

Background: The Toll/IL-1 receptor (TIR) domains are crucial innate immune signaling modules.
Results: The crystal structures of the TIR domains from TcpB and TIRAP reveal similar folds and distinct features.
Conclusion: TcpB may mimic the function of TIRAP through their similar TIR domain structures.
Significance: These findings suggest mechanisms of bacterial mimicry of host signaling adaptor proteins.
Keywords: Bacterial Pathogenesis, Crystal Structure, Innate Immunity, Mal TIRAP, Signal Transduction, Toll IL-1 Receptor (TIR) Domain, TcpB
Abstract
The Toll/IL-1 receptor (TIR) domains are crucial innate immune signaling modules. Microbial TIR domain-containing proteins inhibit Toll-like receptor (TLR) signaling through molecular mimicry. The TIR domain-containing protein TcpB from Brucella inhibits TLR signaling through interaction with host adaptor proteins TIRAP/Mal and MyD88. To characterize the microbial mimicry of host proteins, we have determined the X-ray crystal structures of the TIR domains from the Brucella protein TcpB and the host adaptor protein TIRAP. We have further characterized homotypic interactions of TcpB using hydrogen/deuterium exchange mass spectrometry and heterotypic TcpB and TIRAP interaction by co-immunoprecipitation and NF-κB reporter assays. The crystal structure of the TcpB TIR domain reveals the microtubule-binding site encompassing the BB loop as well as a symmetrical dimer mediated by the DD and EE loops. This dimerization interface is validated by peptide mapping through hydrogen/deuterium exchange mass spectrometry. The human TIRAP TIR domain crystal structure reveals a unique N-terminal TIR domain fold containing a disulfide bond formed by Cys89 and Cys134. A comparison between the TcpB and TIRAP crystal structures reveals substantial conformational differences in the region that encompasses the BB loop. These findings underscore the similarities and differences in the molecular features found in the microbial and host TIR domains, which suggests mechanisms of bacterial mimicry of host signaling adaptor proteins, such as TIRAP.
Introduction
Effective immune responses to exogenous pathogens result in measured cytokine secretion and pathogen clearance, whereas inappropriate responses can lead to uncontrolled inflammation and tissue damage. The Toll-like receptors (TLRs)4 constitute a family of pattern recognition receptors of the innate immune system, which are activated in response to pathogen-associated molecular patterns found on a variety of fungal, bacterial, viral, and environmental stimuli. Extracellular ligand engagement triggers signal transduction cascades involving reorganization of the cytoplasmic Toll/IL-1 receptor (TIR) domains from the receptor molecules. The receptor TIR domain complex acts as a receptive scaffold for the engagement of the adaptor proteins MyD88 and TIRAP (or TRIF and TRAM in alternative signaling pathways), which ultimately results in NF-κB-mediated induction of cytokine- and chemokine-driven inflammatory responses (1).
Microbial pathogens have evolved numerous mechanisms to suppress or evade innate immunity. One such mechanism involves the subversion of the TLR signaling pathways by microbial TIR-interacting proteins (TIPs). Microbial TIPs are thought to function by binding to host TLRs and adaptors containing TIR domains, thus disrupting the TLR signaling complex formation. Bacterial TIPs (bTIPs) include TIR domain proteins expressed by Brucella melitensis (TcpB) (1, 2), Brucella abortus (TcpB/Btp1) (3, 4), Brucella ovis (TcpB) (3), Salmonella enterica (TlpA) (5), Paracoccus dentrificans (PdTLP) (6) uropathogenic Escherichia coli CFT073 (TcpC) (2), and Yersinia pestis (YpTdp) (7). Viral TIPs (vTIPs), as well as endogenous and regulatory TIPs (eTIPs and rTIPs), have also been reported to directly interact with the host TIR domains (8–10). Based on their abilities to affect the severity of infections, microbial TIPs have been proposed as a novel class of virulence factors that function through molecular mimicry of host TIR domain-containing proteins (8, 10, 11). Investigating the structure and function of the TIP-based virulence factors is therefore essential for our understanding of how microbial pathogens evade and suppress TLR-mediated innate immune responses.
The Brucella protein TcpB and uropathogenic E. coli protein TcpC represent two of the most extensively characterized bTIPs to date (2, 9). TcpC, but not TcpB, is secreted and contains intrinsic cell permeability properties, allowing it to traverse the cell membrane to interact with cytosolic TIR domain proteins. Both proteins have been shown to interact with signaling adaptors MyD88 and TIRAP (2) and, in the case of TcpC, the TIR domain from TLR4 (12). Mechanistically, they may function through suppressing the formation of supramolecular signaling complexes composed of the TIR domains of TLRs and adaptor proteins (13).
TcpB protein has been shown to be important in establishing early brucellosis (4). Mice infected with Brucella deficient in TcpB exhibited a delayed onset of brucellosis and inflammation compared with wild type Brucella (2, 4). TcpB disrupts TLR2 and TLR4 signaling initiated at the plasma membrane but not endosomal TLR-mediated signaling (3, 14). It was shown to bind and target TIRAP/Mal for degradation (3, 4) and act as a molecular mimic of TIRAP by binding to MyD88 and phosphoinositides (3, 15, 16). Recently, TcpB was also reported to bind the death domains of MyD88 to negatively regulate signaling (17).
In addition to modulating immune functions, TcpB has been described to stabilize microtubules in a manner similar to that mediated by the cancer therapeutic, paclitaxel/Taxol (18). A long peptide that encompasses the BB loop of TcpB (residues 127–174) has been shown to associate with and stabilize microtubules, with residue Gly-158 identified as critical for microtubule stabilization (18).
To better understand the mechanisms by which bTIPs subvert innate immune responses, we have determined the crystal structures of the TIR domains from both Brucella TcpB and its host partner TIRAP. We further characterize the TIR domain interactions using biochemical and biophysical assays. Our studies reveal similar folds and distinct features of the TcpB and TIRAP TIR domains and provide molecular insights into host-pathogen interactions mediated by the TIR domain-containing proteins.
EXPERIMENTAL PROCEDURES
Co-immunoprecipitation and Immunoblotting
HEK293T cells grown in Dulbecco's modified Eagle's culture medium (10% fetal bovine serum, v/v) were seeded in a 10-cm cell culture dish and transiently co-transfected with different plasmids (maximum 20 μg) using the calcium phosphate method. At 24 h after transfection, the medium was replaced, and cells were cultured for an additional 24 h. Cells were washed with PBS, harvested, and resuspended in 500 μl of lysis buffer (50 mm HEPES, pH 7.6, 1 mm EDTA, 1 mm EGTA, 300 mm NaCl, 0.4 mm PMSF, 1 mm dithiothreitol, 10% glycerol, 20 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm NaF, 0.5% Nonidet P-40), including protease inhibitors (Roche Applied Science). Lysates were centrifuged at 15,000 rpm for 20 min to remove cellular debris. 50 μl of the supernatant was saved for immunoblotting later, and the remaining supernatant was then incubated with anti-FLAG affinity beads (Sigma) overnight at 4 °C. Immunocomplexes were washed three times with PBS containing 500 mm NaCl and eluted from beads by boiling in SDS sample buffer. Samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. After blocking in Tween 20 and Tris-buffered saline (TBS-T) with 5% nonfat milk (w/v) for 60 min at room temperature, blots were incubated with anti-FLAG and anti-Myc antibodies (Sigma) overnight at 4 °C, washed three times with TBS-T, and incubated with appropriate HRP-conjugated secondary antibody (Jackson ImmunoResearch Europe Ltd.) for 60 min at room temperature. After three washes with TBS-T, protein detection was carried out using ECL reagents (PerkinElmer Life Sciences) according to the manufacturer's recommendation.
Luciferase Reporter Assay
HEK293T cells were transfected using LipofectamineTM 2000 (Invitrogen) with NF-κB firefly luciferase (50 ng/ml) and Renilla luciferase reporter constructs (1 ng/ml) as well as titrated amounts of plasmids encoding TIRAP (5, 10, 50, 100, 250, and 500 ng/ml), wild type TcpB (2, 20, and 100 ng/ml), and a mutant TcpB containing mutations of S124A and S127I (TcpBS124A/S127I) (2, 20, and 100 ng/ml) (see “Crystallization” for the design of the mutations). In a second experiment, titrated amounts (2, 20, and 100 ng/ml) of TcpB or TcpBS124A/S127I plasmids were co-transfected with the TIRAP-encoding plasmid (100 ng/ml) to test the inhibitory effects of TcpB and its mutant on the TIRAP-mediated signaling. 48 h post-transfection, the luciferase activities were measured using the dual luciferase reporter assay system (Promega, Madison, WI) and a microplate luminometer (Titertek Berthold, Pforzheim, Germany). All assays were performed in triplicate.
Protein Expression and Purification
The full-length B. melitensis TcpB (NP_540591) and its TIR domain (residues 113–250) were expressed using the pASK IBA3+ vector as described previously (2). Both the wild type and the mutant TcpBS124A/S127I were expressed in BL21(DE3) codon plus RIPL cells (Stratagene). Briefly, the transformed cells were cultured in Luria broth at 20 °C. Cells were induced using 200 μg/liter anhydrotetracycline at an optical density of 0.8 and cultured for an additional 2 h. Cells were harvested, suspended in lysate buffer (20 mm HEPES, pH 7.0, and 150 mm NaCl), and lysed using freeze-thaw cycles followed by sonication. Cleared cell lysates were loaded onto a Strep-tag affinity column (IBA Life Sciences), and the bound protein was purified following the manufacturer's protocol (19). The elution fractions containing purified protein were digested with sequencing grade chymotrypsin (Roche Applied Science) for 2 h at 4 °C and further purified using size exclusion chromatography as described previously (6, 20).
The coding sequence for residues 81–221 of the human TIRAP (NP_001034750) was cloned into a modified pET30a vector that contains a TEV-cleavable N-terminal His6 tag and GB1 fusion protein. The BL21(DE3) codon plus RIPL cells transformed with the expression vector were grown at 37 °C and induced with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside. Cells were harvested and lysed using freeze-thaw cycles and sonicated in 20 mm HEPES, pH 7.0, 150 mm NaCl, and 1 mm β-mercaptoethanol. The fusion protein was purified using Ni2+ affinity chromatography followed by dialysis and incubation with TEV protease at 4 °C in a buffer containing 20 mm HEPES, pH 7.0, 150 mm NaCl, and 3 mm β-mercaptoethanol. The untagged TIRAP TIR domain was purified through a second Ni2+ affinity chromatography followed by a size exclusion chromatography. All purified protein samples were verified by mass spectrometry and N-terminal sequencing.
Crystallization
Small strawlike crystals of TcpB were identified in a sparse matrix screen from a sample of the full-length TcpBS124A/S127I mutant, which had been stored at 4 °C for 3 months. However, these were difficult to reproduce and optimize into diffraction quality crystals. SDS-PAGE analysis of the crystals indicated the presence of the TIR domain-only fragment. Subsequently, the full-length wild type TcpB was subjected to proteolysis using a panel of proteases to recapitulate the crystallizing fragment. Our initial efforts were not successful because the wild type protein was degraded by several proteases. Subsequently, we sought to introduce mutations that might stabilize the TIR domain fold to resist protease degradation. We focused on the βA strand/box 1 motif from the sequence comparison with TIR domains of known structures. The S124A mutation was designed because the equivalent residues from other known TIR domain structures are located at the hydrophobic core, and Ala is the smallest hydrophobic residue, so the risk of disrupting the hydrophobic core is minimal. The S126I mutation was chosen because the equivalent residue in another bacterial TIR domain, PdTIR, is a bulky residue, Trp174. Mutation to Ile may also stabilize the βA strand because we did not know a priori whether the βA strand extends to residue 126. By stabilizing the β-sheet formation at the TIR domain hydrophobic core, it may resist protease degradation. Indeed, treatment of the TcpBS124A/S127I mutant containing double mutations with sequencing grade chymotrypsin (Roche Applied Science) reproducibly yielded a stable fragment of the TcpB TIR domain. This chymotrypsin-treated TcpBS124A/S127I sample was crystallized in 0.2 m sodium chloride, 0.1 m MES, pH 6.0, and 20% PEG 2000 MME (w/v).
During purification of the TIRAP TIR domain, we observed significant solubility differences upon temperature changes, and this was exploited to induce its crystallization. Crystallization plates were set up at 4 °C and placed in a styrofoam box with cold packs and equilibrated to room temperature over a 24–48-h period. Initial crystallization conditions included 50% MPD (v/v), 0.1 m Tris, pH 8.5, and 0.2 m ammonium phosphate monobasic as well as 0.2 m magnesium formate, pH 5.9, and 20% PEG 3350 (w/v). Crystallization conditions were further optimized to include 5% MPD, 100 mm CHES, pH 9.0, 100 mm NaCl, or 0.2 m magnesium formate, pH 5.9, with 5–15% (w/v) PEG 3350, PEG 4000, or PEG 6000. Selenomethionine (SeMet)-labeled TIRAP protein was expressed using methionine auxotrophic strain B834 (EMD Biosciences) in M9 medium, purified, and crystallized as above. Incorporation of SeMet was verified by mass spectrometry.
X-ray Diffraction Data Collection and Structural Determination
X-ray diffraction data collection was carried out at the Advanced Photon Source, Argonne National Laboratory (beam line GM-CAT 23 ID) and the National Synchrotron Light Source, Brookhaven National Laboratory (beamline X29). X-ray diffraction data were processed using the HKL2000 program suite (21) and XDS (22) (Table 1). The structure of TcpB was determined by molecular replacement using the PdTIR structure (Protein Data Bank code 3H16) (6) as a search model with the program Phaser (23). Structural refinement and model rebuilding were carried with PHENIX (24) and COOT (25), respectively.
TABLE 1.
X-ray diffraction data collection and refinement
| TcpBS124A/S127I | hTIRAP native | hTIRAP SeMet | |
|---|---|---|---|
| Data collection | |||
| Space group | P 212121 | P 43212 | P43212 |
| Unit cell (a, b, c) (Å) | 67.5, 67.3, 60.3 | 87.6, 87.6, 80.6 | 87.4,87.4,81.4 |
| Wavelength (Å) | 1.0332 | 1.0332 | 0.97948 |
| Resolution (Å) | 50–2.3 (2.36–2.3)a | 50–2.4 (2.51–2.45)a | 50–2.6 (2.69–2.6)a |
| Total reflections | 172,809 | 68,651 | 110,200 |
| Unique reflections | 12,668 (889) | 12,002 (863) | 9811 (773) |
| Multiplicity | 13.6 (10.3) | 5.7 (5.8) | 11.2 (2.8) |
| Completeness (%) | 99.7 (97.9) | 99.8 (99.5) | 96.8 (78.7) |
| Average I/σ(I) | 21.0 (3.9) | 13.6 (2.9) | 13.7 (2.3) |
| Rmergeb | 0.10 (0.70) | 0.10 (0.86) | 0.21 (0.72) |
| Rpimc | 0.03 (0.23) | 0.05 (0.39) | FOM 0.628 |
| Refinement | |||
| Number of atoms | 2097 | 1072 | |
| Macromolecules | 2044 | 983 | |
| Heteroatoms | 53 | 88 | |
| Protein residues | 261 | 125 | |
| RMSD bond lengths (Å) | 0.006 | 0.011 | |
| RMSD bond angles (degrees) | 0.87 | 1.28 | |
| Rworkd | 0.210 (0.258) | 0.207 (0.259) | |
| Rfreee | 0.263 (0.333) | 0.218 (0.299) | |
| Wilson B-factor (Å2) | 30.8 | 41.6 | |
| Average B-factor (Å2) | 41.0 | 44.2 | |
| Ramachandran favored (%) | 96 | 98 | |
| Ramachandran outliers (%) | 0 | 0 | |
| Molprobity clashscoref | 4.97 | 1.55 | |
| Protein Data Bank code | 4LQC | 4LQD | |
a Values in parenthesis are for the last resolution shell.
b Rmerge = Σh Σi|Ii(h) − 〈I(h)〉|/ΣhΣiIi(h), where Ii(h) and 〈I(h)〉 are the ith and mean measurement of the intensity of reflection h.
c Rpim = Σh ((1/n − 1)½) Σi|Ii(h) − 〈I(h)〉|/ΣhΣiIi(h), where Ii(h) and 〈I(h)〉 are the ith and mean measurement of the intensity of reflection h, and n is the redundancy of reflection h.
d Rwork = Σh‖Fobs (h)| − |Fcalc (h)‖/Σh|Fobs (h)|, where Fobs (h) and Fcalc (h) are the observed and calculated structure factors, respectively. No I/σ cut-off was applied.
e Rfree is the R value obtained for a test set of reflections consisting of a randomly selected 5% subset of the data set excluded from refinement.
f Values from the Molprobity server.
Crystals for the human TIRAP TIR domain diffracted to a resolution of 2.45 Å. Our initial molecular replacement searches using all available TIR domain structures failed to yield a solution. Subsequently, SeMet protein of the human TIRAP TIR domain was purified and crystallized. Five SeMet positions were located using the single-wavelength anomalous dispersion method with the program SHELX (26), and data were collected at the selenium absorption edge, as we reported previously (27). Structural refinement was performed using PHENIX (24). All structures were validated using Molprobity (28) implemented in PHENIX. The structures of both TcpB (Protein Data Bank code 4LQC) and TIRAP (Protein Data Bank code 4LQD) have been deposited in the Protein Data Bank using the RCSB ADIT validation server (29). Figures were produced using the program PyMOL (30).
H/D Exchange Mass Spectrometry
Chymotrypsin-treated TcpBS124A/S127I TIR domain and full-length protein were buffer-exchanged into 20 mm HEPES, 150 mm NaCl, pH 6.7. The coverage map from undeuterated controls was obtained as follows. 2 μl of sample was diluted with 18 μl of 20 mm HEPES, 150 mm NaCl, pH 6.7, at room temperature, followed by 30 μl of ice-cold quench (100 mm phosphate buffer, 1.5 m guanidine HCl, pH 2.4). The samples were immediately injected into a Waters HDX nanoAcquity UPLC with in-line pepsin digestion (porozyme-immobilized pepsin cartridge from Applied Biosystems). The resulting peptides were trapped on an Acquity UPLC BEH 300 C4 peptide trap and separated on an Acquity UPLC BEH C18 column followed by injection into a Waters Synapt G2 mass spectrometer. Peptides were identified using the ProteinLynx Global Server 2.5.1 from Waters.
H/D exchange reactions were performed similarly to the undeuterated controls except that 2 μl of sample was incubated in 18 μl of 20 mm HEPES, 150 mm NaCl, 99.99% D2O, pD 6.7, at room temperature and quenched at various times (10 s, 1 min, 10 min, and 1 h) prior to injection. In order to correct for back-exchange, fully deuterated controls were acquired by incubating 2 μl of sample in 18 μl of 20 mm HEPES, 150 mm NaCl, 5.2 m guanidine DCl, 99.99% D2O, pD 6.7, for 2 h at room temperature prior to quenching and injection. Samples at all time points (including undeuterated controls and fully deuterated controls) were acquired in triplicates. Peptides identified by the ProteinLynx Global Server 2.5.1 were tracked through various deuterium incubation times using Water's DynamX software. The normalized percentage of deuterium uptake (% deuteration) at an incubation time t for a given peptide was obtained as follows: % deuteration = 100 × (mt − m0)/(m100% − m0), where mt is the centroid mass at incubation time t, m0 is the mass of undeuterated control, and m100% is the mass of fully deuterated control.
RESULTS
TcpB Bears Sequence Similarity with PtD and TIRAP
A protein BLAST search using the TcpB/Btp1 sequence from B. melitensis (TcpB) (2) identifies related TIR domain-containing proteins in B. abortus (TcpB/Btp1) (3, 4), B. ovis (TcpB) (3), S. enterica (TlpA) (5), and P. denitrificans (PdTLP) (6). A sequence alignment with the bacterium P. denitrificans protein PdTLP and human adaptor TIRAP shows sequence conservation in the TIR domain box 1–3 regions as well as the DD loop (Fig. 1). Such sequence conservation is corroborated by structural similarities, as discussed below. The region identified for microtubule binding based on the extended TcpB peptide (amino acids 127–173) and the single amino acid mutation (G158A) maps to a segment encompassing the αA, βB, and αB as well as the intervening BB loop.
FIGURE 1.
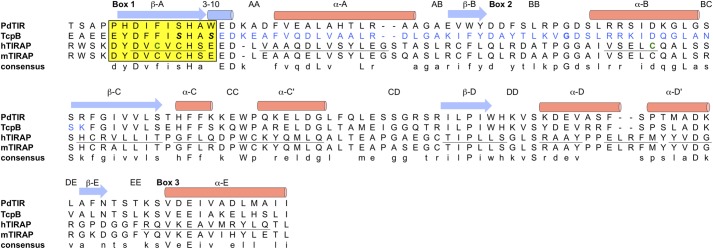
Sequence alignment of the TIR domains from bacterial proteins PdTIR and TcpB and human/mouse TIRAP proteins. The secondary structures for TcpB are indicated by arrows (strand) and cylinders (helices), whereas those for human TIRAP are underlined. The microtubule-binding region of TcpB is highlighted in blue. The TIRAP Cys89 and Cys134 residues involved in disulfide formation are colored green. The box 1 motif is shaded in yellow, and the mutation sites S124A and S127I are highlighted in italic boldface type.
TcpB Wild Type and Mutant Proteins Interact with Human TIRAP
Our initial efforts to crystallize the full-length TcpB protein only yielded crystals of poor quality in 3 months after setting up the crystallization drops. The crystals turned out to only contain the TIR domain of TcpB. This prompted us to adopt a protease trimming strategy with the goal of obtaining crystallizable fragments of TcpB. Whereas protease treatment of the wild type TcpB protein resulted in complete degradation, chymotrypsin treatment of a TcpBS124A/S127I mutant, designed to stabilize the β sheet at the hydrophobic core (see “Crystallization”), reproducibly yielded a stable fragment of the TcpB TIR domain and diffraction quality crystals. To test whether the mutations affect the ability of TcpB to interact with TIRAP, we performed co-immunoprecipitation assays in HEK293T cells. The results show that the TcpB mutants were able to interact with TIRAP as well as the wild type protein (Fig. 2). To further examine whether the mutations affect the function of TcpB to suppress TIRAP-mediated signaling, we employed an NF-κB luciferase reporter assay. As shown in Fig. 3A, TIRAP efficiently activated NF-κB, whereas wild type and mutant TcpB failed to do so. Importantly, wild type and mutant TcpB inhibited TIRAP-mediated activation of the NF-κB reporter (Fig. 3, B and C). Thus, the mutations did not appear to affect the binding of TcpB to TIRAP or its ability to suppress TIRAP-mediated signaling.
FIGURE 2.
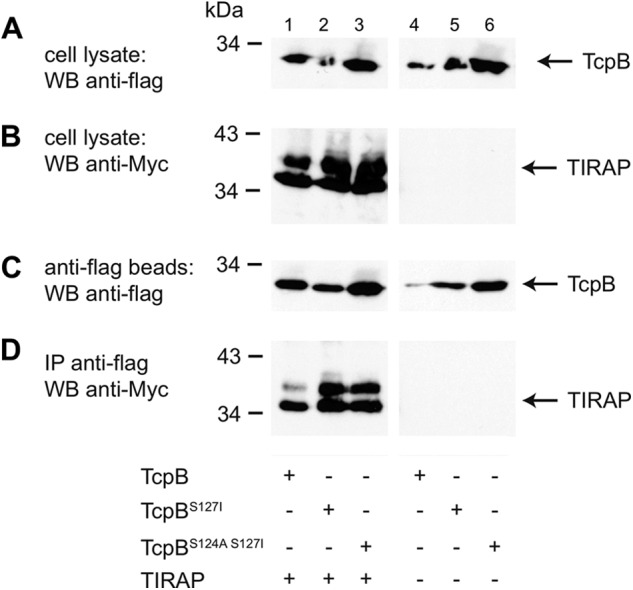
Direct interaction of the TcpB wild type and mutant proteins with human TIRAP. HEK293T cells were transfected with FLAG-tagged TcpB, TcpBS127I, or TcpBS124A/S127I either alone or together with Myc-tagged human TIRAP, as indicated. Transiently transfected cells were lysed after 48 h, and the lysate was subsequently immunoprecipitated with anti-FLAG affinity beads. A and B, expression of TcpB and TIRAP in the cell lysates, respectively. C, amount of precipitated TcpB. D, TIRAP coimmunoprecipitated by the wild type and mutant TcpB. WB, Western blot; IP, immunoprecipitation.
FIGURE 3.
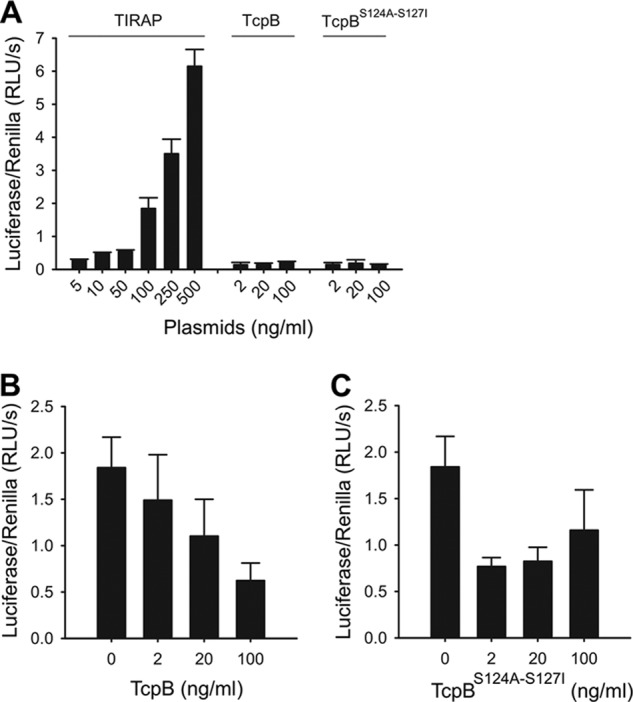
TcpB and TcpBS124A/S127I inhibit TIRAP-mediated NF-κB activation. A, HEK293T cells were transfected with a plasmid encoding for either TIRAP, TcpB, or TcpBS124A/S127I, as indicated, together with NF-κB firefly luciferase and Renilla luciferase reporter constructs. Luciferase activities were determined 48 h post-transfection. Error bars, S.D. of triplicates. B and C, HEK293T cells were transfected with the TIRAP-encoding plasmid (100 ng/ml) and titrated amounts of TcpB (B) or TcpBS124A/S127I (C) as indicated. Luciferase activities were determined 48 h post-transfection. Error bars, S.D. of triplicates.
Crystal Structure of the TcpB TIR Domain
The crystal structure of the TcpBS124A/S127I TIR domain was determined at 2.3 Å resolution (Table 1). It adopts a fold composed of five alternating β-strands and α-helices similar to other TIR domain structures reported to date (Fig. 4A). Residue Ala-124 is completely buried in the hydrophobic core, and residue Ile127 is partially buried and therefore unlikely to significantly alter the structure or surface properties of TcpB (Fig. 4B), consistent with the similar functions of the wild type and mutant TcpB proteins, as described above. A structural similarity search using the Dali server (31) identified TIRAP as one of the closest structural neighbors from mammalian proteins (Table 2), in agreement with a previous report of TcpB as a functional mimicry of TIRAP in its association with phosphoinositides, localization to the plasma membrane, and binding of the cytoskeleton (3).
FIGURE 4.

Comparison of the TIR domain structures. A, a schematic representation of the TcpB TIR domain crystal structure is shown in blue. Inset, TcpB extended BB loop region corresponding to the microtubule-binding peptide. The Gly-158 residue important for microtubule stabilization is shown in green. B, locations of residues 124 and 126 in the TcpB structure (blue). Residue Ala-124 is located at the hydrophobic core and surrounded by residues Phe122, Val177, Leu155, and Phe183. The equivalent residue for Ile126 is Trp174 in PdTIR (pale green). Side chains for the TcpB residues are shown in yellow, and the side chains for the PdTIR residues are shown in green. C, the extended BB loop region corresponding to the microtubule-binding peptide is shown on the left, and the CD loops are shown within a dotted circle on the right. D, comparison of bacterial TIR domain dimers. Schematic representations of TIR domain dimers from TcpB (blue) and PdTIR (green) are superimposed. The loops observed at the dimer interface (DD and EE) and the BB loop important for microtubule interactions are marked.
TABLE 2.
Structural similarity search using the TcpB TIR domain structure
| Z-Score | RMSD | Aligned residues | Identity | |
|---|---|---|---|---|
| Å | % | |||
| PdTIR (3H16) | 24.5 | 0.8 | 126 | 60 |
| TIRAP (3UB3) | 11.0 | 2.5 | 103 | 17 |
| L6TIR (3OZI) | 10.9 | 2.7 | 112 | 21 |
| AtTIR (3JRN) | 10.8 | 2.7 | 108 | 26 |
| TLR2 (1O77) | 10.4 | 3.0 | 114 | 19 |
| TLR1 (1FYV) | 9.4 | 3.0 | 113 | 18 |
| TLR10 (2J67) | 9.4 | 2.9 | 106 | 17 |
| MyD88 (4DOM) | 9.2 | 3.0 | 107 | 17 |
Comparison of the TIR domain structures also reveals distinct features. The conformation of the TIR domain loops from TLRs, TIR adaptor proteins, and microbial TIPs are specific to and conserved within each protein subfamily. Specifically, the BB loop position appears uniquely conserved among the receptors (e.g. TLR1, TLR2, and TLR10), adaptors (e.g. MyD88 and TIRAP) and bTIPs (e.g. TcpB and PdTIR) (Fig. 4C, left). Additionally, a protrusion of the CD loop is observed in the TcpB structure analogous to TIRAP and MyD88 (Fig. 4C, right).
A TcpB BB peptide spanning the αA helix to the αB helix was reported to be important for binding microtubules and maps to one face of the TIR domain. Importantly, mutation of residue Gly-158 at the apex of the BB loop (Fig. 4A) compromised the ability of TcpB to suppress TLR signaling or stabilize microtubules (3, 18), suggesting that the BB loop region is important for both functions.
The TcpB TIR Domain Forms a Symmetric Dimer in the Crystal
There are two unique molecules of the TcpB TIR domain in the crystal lattice, and they form a symmetric dimer mediated by their DD and EE loops with the DE loop in close proximity (Fig. 4D). Strikingly, this dimer configuration of the TcpB TIR domain is essentially the same as the PdTIR dimer (6), with an RMSD of 0.91 Å upon superposition of the two dimers (Fig. 4D). Similar to the PdTIR homodimer, the TcpB TIR domain dimer interface is dominated by hydrogen bonds (Fig. 5A). At the DD loop, residue Lys213 forms main chain and side chain hydrogen bonds with Lys213 and Glu218 from its partner DD loop and αD helix, residue Tyr216 forms a main chain hydrogen bond with its partner EE loop residue Thr234, and residue Asp217 forms a side chain to main chain hydrogen bond with its partner αE residue Val239. On the EE loop side, residue Asn234 forms a side chain hydrogen bond with Ser235 from its partner EE loop, and residue Ser235 forms a side chain hydrogen bond with Asn234 from its partner EE loop. In addition, Trp211 at the βD strand is close to its partner EE loop to stabilize its conformation. In total, there is 1250 Å2 of solvent-accessible surface area buried at this dimer interface, as calculated with the program areaimol (32, 33). Because the TcpB TIR domain dimer interface and microtubule-stabilizing region near the BB loop are on the opposite site of the globular domain, it is plausible that TcpB may form homodimer and engage microtubules simultaneously.
FIGURE 5.

The TcpB TIR domain dimer interface. A, details of the TcpB TIR domain dimer interface are shown with one molecule colored blue and the other green. Hydrogen bonds are shown as gray dotted lines. B, size exclusion chromatograms of the full-length TcpB (purple) and TcpB TIR domain (TcpBS124A/S127I, red) in phosphate-buffered saline. The molecular weight standards are shown above the chromatograms. The molecular masses of the TcpB full-length and TIR domain proteins are 28.0 and 15.6 kDa, respectively. C, the kinetic traces of TcpBS124A/S127I peptides in panels 1 (BB loop), 2 (DD loop), 3 (DE loop), and 4 (EE loop) show protection from deuterium exchange at early time points (colored red in D). Peptides in panels 5 (αB), 6 (αD), and 7 (αE) display protection at later time points (colored orange in D). Peptides in panels 8 (βA), 9 (αB-AB loop), and 10 (αC) display no protection (colored blue and gray in D). D, schematic representation of the TcpB TIR domain dimer with the H/D mass spectrometry mapped peptides colored according to data shown in C.
Three crystal lattice contacts were identified by the PISA (Protein Interfaces, Surfaces, and Assemblies) server (34). These are mediated by the αE helix with the βA and βB strands from the crystallographic symmetry mate, the αA helix with the symmetry mate βB strand, and the αC′ helix with the symmetry mate αD helix. These much smaller hydrophilic interfaces (buried surface areas of 575, 500, and 470 Å2, respectively) may not represent physiological interactions.
Analysis of the TcpB Homodimerization Interface by H/D Exchange
Size exclusion chromatography of the full-length wild type and mutant TcpB samples revealed dimeric and oligomeric species (Fig. 5B). In contrast, the chymotrypsin-treated TcpBS124A/S127I yielded a TIR domain fragment that eluted at a volume consistent with a primarily monomeric state. To characterize the TcpB dimerization interface, we performed H/D mass spectrometry analysis of the TcpB TIR domain and the full-length protein. Comparison of the deuterium exchange rates between the chymotrypsin-treated monomeric TcpB TIR domain and the full-length oligomeric/dimeric TcpB demonstrated differences in primarily two regions (Fig. 5, C and D). The first region, which encompasses peptides 2, 3, and 4, corresponds to the DD, DE, and EE loops (Figs. 5C (left panels) and 6 (A and B)). In the monomeric TcpB TIR domain, these regions are highly exposed to solvent, and the peptides readily took up deuterium; peptides 2 (in the DD loop) and 3 (in the DE loop) displayed close to 100% deuteration within 10 s of incubation. In contrast, the full-length oligomeric TcpB displayed a significant decrease in deuterium uptake at early time points, suggesting that the amide hydrogens in these regions are shielded from the solvent. Furthermore, the structured regions that are immediately adjacent to the DD and EE loops, corresponding to peptides 6 (αD helix) and 7 (αE helix), also exhibited relative differences in deuterium uptake kinetics between the monomeric and oligomeric/dimeric TcpB samples (Fig. 5C, middle panels). At early deuterium incubation times, these regions exhibited low deuterium uptake with no significant differences between the monomeric and dimeric TcpB. However, at longer incubation time points, deuterium uptake was significantly more pronounced in the monomeric form, suggesting that these regions are significantly more dynamic. Overall, the H/D exchange behavior of the TcpB peptides is consistent with the crystallographic dimer interface of the TcpBS124A/S127I TIR domain, analogous to the dimeric PdTIR TIR domain structure.
FIGURE 6.

H/D exchange mass spectrometry of TcpB. A, the differences in percentage deuteration (%D) (%D TcpBS124A/S127I TIR domain − %D TcpBS124A/S127I full-length) at various incubation time points (10 s, 1 min, 10 min, and 1 h) are mapped on the sequence of TcpB. Positive values (blue) indicate greater incorporation for the TcpBS124A/S127I TIR domain. Secondary structure elements are displayed above the sequence. B, pepsin digestion of the TcpBS124A/S127I TIR domain led to the detection of 105 peptides covering 99% of the entire sequence of the TIR domain. The peptide coverage map was generated using MS Tools (43).
The second region with distinct deuterium uptake kinetics for the monomeric versus oligomeric/dimeric TcpB corresponds to the BB loop and the adjacent αB helix. The BB loop peptide (e.g. peptide 1, TLKVGDSL) displayed reduced deuterium uptake at early time points for the full-length protein compared with the monomeric TcpB TIR domain (Fig. 5C, left). Such difference decreased with increasing incubation time and is negligible by 10 min. In addition, the adjacent αB helix exhibits similar decreased dynamics in the oligomeric/dimeric TcpB sample similarly to what was observed for αD and αE helices (Fig. 5C, middle). Because the BB loop is located away from the TcpB TIR domain dimer interface observed in the crystal, it is currently unclear what induced the distinct deuterium uptake kinetics in this region. It is possible that the BB loop may mediate intramolecular interactions within TcpB or play a role in the formation of TcpB oligomers.
Crystal Structure of the TIRAP TIR Domain
Because TcpB was reported to be a microbial mimic of the host adaptor TIRAP, and the TIR domains from these two proteins have high sequence similarity, we studied the structure of the TIRAP TIR domain and compared it with that of the TcpB TIR domain. Consistent with our preliminary report of the TIRAP structure (27) and recent publications from other groups (35, 36), the crystal structure of the human TIRAP TIR domain adopts a non-canonical TIR domain fold highlighted by a disulfide bond formed between Cys89 (βA) and Cys134 (βB) (Fig. 7, A and B). This is concomitant with a transition of the predicted αB helix residues to a long AB loop and an absence of the αB helix (Fig. 7, C and D). The remainder of the TIRAP TIR domain fold is structurally similar to the TIR domains from TcpB and others. A fold similarity search using the Dali server suggested that the TIRAP TIR domain is structurally similar to bacterial TIR domains, such as PdTIR and TcpB TIR (Table 3). The TIRAP TIR domain forms a symmetric dimer in the crystal lattice mediated by its αC′ and αD helices, as reported previously (35, 36).
FIGURE 7.
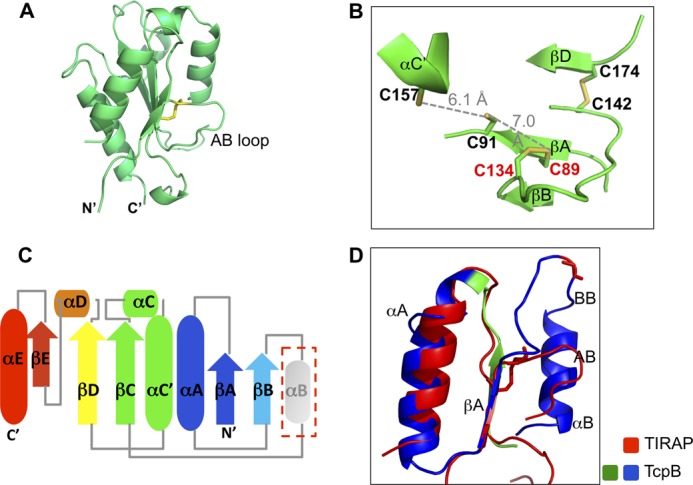
Crystal structure of the TIRAP TIR domain. A, schematic representation of the TIRAP TIR domain with the Cys89–Cys134 disulfide bond shown as sticks. B, close-up view of the disulfides in the TIRAP TIR domain. Residues Cys89, Cys91, Cys134, Cys142, Cys157, and Cys174 are shown as sticks. The distance between Cys91 and Cys157 and that between Cys91 and Cys89 are shown as gray dotted lines. C, schematic drawing of the secondary structure elements for the human TIRAP TIR domain. The absent αB helix is shown in gray. D, superposition of the βA-αB region of the TIRAP and TcpB TIR domains. The extended microtubule-stabilizing peptide from TcpB is colored blue and green, and the corresponding region of human TIRAP is colored red.
TABLE 3.
Structural similarity search using the TIRAP TIR domain structure
| Z-Score | RMSD | Aligned residues | Identity | |
|---|---|---|---|---|
| Å | % | |||
| PdTIR (3H16) | 12.5 | 2.2 | 109 | 12 |
| AtTIR (3JRN) | 11.8 | 2.9 | 112 | 10 |
| L6TIR (3OZI) | 11.2 | 2.6 | 115 | 11 |
| TcpB (4LQC) | 11.0 | 2.5 | 103 | 17 |
| TLR2 (1O77) | 10.1 | 2.7 | 103 | 20 |
| MyD88 (4DOM) | 9.7 | 3.1 | 108 | 17 |
| TLR1 (1FYV) | 9.4 | 3.1 | 110 | 15 |
DISCUSSION
Bacterial pathogens have evolved numerous mechanisms to suppress or evade immune responses directed toward them. One such mechanism involves the direct inhibition of the TLR signaling pathways. In this report, we present structural and biochemical studies of the virulence factor TcpB expressed by Brucella and the human adaptor protein TIRAP. The crystal structure of the TcpB TIR domain reveals that it adopts a canonical TIR fold conserved among the two bacterial TIR structures (TcpB and PdTIR) reported to date. H/D exchange mass spectrometry analysis revealed differences in deuterium uptake kinetics between the oligomeric full-length TcpB and the monomeric TcpB TIR domain, which is consistent with the dimeric crystal structure of the TcpB TIR domain mediated by its DD and EE loops. Additionally, differences in BB loop and αB helix dynamics observed from H/D exchange studies suggest intramolecular autoregulatory interactions between the BB loop region and other segments of TcpB or involvement in formation of oligomeric TcpB complexes.
While the current manuscript was in preparation, the Terradot group (37) and the Kobe group (44) determined essentially identical homodimeric structures of the TcpB TIR domain mediated by the DD and EE loops. Superposition of our structure with that from the Terradot group (4LZP) resulted in an RMSD of 0.87 Å for 252 aligned residues in a dimer (Fig. 8A). Mutagenesis of the relevant DD and EE loop residues reduced the stability of the dimer but did not completely abolish the dimer configuration (37). This was attributed to the stabilization of dimers by the N-terminal helical “tail,” which docks onto a hydrophobic groove across the dimer surface. It remains to be determined what role the N-terminal tail has in regulating TcpB function.
FIGURE 8.

The TcpB homodimer may mediate its association with TIRAP. A, comparison of the homodimeric TIR domain structures for the TcpBS124A/S127I mutant (blue) and the wild type (4LZP; silver). The N-terminal helical “tail” from the 4LZP structure is colored yellow with its N and C termini labeled. The DD and EE loops that mediate dimerization in both structures are marked. The view is rotated ∼90º horizontally from that in Fig. 4D. B, schematic representation of the TIRAP-mediated signaling complex formation. On the left, the TIRAP TIR domain (green) is recruited to the TLR4 TIR domain (gray), and a TIRAP TIR domain dimer formation mediated by its αC′ and αD helices exposes its AB loop (red) for recruitment of MyD88 (orange). The TcpB TIR domain (blue) forms homodimers through its DD and EE loops on the right. Upon association of TcpB with TIRAP, perhaps mediated by the BB loop of TcpB, TIRAP is no longer able to associate with TLR4 and induce downstream signaling.
Because TcpB is a functional mimic of TIRAP, comparison of the TIR domain structures from TcpB and TIRAP may yield important insight into the molecular mechanisms of the functional mimicry. For example, the TIRAP TIR domain dimer mediated by its αC′ and αD helices exposes its long AB loops on the opposite side of the TIR domains. The AB loops may bind MyD88 or TLR4 TIR domains, in analogy to the association of the TcpB TIR domain BB loop region with microtubules. Binding of the TcpB TIR domain dimer to TIRAP may suppress its signaling function through interference with TIRAP localization to the plasma membrane (3), promotion of TIRAP degradation (15), inhibition of productive TIRAP dimer/oligomer formation, or reduced association with upstream or downstream partners. A schematic representation of the latter is outlined in Fig. 8B.
The TcpB G158A mutation that affects its ability to stabilize microtubule is located at the apex of the BB loop and is highly exposed to solvent. This mutation may impact its ability to bind microtubule through modulation of the BB loop flexibility, similar to the “Poc” site I179N mutation in the MyD88 TIR domain (38). The extended BB loop peptide (corresponding to residues 127–174), which was shown to stabilize the microtubule, encompasses the secondary structure elements αB helix, BB loop, and βB strand. This peptide essentially contains the entire solvent-exposed surface opposite to those involved in the TcpB dimer interface. Although the extended BB loop peptide contributes to microtubule stability, an intact TcpB TIR domain is needed for optimal microtubule stabilization (18), suggesting that the remainder of the TcpB TIR domain provides the necessary molecular scaffold for the peptide.
A comparison of the receptor, adaptor, and bacterial TIR domain structures reveals unique positions for loops and helices among the TIR domain subtypes. For example, the BB loops (AB loop in TIRAP) and the CD loops from different TIR domain structures adopt distinct conformations. The BB loop has been suggested to mediate formation of TIR domain complexes. The CD loop was recently shown to mediate TcpC interaction with MyD88 (12), and may be employed in homo- or heterotypic TIR domain interactions involving TIRAP or TcpB as well.
A recent report demonstrated that the TcpC TIR domain-derived peptides were capable of selectively interacting with either TLR4 or MyD88 TIR domains, which represent novel bacteria-derived inhibitory peptides (12). Other inhibitory peptides and peptidomimetics have also been reported to derive from endogenous TIR domains, including TLRs (TLR1, TLR2, and TLR4), and adaptors (MyD88, TIRAP, TRIF, and TRAM) (39–41). Future studies will explore whether similar inhibitory peptides could be derived from the TcpB TIR domain structure. Such peptides or peptidomimetics may provide valuable reagents that specifically target the TIRAP adaptor.
It is clear that the Box 1 motif plays essential roles in the structure and function of TIR domains reported to date. Since our reported observations of a unique fold for the TIRAP TIR domain crystal structure (27), others have published similar crystallographic observations and have described this domain in great detail (35, 36). Most microbial TIR domains, such as those from TcpB, TcpC, and PdTIR, do not contain Cys residues. In contrast, both adaptor molecules MyD88 and TIRAP contain conserved cysteines within the box 1 motif. The TIRAP TIR domain crystal structures are the first to demonstrate a long AB loop with missing αB helix and a box 1 motif-mediated intradomain disulfide bond. Interestingly, mutation of the TLR2 C713S (within its TIR domain Box 1 motif) resulted in an extensively associated TIR domain dimer mediated by an interdomain disulfide bond (42). Our protease digestion analysis of TcpB suggests that mutations S124A and S127I within its TIR domain box 1 motif were necessary for protein stability and crystallization. These mutations may stabilize the βA strand and β sheet formation at the hydrophobic core of the TIR domain. It remains to be determined whether other box 1 motif residues also play a role in modulating intradomain and/or interdomain interactions, the stability of the TIR domains, or TIR domain-mediated signaling.
Acknowledgments
We thank Dr. David S. Waugh (NCI, National Institutes of Health) for the TEV protease expression construct and Dr. Gerhard Wagner (Harvard Medical School) for the GB1-encoding plasmid. We thank Dr. D. Eric Anderson (Mass Spectrometry Facility, NIDDK, National Institutes of Health) for technical support. We are grateful to the beam line scientists at the Advanced Photon Source and National Synchrotron Light Source (Brookhaven National Laboratory) for support.
This work was supported, in whole or in part, by the Division of Intramural Research, NIAID, National Institutes of Health (to T. S. X.).
The atomic coordinates and structure factors (codes 4LQC and 4LQD) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TLR
- T cell receptor
- TIR
- Toll/interleukin-1 receptor
- TIP
- TIR-interacting protein
- TEV
- tobacco etch virus
- SeMet
- selenomethionine
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- H/D
- hydrogen/deuterium
- RMSD
- root mean square deviation.
REFERENCES
- 1. Kenny E. F., O'Neill L. A. (2008) Signalling adaptors used by Toll-like receptors. An update. Cytokine 43, 342–349 [DOI] [PubMed] [Google Scholar]
- 2. Cirl C., Wieser A., Yadav M., Duerr S., Schubert S., Fischer H., Stappert D., Wantia N., Rodriguez N., Wagner H., Svanborg C., Miethke T. (2008) Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat. Med. 14, 399–406 [DOI] [PubMed] [Google Scholar]
- 3. Radhakrishnan G. K., Yu Q., Harms J. S., Splitter G. A. (2009) Brucella TIR domain-containing protein mimics properties of the Toll-like receptor adaptor protein TIRAP. J. Biol. Chem. 284, 9892–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Salcedo S. P., Marchesini M. I., Lelouard H., Fugier E., Jolly G., Balor S., Muller A., Lapaque N., Demaria O., Alexopoulou L., Comerci D. J., Ugalde R. A., Pierre P., Gorvel J.-P. (2008) Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newman R. M., Salunkhe P., Godzik A., Reed J. C. (2006) Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect. Immun. 74, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Low L. Y., Mukasa T., Reed J. C., Pascual J. (2007) Characterization of a TIR-like protein from Paracoccus denitrificans. Biochem. Biophys. Res. Commun. 356, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rana R. R., Simpson P., Zhang M., Jennions M., Ukegbu C., Spear A. M., Alguel Y., Matthews S. J., Atkins H. S., Byrne B. (2011) Yersinia pestis TIR-domain protein forms dimers that interact with the human adaptor protein MyD88. Microb. Pathog. 51, 89–95 [DOI] [PubMed] [Google Scholar]
- 8. Cirl C., Miethke T. (2010) Microbial Toll/interleukin 1 receptor proteins. A new class of virulence factors. Int. J. Med. Microbiol. 300, 396–401 [DOI] [PubMed] [Google Scholar]
- 9. Yadav M., Zhang J., Fischer H., Huang W., Lutay N., Cirl C., Lum J., Miethke T., Svanborg C. (2010) Inhibition of TIR domain signaling by TcpC. MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 6, e1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snyder G. A., Sundberg E. J. (May 22, 2013) Molecular interactions in interleukin and Toll-like receptor signaling pathways. Curr. Pharm. Des. 10.2174/13816128113199990069 [DOI] [PubMed] [Google Scholar]
- 11. Xiao T. S. (2010) Subversion of innate immune signaling through molecular mimicry. J. Clin. Immunol. 30, 638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snyder G. A., Cirl C., Jiang J., Chen K., Waldhuber A., Smith P., Römmler F., Snyder N., Fresquez T., Dürr S. (2013) Molecular mechanisms for the subversion of MyD88 signaling by TcpC from virulent uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 6985–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Neill L. A., Bowie A. G. (2007) The family of five. TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 14. Campos M. A., Rosinha G. M., Almeida I. C., Salgueiro X. S., Jarvis B. W., Splitter G. A., Qureshi N., Bruna-Romero O., Gazzinelli R. T., Oliveira S. C. (2004) Role of Toll-like receptor 4 in induction of cell-mediated immunity and resistance to Brucella abortus infection in mice. Infect. Immun. 72, 176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sengupta D., Koblansky A., Gaines J., Brown T., West A. P., Zhang D., Nishikawa T., Park S.-G., Roop R. M., 2nd, Ghosh S. (2010) Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter, MAL. J. Immunol. 184, 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kagan J. C., Medzhitov R. (2006) Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 17. Chaudhary A., Ganguly K., Cabantous S., Waldo G. S., Micheva-Viteva S. N., Nag K., Hlavacek W. S., Tung C.-S. (2012) The Brucella TIR-like protein TcpB interacts with the death domain of MyD88. Biochem. Biophys. Res. Commun. 417, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radhakrishnan G. K., Harms J. S., Splitter G. A. (2011) Modulation of microtubule dynamics by a TIR domain protein from the intracellular pathogen Brucella melitensis. Biochem. J. 439, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt T. G., Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 20. Radhakrishnan G. K., Splitter G. A. (2010) Biochemical and functional analysis of TIR domain containing protein from Brucella melitensis. Biochem. Biophys. Res. Commun. 397, 59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 27. Snyder G., Jiang J., Chen K., Fresquez T., Smith P., Snyder N., Luchetti T., Cirl C., Miethke T., Tjandra N., Xiao T. (2010) Structural studies of Toll like receptor signaling adapters. J. Immunol. 184, 136.45 [Google Scholar]
- 28. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang H., Guranovic V., Dutta S., Feng Z., Berman H. M., Westbrook J. D. (2004) Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 60, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 30. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 31. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee B., Richards F. M. (1971) The interpretation of protein structures. Estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 33. Potterton L., McNicholas S., Krissinel E., Gruber J., Cowtan K., Emsley P., Murshudov G. N., Cohen S., Perrakis A., Noble M. (2004) Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 60, 2288–2294 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 35. Valkov E., Stamp A., Dimaio F., Baker D., Verstak B., Roversi P., Kellie S., Sweet M. J., Mansell A., Gay N. J., Martin J. L., Kobe B. (2011) Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. U.S.A. 108, 14879–14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin Z., Lu J., Zhou W., Shen Y. (2012) Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS One 7, e34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaplan-Türköz B., Koelblen T., Felix C., Candusso M.-P., O'Callaghan D., Vergunst A. C., Terradot L. (2013) Structure of the Toll/interleukin 1 receptor (TIR) domain of the immunosuppressive Brucella effector BtpA/Btp1/TcpB. FEBS Lett. 587, 3412–3416 [DOI] [PubMed] [Google Scholar]
- 38. Jiang Z., Georgel P., Li C., Choe J., Crozat K., Rutschmann S., Du X., Bigby T., Mudd S., Sovath S., Wilson I. A., Olson A., Beutler B. (2006) Details of Toll-like receptor:adapter interaction revealed by germ-line mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 10961–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kissner T. L., Moisan L., Mann E., Alam S., Ruthel G., Ulrich R. G., Rebek M., Rebek J., Jr., Saikh K. U. (2011) A small molecule that mimics the BB-loop in the Toll interleukin-1 (IL-1) receptor domain of MyD88 attenuates staphylococcal enterotoxin B-induced pro-inflammatory cytokine production and toxicity in mice. J. Biol. Chem. 286, 31385–31396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loiarro M., Capolunghi F., Fantò N., Gallo G., Campo S., Arseni B., Carsetti R., Carminati P., De Santis R., Ruggiero V., Sette C. (2007) Pivotal advance. Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukocyte Biol. 82, 801–810 [DOI] [PubMed] [Google Scholar]
- 41. Loiarro M. (2005) Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J. Biol. Chem. 280, 15809–15814 [DOI] [PubMed] [Google Scholar]
- 42. Tao X., Xu Y., Zheng Y., Beg A. A., Tong L. (2002) An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem. Biophys. Res. Commun. 299, 216–221 [DOI] [PubMed] [Google Scholar]
- 43. Kavan D., Man P. (2011) MSTools. Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58 [Google Scholar]
- 44. Alaidarous M., Ve T., Casey L. W., Valkov E., Ericsson D. J., Ullah M. O., Schembri M. A., Mansell A., Sweet M. J., Kobe B. (2014) Mechanism of bacterial interference with TLR4 signaling by Brucella Toll/interleukin-1 receptor domain-containing protein TcpB. J. Biol. Chem. 289, 654–668 [DOI] [PMC free article] [PubMed] [Google Scholar]