh W. C., Itie A., Elia A. J., Ng M., Shu H. B., Wakeham A., Mirtsos C., Suzuki N., Bonnard M., Goeddel D. V., Mak T. W. (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12, 633–642 [DOI] [PubMed] [Google Scholar]
- 40. Dillon C. P., Oberst A., Weinlich R., Janke L. J., Kang T. B., Ben-Moshe T., Mak T. W., Wallach D., Green D. R. (2012) Survival function of the FADD-CASPASE-8-cFLIPL complex. Cell Rep. 1, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]