

Background: TcpB is a TIR domain-containing protein form Brucella.
Results: TcpB interacts with the host Toll-like receptor and adaptors, and its structure reveals a dimer essential for activity.
Conclusion: TcpB forms a nonfunctional complex with host molecules, thus suppressing signaling.
Significance: The work explains the structural and functional basis of immune suppression by the protein TcpB from a pathogenic bacterium.
Keywords: Adaptor Proteins, Innate Immunity, Protein Structure, Toll IL-1 Receptor (TIR) Domain, Toll-like Receptors (TLR)
Abstract
Upon activation of Toll-like receptors (TLRs), cytoplasmic Toll/interleukin-1 receptor (TIR) domains of the receptors undergo homo- or heterodimerization. This in turn leads to the recruitment of adaptor proteins, activation of transcription factors, and the secretion of pro-inflammatory cytokines. Recent studies have described the TIR domain-containing protein from Brucella melitensis, TcpB (BtpA/Btp1), to be involved in virulence and suppression of host innate immune responses. TcpB interferes with TLR4 and TLR2 signaling pathways by a mechanism that remains controversial. In this study, we show using co-immunoprecipitation analyses that TcpB interacts with MAL, MyD88, and TLR4 but interferes only with the MAL-TLR4 interaction. We present the crystal structure of the TcpB TIR domain, which reveals significant structural differences in the loop regions compared with other TIR domain structures. We demonstrate that TcpB forms a dimer in solution, and the crystal structure reveals the dimerization interface, which we validate by mutagenesis and biophysical studies. Our study advances the understanding of the molecular mechanisms of host immunosuppression by bacterial pathogens.
Introduction
Toll-like receptors (TLRs)5 are a family of pattern recognition receptors that sense danger in the forms of both conserved microbial molecules and host-derived factors that are present when homeostasis is dysregulated (1). The Toll/interleukin-1 receptor (TIR) domain is a key molecular component of TLR-mediated innate immune response pathways. All mammalian TLRs and several cytosolic adaptor proteins contain TIR domains in their C-terminal regions (2). Homo- or heterotypic dimerization of receptor TIR domains is required to initiate downstream signaling via the recruitment of TIR domain-containing adaptor proteins such as MyD88 (myeloid differentiation primary response gene 88), TRIF (TIR domain-containing adaptor protein inducing INF-β), MAL (MyD88 adaptor-like protein), and TRAM (TRIF-related adaptor molecule) (2). Recruitment of one or a combination of these adaptors to TLRs leads to the activation of transcription factors such as NF-κB, AP-1 (activator protein-1), and IRF3 (interferon regulatory transcription factor 3). This enables regulated expression of a suite of innate-immune genes, including those encoding pro-inflammatory cytokines and chemokines that drive immune cell recruitment to the inflammatory lesion (1).
Brucellosis, which results in more than 500,000 deaths annually worldwide, is a zoonotic disease in humans caused by the Gram-negative Brucella (3, 4). Brucella sp. have developed different strategies to evade TLR-mediated host innate immune recognition. One strategy involves the production of virulence factors that interact with and antagonize key signaling components to suppress TLR signaling (3, 5). Recent studies have identified a TIR domain-containing protein known as TcpB, BtpA, or Btp1 produced by Brucella melitensis (hereafter, we use the term TcpB) (6, 7). It has been reported that TcpB restricts pro-inflammatory responses by suppressing the TLR4-, TLR2-, MAL-, and MyD88-mediated activation of NF-κB and inhibiting dendritic cell maturation (8–11). The protein contains two domains, an N-terminal domain reported to bind to phosphoinositides (10) and a C-terminal TIR domain (TcpB-TIR). Radhakrishnan et al. (10) suggested that TcpB mimics MAL by using its N-terminal domain to bind to lipids in the plasma membrane and competes with MAL for binding to MyD88. However, Sengupta et al. (9) reported that TcpB interacts with MAL and not MyD88 and that TcpB does not interfere with the MAL-MyD88 interaction. Notably, TcpB binding to MyD88 has been reported in other studies (7, 11), with a recent study by Chaudhary et al. (12) suggesting that TcpB binds to MyD88 with higher affinity compared with MAL. The data reported so far do not agree on the mechanism by which TcpB interacts with mammalian TIR domain-containing proteins (6). Although TIR domains generally interact homo- or heterotypically with other TIR domains (2), all previous TcpB interaction studies used full-length TcpB. Furthermore, the interaction of TcpB with MyD88 was suggested to be independent of the MyD88 TIR domain (12).
Although TIR domain homo- and heterotypic association is clearly crucial for TLR signaling, the majority of isolated TIR domains exist as monomers in solution (2). Crystal structures of TIR domains from mammalian, plant, and bacterial TIR domain-containing proteins have significant structural differences, and analyses of the crystal lattices have not revealed any common homotypic TIR-TIR domain interfaces (2, 13–21). However, mutational studies have validated a number of proposed interfaces, often involving different loop regions (2). Although the available data suggest that microbial TIR domain-containing proteins may also be functional as dimers (2, 22), the mechanisms of action are not clear.
To elucidate the molecular basis of function of the microbial TIR domain-containing proteins, we used the B. melitensis TcpB protein as a model to study the interaction between the bacterial and mammalian TIR domain-containing proteins. Using co-immunoprecipitation assays, we show that TcpB can interact with MAL, MyD88, and TLR4, with the interaction interface for both MAL and TLR4 involving the TcpB BB loop. TcpB does not interact with TRAM or IRAK-2, suggesting that TcpB specifically targets the MAL-dependent signaling pathway. Furthermore, competition assays demonstrate that TcpB disrupts the receptor-adaptor interaction between TLR4 and MAL, but not the adaptor-adaptor interaction between MAL and MyD88. We also show that TcpB exists as a stable dimer in solution and that both the TIR and N-terminal domains are involved in self-association. Structural and functional analysis of the TcpB TIR domain reveals a homotypic interface that is required for potent NF-κB suppression mediated by TcpB. This study provides new insights into the mechanism of bacterial TIR domain-containing protein immunomodulation of TLR signaling at the molecular and structural level.
EXPERIMENTAL PROCEDURES
Plasmids
For recombinant protein expression in Escherichia coli, the cDNA of the full-length TcpB (TcpB-fl) was generated by PCR-based gene synthesis and used for cloning TcpB-fl, the TcpB-TIR domain (TcpB120–250), a longer construct containing the TcpB-TIR domain (TcpB70–250) and the TcpB N-terminal domain (TcpB1–119) sequences into the pMCSG7 vector (23), and subsequent transformation into E. coli BL21 (DE3) for protein expression. The TcpB residue numbering used in the paper corresponds to the UniProt accession number Q8YF53. For protein expression in mammalian cells, a plasmid encoding the cDNA of TcpB-fl (obtained from Thomas Miethke, Technical University, Munich, Germany (11)) was used to clone TcpB and its mutants into the pEF6/V5 vector (Invitrogen), to be subsequently used in luciferase NF-κB reporter assays and co-immunoprecipitation studies. HA-MyD88, HA-MAL, HA-TLR4, FLAG-MAL, FLAG-TRAM, Myc-IRAK-2, NF-κB reporter gene, and phRL-TK reporter gene were obtained from Andrew Bowie (Trinity College, Dublin, Ireland).
Immunoprecipitation and Immunoblotting
HEK293 cells (1 × 106 cells/well) were seeded into 6-well plates, 24 h before transfection with Lipofectamine 2000. Cells were harvested after 24 h using 350 μl of radioimmunoprecipitation assay lysis buffer (50 mm Tris, pH 8, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate). To test the interaction of TcpB and TcpBG158A with MAL, MyD88, TLR4, TRAM, and IRAK-2, 1 μg of full-length V5-TcpB or V5-TcpBG158A was co-transfected with 1 μg of HA-MAL, HA-MyD88, HA-TLR4, FLAG-TRAM, Myc-IRAK-2, or HA-tagged control protein (all correspond to full-length proteins). 50 μl of lysate was retained for analysis of protein expression. The remainder was incubated with 2 μl of anti-V5 for 2 h at 4 °C with rotation. 25 μl of prewashed (washing buffer: 10 mm Hepes, pH 8.0, 1.5 mm MgCl2, 10 mm KCl, 150 mm NaCl) Dynabeads® protein G (Invitrogen) magnetic beads were added to the lysate and further incubated for 2 h at 4 °C with rotation. The lysates were then washed three times with high salt concentration buffer (10 mm Hepes, pH 8.0, 1.5 MgCl2, 10 mm KCl, 300 mm NaCl). To test the interference of TcpB with receptor-adaptor or adaptor-adaptor interactions, 1 μg of FLAG-MAL or FLAG-TRAM was co-transfected with 1 μg of HA-MyD88 or HA-TLR4. The lysate was incubated with 2 μl of anti-FLAG-M2 for 2 h at 4 °C with rotation. 25 μl of prewashed (washing buffer: 10 mm Hepes, pH 8.0, 1.5 mm MgCl2, 10 mm KCl, 150 mm NaCl) Dynabeads® protein G (Invitrogen) magnetic beads were added to the lysate and further incubated for 2 h at 4 °C with rotation. Increasing amounts (1, 10, and 100 μg) of recombinant TcpB-fl, TcpB120–250, TcpB1–119, or GST proteins were added to the lysate and further incubated for 3 h at 4 °C with rotation. The lysates were then washed three times with high salt concentration buffer. Samples were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane for immunoblotting.
NF-κB Luciferase Reporter Assay
To compare the NF-κB suppression activity of wild-type versus mutant full-length TcpB, HEK293-TLR4-MD2 cells were seeded into 24-well plates (8 × 104 cells/well). 24 h later, cells were co-transfected with 100 ng of NF-κB-luciferase reporter gene, 20 ng/well of phRL-TK reporter gene, and 200 ng of TcpB or TcpB mutants using Lipofectamine 2000. The total amount of DNA per transfection was kept constant at 460 ng by the addition of pGL-2B empty vector. After 24 h, cells were stimulated with 100 ng/ml LPS. After a further 8 h, cells were lysed in passive lysis buffer (Promega), and whole cell lysates were analyzed for firefly and Renilla luciferase activities using a dual luciferase assay kit (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity, and data are expressed as relative luciferase activity and are representative of three independent experiments, each performed in duplicate. Statistical analysis was carried out in GraphPad Prism version 6 (San Diego, CA) using two-way analysis of variance with Dunnett's multiple comparisons test.
Protein Production, Purification, and Crystallization
The TcpB120–250 protein was expressed using auto-induction media (24) and purified using nickel affinity chromatography followed by size exclusion chromatography. Peak fractions were pooled. SDS-PAGE analysis showed more than 95% protein purity. The protein was concentrated to 22 mg/ml and subjected to crystallization trials. The protein was crystallized in 0.1 m Bis-Tris, pH 5.5, 0.2 m NaCl, and 25% w/v PEG3350 using the hanging drop vapor diffusion method (25).
Data Collection and Structure Determination
Diffraction data were collected using the MX2 beamline at the Australian Synchrotron (Melbourne, Australia) using Blu-Ice software (26). Data indexing/integration and scaling were performed using XDS (27) and SCALA (28), respectively. The structure was solved by molecular replacement using Phaser (29), and the PdTIR (Paracoccus denitrificans TIR domain-containing protein) structure (Protein Data Bank code 3h16) (19) as a template model. PdTIR structure was the best model because of its high (61%) amino acid sequence identity with TcpB120–250. Automated model building was performed using Autobuild within the Phenix package (30, 31). Iterative model building in Coot (32) was carried out between rounds of refinement in Phenix and Buster (30, 33). Automated noncrystallographic symmetry with local structural similarity restraints was used throughout refinement in Buster (34). Structure validation was performed using MolProbity (35), and the structure was analyzed using Coot and PyMOL. The figures were prepared using PyMOL. The coordinates and structure factors have been deposited in the Protein Data Bank (code 4c7m).
Multiangle Light Scattering
Multiangle light scattering (MALS) coupled with size exclusion chromatography was performed using a DAWN HELEOS II 18-angle light scattering detector coupled with an Optilab rEX refractive index detector (Wyatt Technology, Santa Barbara, CA) and combined inline with a pre-equilibrated Superdex 200 10/300 size exclusion column (GE Healthcare), using a buffer containing 50 mm Hepes, pH 8.0, 250 mm NaCl, and 1 mm DTT (36). All experiments were performed at room temperature, and ∼300 μg of each sample was loaded. Molecular mass calculations were calculated at the top of the elution peaks using the Astra 6.1 software (Wyatt Technology) based on extrapolation from Debye plots (Zimm formalism) using a dn/dc value of 0.1850 ml g−1 (37).
Circular Dichroism
CD spectra were measured using the Jasco J710 spectropolarimeter (Tokyo, Japan) for TcpB-fl, TcpB120–250, TcpB1–119, and TcpB70–250 at 25 °C. Samples were predialyzed into 20 mm sodium phosphate, pH 8.0, and 100 mm NaCl. The data were collected using 0.5-nm wavelength increments from 260 to 195 nm at 20 nm/min, with a spectral bandwidth of 1.0 nm, 0.1-mm path length cell, 0.5-s response time, and five accumulations.
Small Angle X-ray Scattering (SAXS)
Purified TcpB-fl, TcpB120–250, TcpB70–250, and TcpB1–119 were gel-filtered using a buffer containing 50 mm Hepes, pH 8.0, 250 mm NaCl, and 1 mm DTT, at 4 °C. Concentrations were obtained by UV absorbance at 280 nm. Data were collected at the SAXS/WAXS beamline of the Australian Synchrotron (Melbourne, Australia) on a Pilatus 1M detector at a sample to detector distance of 1.6 m. SAXS measurements were collected at 20 °C from dilution series made with post-peak gel filtration buffer. 90 μl of sample was injected through a 1.5-mm-diameter quartz capillary at a rate of 1 μl/s, capturing an image every 1 s. Consistent, successive exposures were normalized to transmitted intensity, reduced, scaled to absolute intensity using pure water, averaged, and buffer-subtracted using ScatterBrain. The ATSAS 2.5 software package was used for subsequent analyses (38–40). Guinier analysis was performed for q.Rg < 1.3 using AUTORG in PRIMUS, and data sets were examined for concentration dependence and linearity. P(r) distributions were obtained for all constructs by indirect transformation in AUTOGNOM. Normalized Kratky plots were calculated as q.Rg versus q.Rg.I(q)/I(0), and they were analyzed for increases at high q indicative of flexibility. For constructs showing limited flexibility, molecular masses were estimated from the P(r) distributions using SAXSMoW (41), and those for highly flexible constructs were calculated from absolute I(0). Predicted scattering from the crystallographic dimer was calculated with CRYSOL (42) using default parameters. In cases where flexibility was limited, and no concentration dependence was observed, ab inito models were generated from 12 DAMMIF (43) runs under P2 symmetry restraints and clustered and averaged in DAMAVER (44).
RESULTS
TcpB Interacts with MAL, MyD88, and TLR4
We used co-immunoprecipitation analyses to test in parallel the interaction of V5-TcpB with HA-MAL, HA-MyD88, HA-TLR4, FLAG-TRAM, and Myc-IRAK-2 (all full-length proteins, with specific tags as indicated). We began by confirming known interactions between MAL and MyD88, MAL and TLR4, TRAM and MyD88, and TRAM and TLR4 (Fig. 1). V5-TcpB interacted specifically with HA-MAL, HA-MyD88, and HA-TLR4 (Fig. 2, A and B), but not FLAG-TRAM, Myc-IRAK-2 (Fig. 2C), or an HA-tagged control protein (Fig. 2B). These results suggest that, in the case of TLR4 signaling, TcpB specifically targets the MAL/MyD88-dependent signaling, but not TRAM-dependent signaling. In previous studies, the TcpB BB loop was suggested to be critical for function (10). To test this suggestion, we mutated the conserved glycine 158 to alanine in the BB loop (V5-TcpBG158A) and tested the interaction with HA-MAL, HA-MyD88, and HA-TLR4. Critically, the G158A mutation abolished the interaction with HA-TLR4 (Fig. 2B) and significantly reduced the interaction with HA-MAL (Fig. 2A), but the interaction with HA-MyD88 was unaffected (Fig. 2A), suggesting that only the interaction with MAL and TLR4 requires an intact TcpB BB loop.
FIGURE 1.

Western blot analysis of controls for co-immunoprecipitation analysis. Using Lipofectamine 2000, HEK293 cells (1 × 106 cells/well) were co-transfected with 1 μg of FLAG-MAL and 1 μg of HA-MyD88 (A, lane 4) or 1 μg of Myc-IRAK-2 (A, lane 5); 1 μg of FLAG-TRAM and 1 μg of HA-MyD88 (A, lane 9) or 1 μg of Myc-IRAK-2 (A, lane 10); 1 μg of FLAG-MAL and 1 μg of HA-TLR4 (B, lane 4) or HA-tagged control (B, lane 5); and 1 μg of FLAG-TRAM and 1 μg of HA-TLR4 (B, lane 9) or HA-tagged control (B, lane 10). The lysates were immunoprecipitated with anti-FLAG-M2 antibody bound to prewashed Dynabeads® protein G magnetic beads, followed by a 2-h incubation at 4 °C with rotation. Beads were then subjected to washing, SDS-PAGE, and immunoblotting with the indicated antibodies. Data in A and B were generated in the same experiment, and both panels are representative of two independent experiments. Notably, MAL-IRAK-2, MyD88-IRAK-2, and TRAM-MyD88 interactions have been reported previously (57–59).
FIGURE 2.

Co-immunoprecipitation of TcpB with MAL, MyD88, and TLR4. HEK293 cells (1 × 106 cells/well) were co-transfected using Lipofectamine 2000 with 1 μg of V5-TcpB or V5-TcpBG158A and 1 μg of HA-MAL (A, lanes 6 and 7), HA-MyD88 (A, lanes 8 and 9), HA-TLR4 (B, lanes 5 and 6), HA-tagged control protein (B, lanes 7 and 8), FLAG-TRAM (C, lanes 7 and 8) or Myc-IRAK-2 (C, lanes 9 and 10). After 24 h, the lysates were immunoprecipitated with anti-V5 antibody bound to prewashed Dynabeads® protein G magnetic beads, followed by a 2-h incubation at 4 °C with rotation. Beads were then subjected to washing, SDS-PAGE, and immunoblotting with the indicated antibodies. Data in A–C were generated in the same experiment, and all panels are representative of two independent experiments.
TcpB Disrupts the Interaction between MAL and TLR4
Because TcpB can interact with TIR domains from multiple proteins, it seems likely that its mechanism of inhibition of TLR4 signaling would involve disruption of receptor-adaptor or adaptor-adaptor TIR domain interactions. Therefore, we used competition assays to investigate the effects of TcpB on these interactions. To assess the effect of TcpB on MAL-MyD88, MAL-TLR4, TRAM-MyD88, or TRAM-TLR4 interactions, increasing amounts of recombinant full-length TcpB (TcpB-fl), TcpB-TIR (TcpB120–250), or TcpB N-terminal domain (TcpB1–119) were incubated with the lysate of cells co-transfected with FLAG-MAL and HA-MyD88, FLAG-MAL and HA-TLR4, FLAG-TRAM and HA-MyD88, or FLAG-TRAM and HA-TLR4. The addition of increasing amounts of TcpB-fl, TcpB-TIR120–250, or TcpB1–119 had no effect on the interaction between MAL and MyD88 (Fig. 3A), TRAM and MyD88, and TRAM and TLR4 (Fig. 3, C and D). By contrast, under the same conditions, both TcpB-fl and TcpB-TIR120–250 abolished the interaction between MAL and TLR4 when the cell lysate was incubated with 10 μg or more of recombinant protein (Fig. 3B). For the TcpB1–119 titration, an interaction was still apparent at the highest concentration of recombinant protein (Fig. 3B), suggesting that this region of TcpB does not impair the MAL-TLR4 interaction. Recombinant GST protein, used as a negative control, did not disrupt MAL-MyD88, TRAM-MyD88, MAL-TLR4, or the TRAM-TLR4 interactions (Fig. 3).
FIGURE 3.

Effect of recombinant TcpB on the interaction between MAL-MyD88, MAL-TLR4, TRAM-MyD88, and TRAM-TLR4. HEK293 cells (1 × 106 cells/well) were co-transfected using Lipofectamine 2000 with 1 μg of FLAG-MAL with 1 μg of HA-MyD88 (A) or 1 μg of HA-TLR4 (B) and with 1 μg of FLAG-TRAM with 1 μg of HA-MyD88 (C) or 1 μg of HA-TLR4 (D). After 24 h of transfection, the lysates were immunoprecipitated with anti-FLAG-M2 bound to prewashed Dynabeads® protein G magnetic beads, followed by a 2-h incubation at 4 °C with rotation. Increasing amounts (1, 10, and 100 μg) of recombinant TcpB-fl (A–D, lanes 2–4), TcpB120–250 (A–D, lanes 5–7), TcpB1–119 (A–D, lanes 9–11), or GST (A–D, lanes 12–14) were added and incubated for 3 h at 4 °C with rotation. Beads were then subjected to washing, SDS-PAGE, and immunoblotting with the indicated antibodies. Data are representative of two independent experiments.
The Crystal Structure of TcpB Reveals Unique Loop Conformations and Surface Features
To obtain structural insights into the TcpB-mediated suppression of TLR4 signaling, we determined the crystal structure of TcpB-TIR. Several constructs were expressed, purified, and subjected to crystallization trials (25). One of the constructs, encompassing residues 120–250 (TcpB120–250), yielded plate-like crystals that diffracted x-rays to 2.6 Å resolution, and the structure was refined to final Rwork/Rfree values of 0.23/0.24. The small difference between Rwork and Rfree may reflect the usage of the local structure similarity approach in automated noncrystallographic symmetry restraints during the refinement process (34). The crystals have the symmetry of the space group P21 with four molecules (A, B, C, and D) in the asymmetric unit (Table 1). Each of the molecules has a typical TIR domain fold with a central five-stranded parallel β-sheet (βA–βE) surrounded by five α-helices (αA–αE) connected by loops (Fig. 4A; the naming of secondary structure elements follows the nomenclature used for the structures of PdTIR and the TLR1 TIR domain (13, 19)). A portion of the αC helix of chain A (residues 188–189), the CD loop of chains B (residues 199–204), C (residues 199–203), and D (residues 198–204), and the BB loop of chain C (residues 156–160) have poor electron density and therefore could not be modeled. All of the four molecules have similar structures with an RMSD <0.65 Å for 116 equivalent Cα atoms.
TABLE 1.
Crystallographic data
| Diffraction data statistics | |
| Space group | P21 |
| Unit cell dimensions (Å) | |
| a | 51.97 |
| b | 73.68 |
| c | 74.76 |
| β | 93.29° |
| Molecules per asymmetric unit | 4 |
| Resolution range (Å) | 52.43- 2.71 (2.57–2.71)a |
| No. of unique observations | 18,090 |
| Completeness (%) | 100.0 (99.7) |
| Multiplicity | 3.7 (3.7) |
| Rmerge (%)b | 8.9 (79.0) |
| Average I/σ(I) (%) | 10.6 (1.9) |
| Refinement statistics | |
| Resolution (Å) | 42.42–2.57 |
| No. of reflections, work set | 17070 |
| No. of reflections, test set | 1009 |
| Rwork (%)c | 23.17 |
| Rfree (%)d | 24.01 |
| No. of protein atoms | 4,061 |
| No. of water molecules | 8 |
| Overall B factor (Å2) | 59.46 |
| RMSDs from ideal values | |
| Bonds (Å) | 0.010 |
| Angles (°) | 1.05 |
| Ramachandran plot (%)e | |
| Favored | 98.98 |
| Disallowed | 0 |
a The numbers in parentheses are for the highest resolution shell.
b Rmerge = Σhkl(Σi(|Ihkl,i − <Ihkl>|))/Σhkl,i <Ihkl>, where Ihkl,i is the intensity of an individual measurement of the reflection with Miller indices h, k, and l, and <Ihkl> is the mean intensity of that reflection.
c Rwork = Σhkl(||Fobs hkl| − |Fcalc hkl||)/|Fobs hkl|, where |Fobs hkl| and |Fcalc hkl| are the observed and calculated structure factor amplitudes.
d Rfree is equivalent to Rwork but is calculated with reflections (5.6%) omitted from the refinement process.
e Calculated using MolProbity (35).
FIGURE 4.

Crystal structure of the TcpB TIR domain and comparison with available TIR domain structures. A, cartoon representation of the TcpB120–250 crystal structure, colored by secondary structure elements (α-helices, blue; β-sheet, purple; loop regions, pink). B–E, ribbon representations of the superposition of TcpB120–250 (cyan) with PdTIR (B, orange) (Protein Data Bank code 3h16) and the TIR domains from TLR2 (C, blue) (Protein Data Bank code 1fyw), MyD88 (D, green) (Protein Data Bank code 4dom), and MAL (E, yellow) (Protein Data Bank code 2y92).
A search using DALI (45) identified PdTIR (Protein Data Bank code 3h16) as the closest structural homologue with an RMSD value of 1.2 Å for 127 Cα atoms and a Z score of 22.4. Mammalian TIR domain structures from MAL (Protein Data Bank code 2y92; RMSD value of 2.5 Å for 104 Cα atoms and Z score of 10.4), MyD88 (Protein Data Bank code 4dom; RMSD value of 2.9 Å for 105 Cα atoms and Z score of 9.3), and TLR2 (Protein Data Bank code 1fyw; RMSD value of 2.8 Å for 109 Cα atoms and Z score of 9.3) have less structural similarity. The structure reveals distinct loop conformations compared with other TIR domain structures (Fig. 4, B–E). Superposition of the structures of TcpB120–250 and PdTIR reveals that the BB loop adopts a similar conformation in both structures, whereas the conformations of the CD and DE loops are significantly different (Fig. 4B). TcpB120–250 also lacks the 310 helix between βB strand and αB helix observed in the PdTIR and MyD88 TIR domain structures (Fig. 5) (18, 19). In comparison to the mammalian TIR domain structures, TcpB120–250 adopts significantly different AB, BB, CD, DE, DD, and EE loop conformations (Fig. 4, C–E). Analysis of the surface electrostatic potential of TcpB120–250 reveals that it contains extensive positively charged patches that are absent in the TIR domains of MAL or PdTIR, which both have strongly electronegative surfaces (Fig. 6). It has been suggested that complementary-charged surfaces mediate TIR domain interactions (16, 46).
FIGURE 5.
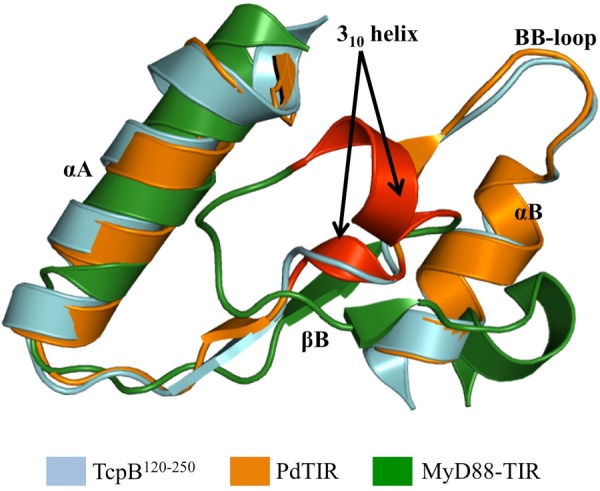
Superposition of TIR domains to highlight the differences in 310 helices. Superposition of the structure of TcpB120–250 (cyan), PdTIR (orange), and the TIR domain of MyD88 (green) is shown. The 310 helices are colored red, and this structural feature is only found in the structures of PdTIR and the MyD88 TIR domain and is missing from the TcpB120–250 structure.
FIGURE 6.

Comparison of electrostatic properties. The solvent-accessible surfaces of TcpB120–250 (A), PdTIR (B), TLR4 TIR domain (C), MyD88 TIR domain (D), and MAL TIR domain (E) are colored according to electrostatic potential, ranging from blue (positive charge) through white to red (negative charge) in the range ± 0.5 kT/e. The electrostatic potential was calculated using the APBS tool within the PyMOL software (60). The upper panel is a cartoon representation of the TcpB120–250 structure in A, to help with the orientation of the structures. The TLR4 TIR domain structure was modeled using Phyre2 (49). TcpB120–250 has a distinctive positive charge across an area covering the βB, BB loop, and αB, compared with scattered negatively charged patches in PdTIR. The TIR domains from the mammalian proteins MyD88, MAL, and TLR4 are characterized by extensive positively or negatively charged patches across distant regions.
TcpB Exists as a Dimer in Solution
We examined the oligomeric state of recombinant TcpB constructs (TcpB-fl, TcpB120–250, TcpB70–250, and TcpB1–119) in solution by size exclusion chromatography coupled with MALS and by SAXS. Full-length TcpB behaved as a stable dimer according to MALS, with an experimental molecular mass of 55.7 kDa, consistent with a theoretical dimer molecular mass of 56.0 kDa (Fig. 7A). SAXS analysis of this protein was precluded by aggregation. Conversely, TcpB120–250 was found to be in a dynamic equilibrium between a monomer and a dimer. Using MALS, the elution peak showed a molecular mass of 23.7 kDa at the top of the peak (Fig. 8A), between the expected mass of the dimeric (30.0 kDa) and monomeric (15.0 kDa) states. A decrease in molecular mass from 24.9–21.7 kDa was observed across the peak, suggesting that the TcpB120–250 is in a dynamic equilibrium between a monomer and a dimer (47). The SAXS-derived molecular mass was seen to increase from 24.6 to 29.2 kDa across a dilution series, suggesting a concentration-dependent monomer-dimer equilibrium (Fig. 9, A–C). Similarly, TcpB1–119 also displayed evidence of monomer-dimer equilibrium. The MALS elution peak showed a molecular mass of 17.5 kDa at the top of the peak, and the range of molecular masses observed across the peak corresponds to 19.1–13.3 kDa. This is between the expected mass of the dimeric (26.0 kDa) and monomeric (13.0 kDa) states (Fig. 7B), whereas SAXS yielded a similarly intermediate molecular mass of 22.0 ± 0.2 kDa. Unlike TcpB120–250, TcpB1–119 did not show concentration dependence across the range studied by SAXS (Fig. 9C). Using MALS, the size exclusion chromatography elution peak of TcpB70–250 showed a molecular mass of 34.1 kDa, close to a dimeric (41.0 kDa) state (Fig. 7C), whereas the molecular mass based on SAXS was found to be 41.2 ± 0.2 kDa across a 4-fold dilution series, and no concentration dependence was observed (Fig. 9C). Guinier plots were linear (Fig. 9B) for the three constructs analyzed. Overall, these results suggest that both the TIR and the N-terminal domains are necessary for TcpB to form a stable dimer.
FIGURE 7.

MALS analyses of TcpB-fl, TcpB1–119, and TcpB70–250. The traces show protein concentration (arbitrary units; thin lines) and the molecular mass distribution across the peaks (thick lines) for TcpB-fl (A), TcpB1–119 (B), and TcpB70–250 (C). Approximately 300 μg of each protein was used in a buffer containing 50 mm Hepes, pH 8.0, 250 mm NaCl, and 1 mm DTT.
FIGURE 8.

MALS analyses of wild-type and mutant TcpB120–250. The traces show protein concentration (arbitrary units; thin lines) and the molecular mass distribution across the peaks (thick lines) for wild-type TcpB120–250 (A) and the D217A (B), Y216A (C), K213A (D), K213E (E), S235A (F), N233A (G), L236A (H), W211A (I), R220A (J), R220E (K), and G158A (L) mutants. Approximately 300 μg of each purified protein was used in a buffer containing 50 mm Hepes, pH 8.0, 250 mm NaCl, and 1 mm DTT.
FIGURE 9.

Small-angle x-ray scattering data. A, experimental scattering from the TcpB70–250 and TcpB120–250 proteins is plotted in black, with 1σ error bars shown in gray, as I(q) versus q on an arbitrary, logarithmic scale. The fits of representative models from the two clusters of ab initio reconstructions are shown in purple on the TcpB70–250 curve, and calculated scattering from the crystallographic dimer is shown in cyan, fitting TcpB120–250 to a χ value of 5.8. B, highest concentration data sets for all three constructs, transformed as ln I(q) versus q2, demonstrating linearity over the ranges where q.Rg < 1.3. C, molecular masses are plotted for TcpB120–250, TcpB70–250, and TcpB1–119. Dotted lines indicate predicted molecular masses of dimeric and monomeric species where noted and are colored by construct. D, Representative (inner) and averaged (outer) ab initio models are shown in purple for the two clusters restored from TcpB70–250 modeling, as surface representation with van der Waals radii at 3 Å. The dimeric crystal structure in cartoon representation (cyan) is manually superimposed onto the globular domain. E, normalized, dimensionless Kratky plots are shown for the three constructs of TcpB and for a stable, globular standard, glucose isomerase. Increases at higher scattering angles indicating flexibility are observed in all constructs, with TcpB1–119 showing the most disorder. F, UV CD spectra (195–260 nm) of TcpB-fl (green), TcpB70–250 (purple), TcpB120–250 (cyan), and TcpB1–119 (red).
TIR-TIR Domain Interfaces in the TcpB Crystals
The four molecules in the asymmetric unit of the TcpB120–250 crystals form two equivalent 2-fold symmetrical dimers (chains A:C and B:D), with total buried surface areas of 1,266 and 1,283 Å, respectively (calculated using PISA (48)). The structural elements involved in these interfaces include the DD and EE loops and the αD and αE helices (Fig. 10A). A network of hydrogen bonds and salt bridges stabilizes the interface (Fig. 10, B and C). Key interacting residues at the interface are conserved among a number of bacterial TIR domains (Fig. 11A). At the core of the interface, the EE loops are connected by hydrogen bonds between the hydroxyl oxygens from the side chain of two Ser235 residues and supported by the hydrophobic contacts between the two Leu236 residues (Fig. 10B). The Ser235 residues also form hydrogen bonds with the side chain of Asn233, and the overall conformation of the EE loops is stabilized by the side chain of the Trp211 residues (Fig. 10B). In the DD loops, the two Lys213 residues form hydrogen bonds with each other and with the side chains of Glu218 (αD helix) and Glu181 (αC helix) (Fig. 10C). At the edge of the interface, the side chain of Asp217 (αD helix) forms hydrogen bonds with the side chain of Arg220 (αD helix) from the same chain and with the main chain amide nitrogen of Val239 (αE helix) from a neighboring chain (Fig. 10C). The main chain carbonyl oxygen of Thr234 (EE loop) also forms a hydrogen bond with the main chain nitrogen amide of Tyr216 (αD helix) from a neighboring chain (Fig. 10C).
FIGURE 10.

The TcpB120–250 TIR-TIR domain interface observed in the crystal structure. A, cartoon representation of the dimer interface. The interface involves the DD and EE loops and the αD and αE helices. B and C, detailed overview of the dimer interface. The side chains of key residues involved in close contacts are displayed in wire frame-representation. A 40% transparency has been applied to the dimer ribbon diagram.
FIGURE 11.

Multiple sequence alignment of TIR domains. A, the amino acid sequences of TcpB-TIR (residues 119–250) and PdTIR (residues 167–299), and the TIR domains from the uropathogenic E. coli TIR domain-containing protein (TcpC-TIR) (residues 171–307), the S. enterica serovar enteritidis TIR domain-containing protein (TlpA-TIR) (residues 160–293), and S. aureus TIR domain-containing protein (Sa-TIR) (residues 143–280). B, the TIR domain from MAL (residues 86–256), MyD88 (residues 161–296), TLR4 (residues 587–752), TLR2 (residues 641–784), TLR1 (637–786), and TLR10 (residues 634–811) were aligned using MUSCLE (61). The alignment was formatted using ESPript (62). The positions of secondary structure elements of TcpB-TIR are shown above the alignment. Conserved residues are shown in white on red background. Similar residues are shown in red and surrounded by red boxes. Key residues in TcpB-TIR dimer contact are shown in blue.
Mutational Analysis of the TcpB TIR Domain Dimer
To examine whether the TcpB120–250 dimer interface observed in the crystals corresponds to the dynamic dimer observed in solution, we analyzed the effects of amino acid substitutions in this interface on recombinant TcpB120–250 self-association, using MALS. Because the arrangement of molecules in the crystal structure suggests the BB loop does not participate in dimerization, we mutated the conserved Gly158 residue to alanine as a negative control. The G158A mutation did not affect the TcpB120–250 dynamic equilibrium, yielding a molecular mass of 23.1 kDa (Table 2 and Fig. 8L). Of eight residues in the crystal interface that were mutated, mutations of Asp217, Tyr216, Lys213, Ser235, Leu236, Trp211, and Arg220 substantially shifted the equilibrium toward the monomeric state (Table 2 and Fig. 8, B–F and H–K). Several of these residues, including Tyr216, Lys213, Ser235, and Leu236, occupy central positions in the crystal interface. The Trp211 and Arg220 residues are not directly involved in interactions between molecules but are important for maintaining the DD and EE loops, the αD helix, and the surrounding surface in the correct conformations. Mutation of Asn233 only slightly affected the dimer equilibrium (Table 2 and Fig. 8G).
TABLE 2.
MALS analysis of the TcpB TIR domain mutations in the dimer interface
| Protein variant | Location in the crystal structure | Experimental molecular massa |
|---|---|---|
| kDa | ||
| Wild-type TcpB120–250 | 23.7 ± 0.05 (24.9–21.7) | |
| D217A | αD helix | 18.5 ± 0.04 (21.1–16.1) |
| Y216A | αD helix | 19.6 ± 0.08 (21.7–17.0) |
| K213A | DD loop | 17.5 ± 0.08 (20.1–16.0) |
| K213E | DD loop | 18.4 ± 0.07 (21.6–16.4) |
| S235A | EE loop | 19.4 ± 0.06 (19.9–17.4) |
| N233A | EE loop | 21.5 ± 0.05 (24.7–19.9) |
| L236A | EE loop | 18.9 ± 0.05 (20.0–16.4) |
| W211A | DD loop | 18.1 ± 0.04 (19.9–16.5) |
| R220A | αD helix | 18.6 ± 0.06 (22.3–16.8) |
| R220E | αD helix | 19.4 ± 0.08 (21.0–17.8) |
| G158A | BB loop | 23.1 ± 0.06 (24.4–20.0) |
a The theoretical molecular mass of the wild-type TcpB120–250 monomer is 15.0 kDa. The position of the mutations in the dimer interface and the experimental molecular masses at the top of the elution peak of the mutants obtained from MALS are presented. Errors are calculated by Astra software and relate to the uncertainty in the molecular mass calculation. The values in parentheses correspond to the range in the calculated molecular mass across the peak (Fig. 8).
Mutations in the Dimer Interface Reduce TcpB-mediated Inhibition of NF-κB Activation
To test the functional relevance of the TcpB TIR domain dimerization, we analyzed the effect of the dimer interface mutants on NF-κB activation using LPS-responsive HEK293 cells stably expressing TLR4-MD2. In these experiments, we used only TcpB-fl, because we were unable to successfully express TcpB120–250 in HEK293 cells. Because the BB loop mutation (G158A) was shown to have an effect on TcpB function (10), this mutation was included in our experiment as a positive control. Whereas wild-type TcpB-fl effectively suppressed TLR4-mediated activation of an NF-κB reporter, the K213E, N233A, L236A, and R220A mutants all lost this inhibitory activity. As expected, the G158A mutant also showed a similar phenotype. By contrast, the D217A and Y216A mutants retained similar inhibitory activities to wild-type TcpB-fl (Fig. 12). The K213A, S235A, W211A, and R220E mutants showed an intermediate phenotype between wild-type TcpB-fl and the G158A mutant (Fig. 12).
FIGURE 12.

The dimer interface is important for TcpB function. HEK293-TLR4-MD2 cells (8 × 104 cells/well) were co-transfected for 24 h with 200 ng of plasmids encoding V5-TcpB (wild type), V5-TcpB mutants (D217A, Y216A, K213A, K213E, S235A, N233A, L236A, W211A, R220A, R220E, and G158A), or empty vector pGL-2B (EV) with NF-κB luciferase reporter gene (100 ng) and phRL-TK reporter gene (20 ng) using Lipofectamine 2000. The final amount of DNA (460 ng) was kept constant in all transfections by adding empty vector pGL-2B. Cells were stimulated with 100 ng/ml LPS for 8 h and lysed with Promega lysis buffer. The data (average of n = 3) are displayed as luciferase activity, relative to Renilla luciferase activity. Error bars represent S.E. of three independent experiments. ****, p < 0.0001 compared with empty vector plus LPS.
Structural Characterization in Solution Confirms the Dimer Formation of TcpB TIR Domain
We compared the SAXS data for TcpB120–250 to the dimeric molecule observed in the TcpB120–250 crystal structure (Fig. 9A). Despite the concentration dependence previously described, the theoretical scattering from the structure is relatively consistent. Some deviation in the <0.05 and 0.1–0.2 Å−1 q range is not unexpected in the presence of interparticle effects. We therefore generated ab initio models for TcpB70–250 with P2 symmetry restraints (Fig. 8D). Two clusters of reconstructions were observed, with incidences of 50% each from 12 DAMMIF (43) runs. Both clusters share a globular domain consistent with the size and shape of the crystal structure and have extended features accounting for the C-terminal end of TcpB1–119. In one set of reconstructions, these features appear on both sides of the globular domain, whereas in others this occurs only on a single side (Fig. 9D). For TcpB1–119, a steady increase is observed at higher scattering angles in a normalized Kratky plot (Fig. 9E); this behavior is consistent with a highly flexible and disordered peptide chain. Similar analysis for TcpB70–250 and TcpB120–250 indicates a lesser degree of flexibility (Fig. 9E). Circular dichroism data for the TcpB1–119 showed a strong negative peak near 200 nm and a weaker peak near 220 nm, which suggests that TcpB1–119 contains a mixture of α-helical and random coil structure (Fig. 9F). Modeling analyses using Phyre2 (49) suggest that both TcpB1–119 and the N-terminal domains of other similar bacterial TIR domain-containing proteins may form elongated anti-parallel or parallel α-helical coiled-coil structures (data not shown).
DISCUSSION
TcpB Interaction with MAL, MyD88, and TLR4
In this study, we used the B. melitensis TIR domain-containing protein (TcpB/BtpA/Btp1) as a model system to shed light on the molecular mechanism of immunosuppression by bacterial TIR domain-containing proteins, using cell-based interaction studies and x-ray crystallography. TcpB belongs to a family of bacterial TIR domain-containing proteins that includes proteins from Salmonella enterica serovar enteritidis (TlpA), uropathogenic E. coli (TcpC) and Staphylococcus aureus (11, 50, 51). Some of these proteins have been shown to suppress TLR4 and TLR2 signaling pathways (6). Several studies described their potential binding partners in the host. For example, TcpB and TcpC were shown to interact with MyD88 (7, 11). Peptides corresponding to the BB and DD loops of TcpC were shown to interact with TLR4 and MyD88, respectively (11, 18). The TIR domain-containing protein from the nonpathogenic bacterium P. denitrificans (PdTLP) was shown to interact with MyD88 and TLR4 (52). Using co-immunoprecipitation analyses, we found that TcpB interacts with MAL, MyD88, and TLR4 (Fig. 2, A and B), with the TcpB BB loop being critical for interactions with MAL and TLR4 (Fig. 2, A and B). TcpB did not interact with TRAM and IRAK-2 (Fig. 2C). Notably, it has been reported that MAL and MyD88 are both involved in TcpB-mediated inhibition of NF-κB activation (8, 10). However, TcpB did not affect TLR9-mediated activation of NF-κB (a MyD88-dependent receptor) (8) but did antagonize TLR5 signaling (7). Recently, co-immunoprecipitation studies identified an interaction between TLR5 and MAL (53). Together with our co-immunoprecipitation studies, it is therefore reasonable to suggest that TcpB also targets other MAL-mediated TLR signaling pathways.
A recent report suggested that regions outside the TIR domain of MyD88 are involved in the interaction with TcpB and that TcpB binding enforces an inactive state of MyD88 (12). Our data suggest that the bacterial TIR domain-containing proteins may form complexes with multiple host proteins. A similar mechanism was reported for vaccinia virus A46R, which has been shown to target multiple host TIR domain-containing proteins (54).
Sengupta et al. (9) reported that TcpB interacts with MAL and does not interfere with the MAL-MyD88 interaction. We similarly observed that recombinant TcpB-fl, TcpB120–250, and TcpB1–119 were unable to interfere with the MAL-MyD88 interaction (Fig. 3A). Based on our observation that TcpB could interact with MAL and TLR4 at two opposite sides of the TcpB TIR domain dimer (see below), it is likely that TcpB rather interferes with the receptor-adaptor interactions. Increasing amounts of recombinant TcpB-fl or TcpB120–250 proteins, but not TcpB1–119, interfered with the MAL-TLR4 interaction (Fig. 3B). Our observations are thus consistent with the previously suggested mechanism of molecular mimicry, whereby TcpB mimics MAL function by binding to the plasma membrane using its N-terminal domain and competes with MAL for binding to TLR4 (Fig. 13) rather than with MyD88 as suggested by Radhakrishnan et al. (10).
FIGURE 13.
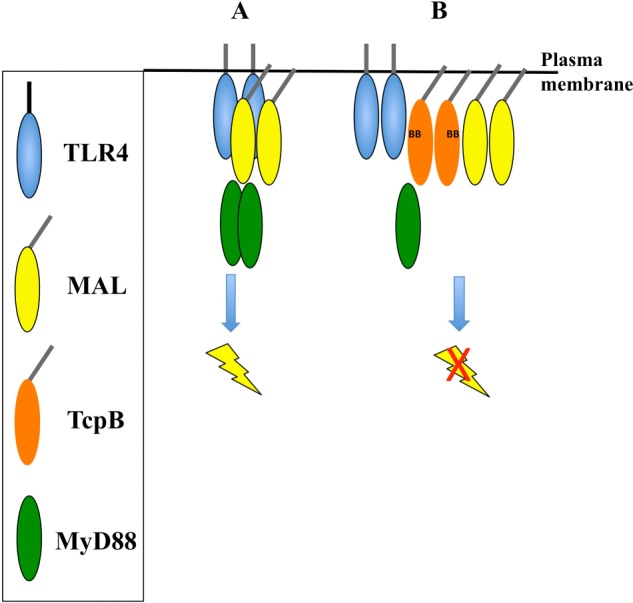
Schematic modeling of the TcpB inhibition of TLR4 signaling. Oval shapes represent TcpB (orange), TLR4 (blue), MAL (yellow), and MyD88 (green). The model emphasizes the key findings from our study and is a simplified diagram of the signaling system. For example, the exact domains involved in the interactions between these proteins have not yet been clearly defined, and thus the model does not show specific domain interactions. A, a simplified representation of the TLR4 signaling pathway, showing the activation and dimerization of TLR4 creates a platform to form a complex with MAL and MyD88. B, the presence of TcpB causes suppression of the TLR4 signaling pathway, by forming a complex with TLR4, MAL, and MyD88.
TcpB Dimerization
We demonstrate that the TIR domain from TcpB has a tendency to homodimerize, with the crystal structure and mutational studies suggesting that the DD and EE loops and the αD and αE helices are involved in the interface (Fig. 10A). In this arrangement, the BB loops from the two interacting molecules are exposed to the solvent for possible interaction with host molecules. Therefore, it is conceivable that MAL and TLR4 could simultaneously interact with the TcpB TIR domain dimer, using the BB loop-containing interfaces at the opposite sides of the dimer. It is unlikely that TcpB has a similar MyD88-binding interface to TcpC (18), because the TcpB TIR domain DD loop participates in dimer formation. Notably, the DD loops from the TIR domains of TcpB and TcpC share low sequence similarity (Fig. 11A), possibly supporting different functions. Importantly, the dimer interface we identified in the TcpB120–250 is also found in PdTIR crystals (19); to our knowledge, this is the first observation of a common interface shared between two different TIR domain structures and suggests some parallels in the molecular basis of function among bacterial TIR domains.
Although in vivo studies have shown that a number of TIR domain-containing proteins dimerize, the majority of isolated recombinant TIR domains have been reported to be monomers in solutions, and other regions in the molecules were found to be required for the proteins to form stable dimers (2, 46, 55). In solution, our study shows that isolated domains of TcpB are in reversible equilibrium between monomers and dimers, and both domains are required to form a stable TcpB dimer (Figs. 7, 8, and 9C). We confirmed, by mutational analysis of key interacting residues, that the dimer interface observed in the TcpB120–250 crystals is responsible for TcpB120–250 dimerization in solution (Table 2 and Fig. 7). These analyses also show that the BB loop does not participate in dimer formation, which further supports the suggested MAL and TLR4-binding interface. We further demonstrate, by using NF-κB reporter assays, that a number of mutations in the dimer interface affected the ability of TcpB-fl to inhibit NF-κB activation, although they did not necessarily disrupt dimerization completely (Fig. 10). The apparent discordance for a small number of the mutations (e.g., Y216A, D217A) in affecting dimer formation versus reporter gene inhibition may reflect the necessary usage of truncated (MALS) versus full-length TcpB proteins (reporter assays) in the different assays.
Fekonja et al. (22) reported that bacterial TIR domain-containing proteins use dimeric N-terminal coiled-coil domains not only for membrane attachment, but also to enhance TIR domain dimer formation to aid in the inhibition of TLR signaling. Our data suggest that TcpB1–119 by itself is highly flexible, with some limited α-helical character and has a tendency to dimerize (Figs. 9, C–E, and 7B). SAXS data further demonstrate the dimer formation of TcpB, with one of the two reconstructions suggesting that the N-terminal tails of the TIR domain could be on the opposite sides of the dimer (Fig. 9D), which would be consistent with the TcpB TIR domain dimer observed in the crystals (Fig. 10A). Based on structural modeling, we speculate that parts of TcpB1–119 could adopt an antiparallel or parallel coiled-coil structure. Future studies will unveil the detailed structural characteristics of the N-terminal domains from bacterial TIR domain-containing proteins.
Conclusions
Understanding the molecular basis of the TIR domain association between microbial and host TIR domain-containing proteins has been a challenge, with the lack of structural information on heterotypic complexes making it difficult to model a coherent mechanism of action. Our study has provided significant insights into the mechanism of TcpB interaction with host signaling molecules at both molecular and structural levels. We find that TcpB dimerization is important for function. We believe that the dimer interface reported in this study and the involvement of the BB loop in the interaction of TcpB with host molecules could be used as targets for developing peptides as anti-inflammatory agents. Ultimately, such an approach could lead to the development of novel therapeutics for the treatment of chronic inflammatory diseases.
While our manuscript was in preparation, Kaplan-Türköz et al. (56) published the 3.15 Å resolution crystal structure of the TIR domain of TcpB. The structure shows the analogous dimeric arrangement we see in our crystals. Mutagenesis of the interface residues in the context of full-length protein similarly supports our suggestions of the dimerization interface (for example, the S235A mutation). Because the crystallized protein contained some additional N-terminal residues (starting at residue 75), the authors find that the N-terminal region of one monomer packs against the dimer. This feature is consistent with the conclusions of our biophysical experiments. Comparison of this dimeric structure to the SAXS data for TcpB70–250 yielded poor fits both prior to and after modeling of missing residues (analysis not shown). It appears that structural information on extended constructs will be required to understand the structural basis of the role of the N-terminal domain of TcpB in its dimerization.
Acknowledgments
We thank Thomas Miethke for the TcpB cDNA, Andrew Bowie for mammalian protein-expressed plasmids, Gregory Kelly for help with cell-based experiments, and Laurent Terradot for coordinates before release by the Protein Data Bank. We acknowledge the use of the MX2 and SAXS/WAXS beamlines at the Australian Synchrotron (Melbourne, Australia) and the UQ-ROCX Diffraction Facility.
This work was supported in part by National Health and Medical Research Council Grant APP1003326 (to B. K. and A. M.).
The atomic coordinates and structure factors (code 4c7m) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TLR
- Toll-like receptor
- RMSD
- root mean square deviation
- MALS
- multiangle light scattering
- TIR
- Toll/interleukin-1 receptor
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity. Update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 2. Ve T., Gay N. J., Mansell A., Kobe B., Kellie S. (2012) Adaptors in Toll-like receptor signaling and their potential as therapeutic targets. Curr. Drug Targets 13, 1360–1374 [DOI] [PubMed] [Google Scholar]
- 3. Atluri V. L., Xavier M. N., de Jong M. F., den Hartigh A. B., Tsolis R. M. (2011) Interactions of the human pathogenic Brucella species with their hosts. Annu. Rev. Microbiol. 65, 523–541 [DOI] [PubMed] [Google Scholar]
- 4. Pappas G., Papadimitriou P., Akritidis N., Christou L., Tsianos E. V. (2006) The new global map of human brucellosis. Lancet Infect. Dis. 6, 91–99 [DOI] [PubMed] [Google Scholar]
- 5. Elde N. C., Malik H. S. (2009) The evolutionary conundrum of pathogen mimicry. Nat. Rev. Microbiol. 7, 787–797 [DOI] [PubMed] [Google Scholar]
- 6. Rana R. R., Zhang M., Spear A. M., Atkins H. S., Byrne B. (2013) Bacterial TIR-containing proteins and host innate immune system evasion. Med. Microbiol. Immunol. 202, 1–10 [DOI] [PubMed] [Google Scholar]
- 7. Salcedo S. P., Marchesini M. I., Degos C., Terwagne M., Von Bargen K., Lepidi H., Herrmann C. K., Santos Lacerda T. L., Imbert P. R., Pierre P., Alexopoulou L., Letesson J. J., Comerci D. J., Gorvel J. P. (2013) BtpB, a novel Brucella TIR-containing effector protein with immune modulatory functions. Front. Cell. Infect. Microbiol. 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salcedo S. P., Marchesini M. I., Lelouard H., Fugier E., Jolly G., Balor S., Muller A., Lapaque N., Demaria O., Alexopoulou L., Comerci D. J., Ugalde R. A., Pierre P., Gorvel J.-P. (2008) Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sengupta D., Koblansky A., Gaines J., Brown T., West A. P., Zhang D., Nishikawa T., Park S. G., Roop R. M., 2nd, Ghosh S. (2010) Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter, MAL. J. Immunol. 184, 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radhakrishnan G. K., Yu Q., Harms J. S., Splitter G. A. (2009) Brucella TIR domain-containing protein mimics properties of the toll-like receptor adaptor protein TIRAP. J. Biol. Chem. 284, 9892–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cirl C., Wieser A., Yadav M., Duerr S., Schubert S., Fischer H., Stappert D., Wantia N., Rodriguez N., Wagner H., Svanborg C., Miethke T. (2008) Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat. Med. 14, 399–406 [DOI] [PubMed] [Google Scholar]
- 12. Chaudhary A., Ganguly K., Cabantous S., Waldo G. S., Micheva-Viteva S. N., Nag K., Hlavacek W. S., Tung C. S. (2012) The Brucella TIR-like protein TcpB interacts with the death domain of MyD88. Biochem. Biophys. Res. Commun. 417, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y., Tao X., Shen B., Horng T., Medzhitov R., Manley J. L., Tong L. (2000) Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408, 111–115 [DOI] [PubMed] [Google Scholar]
- 14. Ohnishi H., Tochio H., Kato Z., Orii K. E., Li A., Kimura T., Hiroaki H., Kondo N., Shirakawa M. (2009) Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khan J. A., Brint E. K., O'Neill L. A., Tong L. (2004) Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J. Biol. Chem. 279, 31664–31670 [DOI] [PubMed] [Google Scholar]
- 16. Valkov E., Stamp A., Dimaio F., Baker D., Verstak B., Roversi P., Kellie S., Sweet M. J., Mansell A., Gay N. J., Martin J. L., Kobe B. (2011) Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. U.S.A. 108, 14879–14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin Z., Lu J., Zhou W., Shen Y. (2012) Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS One 7, e34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Snyder G. A., Cirl C., Jiang J., Chen K., Waldhuber A., Smith P., Römmler F., Snyder N., Fresquez T., Dürr S., Tjandra N., Miethke T., Xiao T. S. (2013) Molecular mechanisms for the subversion of MyD88 signaling by TcpC from virulent uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 6985–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan S. L., Low L. Y., Hsu S., Li S., Liu T., Santelli E., Le Negrate G., Reed J. C., Woods V. L., Jr., Pascual J. (2009) Molecular mimicry in innate immunity. Crystal structure of a bacterial TIR domain. J. Biol. Chem. 284, 21386–21392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan S. L., Mukasa T., Santelli E., Low L. Y., Pascual J. (2010) The crystal structure of a TIR domain from Arabidopsis thaliana reveals a conserved helical region unique to plants. Protein Sci. 19, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernoux M., Ve T., Williams S., Warren C., Hatters D., Valkov E., Zhang X., Ellis J. G., Kobe B., Dodds P. N. (2011) Structural and functional analysis of a plant resistance protein TIR domain reveals interfaces for self-association, signaling, and autoregulation. Cell Host Microbe 9, 200–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fekonja O., Bencina M., Jerala R. (2012) Toll/interleukin-1 receptor domain dimers as the platform for activation and enhanced inhibition of Toll-like receptor signaling. J. Biol. Chem. 287, 30993–31002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eschenfeldt W. H., Lucy S., Millard C. S., Joachimiak A., Mark I. D. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 25. Alaidarous M., Ve T., Ullah M. O., Valkov E., Mansell A., Schembri M. A., Sweet M. J., Kobe B. (2013) Cloning, expression, purification, crystallization and preliminary x-ray crystallographic analysis of the TIR domain from the Brucella melitensis TIR-domain-containing protein TcpB. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 1167–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-Ice and the distributed control system. Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D. (2008) Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 [DOI] [PubMed] [Google Scholar]
- 32. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 34. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2012) Exploiting structure similarity in refinement. Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Dijk J. A., Smit J. A. (2000) Size-exclusion chromatography-multiangle laser light scattering analysis of β-lactoglobulin and bovine serum albumin in aqueous solution with added salt. J. Chromatogr. A 867, 105–112 [DOI] [PubMed] [Google Scholar]
- 37. Folta-Stogniew E., Williams K. R. (1999) Determination of molecular masses of proteins in solution. Implementation of an HPLC size exclusion chromatography and laser light scattering service in a core laboratory. J. Biomol. Tech. 10, 51–63 [PMC free article] [PubMed] [Google Scholar]
- 38. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS, p 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 40. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 324–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer H., De Oliveira Neto M., Napolitano H. B., Polikarpov I., Craievich A. F. (2009) Determination of the molecular weight of proteins in solution from a single small-angle x-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 42. Svergun D., Barberato C., Koch M. H. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 43. Svergun D. I., Franke D. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valkov V. V., Svergun D. I. (2003) Uniqueness of ab-intio shape determination in small-angle acattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dunne A., Ejdeback M., Ludidi P. L., O'Neill L. A., Gay N. J. (2003) Structural complementarity of Toll/interleukin-1 receptor domains in Toll-like receptors and the adaptors Mal and MyD88. J. Biol. Chem. 278, 41443–41451 [DOI] [PubMed] [Google Scholar]
- 47. Chang T. H., Chang S. J., Hsieh F. L., Ko T. P., Lin C. T., Ho M. R., Wang I., Hsu S. T., Guo R. T., Chang W., Wang A. H. (2013) Crystal structure of vaccinia viral A27 protein reveals a novel structure critical for its function and complex formation with A26 protein. PLoS Pathog. 9, e1003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 49. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web. A case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 50. Newman R. M., Salunkhe P., Godzik A., Reed J. C. (2006) Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect. Immun. 74, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cirl C., Miethke T. (2010) Microbial Toll/interleukin 1 receptor proteins. A new class of virulence factors. Int. J. Med. Microbiol. 300, 396–401 [DOI] [PubMed] [Google Scholar]
- 52. Low L. Y., Mukasa T., Reed J. C., Pascual J. (2007) Characterization of a TIR-like protein from Paracoccus denitrificans. Biochem. Biophys. Res. Commun. 356, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi Y. J., Jung J., Chung H. K., Im E., Rhee S. H. (2013) PTEN regulates TLR5-induced intestinal inflammation by controlling Mal/TIRAP recruitment. FASEB J. 27, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stack J., Haga I. R., Schröder M., Bartlett N. W., Maloney G., Reading P. C., Fitzgerald K. A., Smith G. L., Bowie A. G. (2005) Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201, 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burns K., Martinon F., Esslinger C., Pahl H., Schneider P., Bodmer J. L., Di Marco F., French L., Tschopp J. (1998) MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 273, 12203–12209 [DOI] [PubMed] [Google Scholar]
- 56. Kaplan-Türköz B., Koelblen T., Felix C., Candusso M. P., O'Callaghan D., Vergunst A. C., Terradot L. (2013) Structure of the Toll/interleukin 1 receptor (TIR) domain of the immunosuppressive Brucella effector BtpA/Btp1/TcpB. FEBS Lett. 587, 3412–3416 [DOI] [PubMed] [Google Scholar]
- 57. Fitzgerald K. A., Palsson-McDermott E. M., Bowie A. G., Jefferies C. A., Mansell A. S., Brady G., Brint E., Dunne A., Gray P., Harte M. T., McMurray D., Smith D. E., Sims J. E., Bird T. A., O'Neill L. A. (2001) Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413, 78–83 [DOI] [PubMed] [Google Scholar]
- 58. Muzio M., Ni J., Feng P., Dixit V. M. (1997) Pillars article. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615 [DOI] [PubMed] [Google Scholar]
- 59. Ohnishi H., Tochio H., Kato Z., Kawamoto N., Kimura T., Kubota K., Yamamoto T., Funasaka T., Nakano H., Wong R. W., Shirakawa M., Kondo N. (2012) TRAM is involved in IL-18 signaling and functions as a sorting adaptor for MyD88. PLoS One 7, e38423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Edgar R. C. (2004) MUSCLE. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gouet P., Robert X., Courcelle E. (2003) ESPript/ENDscript. Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31, 3320–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]