

Background: N-Linked glycosylation is catalyzed by oligosaccharyltransferase (OST).
Results: A so far unrecognized sequence motif in the external loop 5 (EL5) of bacterial OST is essential for catalysis.
Conclusion: EL5 is involved in acceptor substrate binding and in critical interactions with the lipid-linked oligosaccharide.
Significance: The study defines the dual role of EL5 during catalysis in protein N-glycosylation.
Keywords: Campylobacter, Cysteine-mediated Cross-linking, Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, Glycoprotein Biosynthesis, Glycosyltransferases, Membrane Enzymes, Oligosaccharide, Protein Structure
Abstract
Asparagine-linked glycosylation is a post-translational protein modification that is conserved in all domains of life. The initial transfer of a lipid-linked oligosaccharide (LLO) onto acceptor asparagines is catalyzed by the integral membrane protein oligosaccharyltransferase (OST). The previously reported structure of a single-subunit OST enzyme, the Campylobacter lari protein PglB, revealed a partially disordered external loop (EL5), whose role in catalysis was unclear. We identified a new and functionally important sequence motif in EL5 containing a conserved tyrosine residue (Tyr293) whose aromatic side chain is essential for catalysis. A synthetic peptide containing the conserved motif can partially but specifically rescue in vitro activity of mutated PglB lacking Tyr293. Using site-directed disulfide cross-linking, we show that disengagement of the structurally ordered part of EL5 is an essential step of the glycosylation reaction, probably by allowing sequon binding or glyco-product release. Our findings define two distinct mechanistic roles of EL5 in OST-catalyzed glycosylation. These functions, exerted by the two halves of EL5, are independent, because the loop can be cleaved by specific proteolysis with only slight reduction in activity.
Introduction
Glycosyltransferases utilize activated donor sugar substrates for the transfer to various targets, thereby forming glycosidic bonds. The transfer of glycans to the side chain of asparagines, a process termed N-linked protein glycosylation, is of major importance for protein folding, cell viability, and organism development (1–3). Protein N-glycosylation occurs in all three domains of life (4–6), and although there exist a number of variations, the general mechanism is conserved; a lipid-linked oligosaccharide (LLO)4 is assembled in a multistep process before being translocated from the cytosolic to the luminal side of the endoplasmic reticulum membrane of eukaryotes or to the periplasmic/external side of the plasma membrane of prokaryotes. The integral membrane enzyme oligosaccharyltransferase (OST) then catalyzes the en bloc transfer of the oligosaccharide to asparagine residues located in the consensus sequon Asn-Xaa-Ser/Thr (with Xaa ≠ Pro) of acceptor polypeptides. The OST enzyme in higher eukaryotes is a multiprotein complex with Stt3 as the catalytic subunit (7, 8), but in kinetoplastids and prokaryotes, OST is a single-subunit enzyme that is homologous to Stt3 (9–11). The crystal structure of a full-length, bacterial Stt3 homolog, the PglB protein of Campylobacter lari, revealed the architecture of this class of enzymes (12). PglB contains an N-terminal transmembrane (TM) domain featuring 13 TM segments and a soluble domain facing the periplasm. The TM domain contains two large external loops (EL1 and EL5) that provide non-covalent interactions between the TM and the periplasmic domains. Whereas the role of EL1 appears to be mainly a structural one, EL5 was proposed to be involved in catalytic steps of the process. Recent functional studies have provided a quantitative basis for sequon recognition and binding by PglB; the +2 sequon Ser/Thr of acceptor substrates is recognized by a binding pocket provided by the WWD motif (a diagnostic motif among Stt3 homologs) and a neighboring Ile residue (Ile572), suggesting that the +2 Ser/Thr defines the specificity of N-linked glycosylation sites (13). The C. lari PglB enzyme was also useful for studying the mechanism of activation of the acceptor Asn side chain of sequon (14).
Arguably the least understood feature of the PglB structure (and OST mechanism) is the role of the external loop EL5. Both for substrate binding and Asn activation, the C-terminal half of this loop appears to play an important role by pinning the bound sequon against the periplasmic domain, providing the essential residue Glu319 to the catalytic site and positioning the acceptor Asn correctly for glycosylation (12, 13). The function of the N-terminal half of EL5, which was disordered in the structure of peptide-bound PglB (supplemental Fig. S1, top state), remained unclear.
To explore the function of the N-terminal half of EL5 and to investigate conformational rearrangements in the C-terminal half during catalysis, we performed a detailed structure-function analysis of C. lari PglB. We used a synthetic, fluorescently labeled peptide that contained a bacterial glycosylation sequon and a previously established in vitro assay that not only provided high precision in determining glycosylation turnover but also allowed us to observe very low reaction rates of disfavored PglB mutants and determine changes in sequon binding affinities (13). We thus identified a previously unrecognized, catalytically essential motif, termed Tyr-plug, in the N-terminal half of EL5 that contains a conserved tyrosine residue (Tyr293). The motif is essential for catalysis but not for peptide binding.
We had speculated earlier that once the N-glycosidic linkage is formed, the glyco-polypeptide will need to dissociate from the enzyme, which will probably require a disengagement of EL5 during the catalytic cycle (supplemental Fig. S1). To investigate this hypothesis, we engineered disulfide cross-links in PglB by introducing cysteines in EL5 and the enzyme core at three distinct locations. The resulting PglB mutants were expressed and purified in large quantities, and sequon binding and glycosylation turnover rates were determined under oxidizing and reducing conditions. The results allowed us to define the dynamics of EL5 during catalysis.
EXPERIMENTAL PROCEDURES
Construction of Plasmids
Point mutations in the pglB gene of C. lari were generated by the QuikChangeTM method either on a pBAD plasmid (12) for in vitro glycosylation studies or on a pMLBAD plasmid (15) for in vivo glycosylation assays. For in vitro glycosylation, the two endogenous glycosylation sites were removed (N535Q and N556Q) to prevent autoglycosylation of PglB, and all subsequent mutations were based on this construct. We refer to wild type PglB in our in vivo and in vitro experiments irrespective of the presence of endogenous glycosylation sites because the catalytic site and the peptide binding site are not affected by these mutations. Plasmid DC3 was obtained by digesting plasmid pACYCpglmut (11) with enzyme BbvCI and ligation of the insert containing the pglB gene into the backbone of plasmid pACYCpgl::pglJ (16) digested with the same enzyme. Plasmid pCL67 was obtained by digesting a plasmid containing a synthesized GFP loop with a glycosylation site (Invitrogen) with enzymes MfeI and DraI and ligation of the 246-bp insert into plasmid pSN28 (17), where the NheI site was inactivated (by QuikChangeTM) and which was treated with the same enzymes.
To generate plasmids with EL5 mutations, the restriction sites for enzymes SwaI (S280N) and XhoI (silent) were introduced into the pMLBAD plasmid containing the pglB gene by the QuikChangeTM method. Plasmid EL5 mutant 1 was obtained by cloning of a PCR fragment amplified with oligonucleotides 1 and 2 using a Wolinella succinogenes genome sequencing cosmid (18) as template. The PCR product was digested with enzymes SwaI and XhoI and ligated into the pMLBAD plasmid digested with the same enzymes. The sequence of EL5 mutants 2, 3, 4, 6, and 8 was generated by gene synthesis (Invitrogen), and the pMLBAD plasmids for the corresponding EL5 variants were constructed by subcloning of provided vectors using enzymes SwaI and XhoI. The pBAD construct for EL5 mutant 8 was subcloned from the pMLBAD construct using enzymes NcoI and EcoRI. The mutation S280N, generated for the SwaI restriction site was removed from all final plasmids by the QuikChangeTM method.
EL5 mutant 7 was obtained by overlap PCR to remove PglB residues 237–280. The upstream PCR fragment was generated with oligonucleotides 3/4, and the downstream fragment was generated with oligonucleotides 5/6. Overlap PCR was performed using the two purified PCR fragments and oligonucleotides 7/8. The overlap PCR fragment was digested with enzymes NcoI and EcoRI for ligation into the pBAD plasmid digested with the same enzymes; the fragment was digested with enzymes MfeI and PstI for ligation into the pMLBAD plasmid digested with enzymes EcoRI and PstI.
Constructs for EL5 mutant 5 were simultaneously created for the pBAD and pMLBAD plasmid using overlap PCR. For pMLBAD, the upstream PCR fragment was generated with oligonucleotides 9/10, and the downstream fragment was generated with oligonucleotides 11/12. For pBAD, the upstream PCR fragment was generated with oligonucleotides 13/10, and the downstream fragment was generated with oligonucleotides 11/14. Overlap PCR was performed using the purified PCR fragments and oligonucleotides 9/12 and 13/14, respectively. The overlap PCR fragments were ligated into zero blunt end TOPO vectors (Invitrogen), and the final expression plasmids were subcloned using enzymes EcoRI/PstI for the pMLBAD and NcoI/EcoRI for the pBAD vector, respectively.
All plasmids were validated by DNA sequencing. A description of the oligonucleotide primers used and the plasmids constructed can be found in supplemental Tables S1 and S2.
In Vivo Glycosylation Assay
In vivo complementation analysis of PglB mutants was performed as described before (12). Briefly, Escherichia coli SCM6 cells were transformed with three separate plasmids carrying 1) the C. jejuni pglBmut cluster (containing an inactivated pglB gene) (11), 2) the glycosylation acceptor protein 3D5 containing a DQNAT acceptor site (19), and 3) C. lari PglB, wild type or mutants. Expression and glycosylation of the acceptor protein 3D5 was monitored by SDS-PAGE of periplasmic cell extracts and visualized by mobility shift to increased size in an Immunoblot using an anti-c-Myc antibody (Calbiochem) or by the reactivity of the glycoprotein in an anti-glycan Immunoblot using hR6 antiserum. Immunodetection of PglB expression was performed from whole cell extracts using an anti-HA antiserum (Santa Cruz Biotechnology, Inc.).
Expression and Purification of PglB Mutants
Overexpression and purification of PglB mutants was performed as described previously (13). The desalted protein was analyzed by size exclusion chromatography (Superdex 200, GE Healthcare) and, if needed, concentrated up to 26 mg/ml in a 100 kDa molecular mass cut-off Ultra-15 concentrator (Amicon, Millipore). The concentrated sample was reanalyzed by size exclusion chromatography, and protein concentrations were determined by absorbance at 280 nm using diluted samples.
EL5 cross-linking variants were prepared from whole cells. Cells of mutant Pos1 (N321C/L374C) and Pos2 (F311C/F579C) were resuspended in a buffer containing 10 mm Tris-HCl, pH 8.0, 250 mm NaCl, and 10 mm β-mercaptoethanol at a cell mass to buffer ratio of 1:7 (w/v). The suspension was supplemented with 1% (w/v) N-dodecyl-β-d-maltopyranoside (Anatrace) and sonicated for 5 min on ice. Sonication was repeated after 5 and 15 min, and the protein was solubilized for 1 h at 4 °C. Nickel-nitrilotriacetic acid (Ni-NTA) equilibration and washing buffers contained 10 mm β-mercaptoethanol, which was removed in a last washing step prior to elution. The Ni-NTA eluate was supplemented with 10 mm MnCl2 and 1 mm CuCl2, incubated for 20 min at 4 °C, and desalted into a buffer containing 10 mm Mes-NaOH, pH 6.5, 100 mm NaCl, 0.5 mm EDTA, 3% (v/v) glycerol, and N-dodecyl-β-d-maltopyranoside.
Cells of EL5 cross-linking mutant Pos3 (F308C/K528C) were resuspended as described above, omitting the addition of β-mercaptoethanol during solubilization and purification. The Ni-NTA-bound protein was washed for 20 column volumes, followed by 5 column volumes of imidazole-free wash and a 5-column volume wash containing 1 mm CuCl2. After a 15-min incubation at 4 °C, CuCl2 was washed out, and the protein was eluted and desalted as described above.
For glycosylation turnover and sequon binding measurements of reduced EL5 mutants, cross-linked samples were treated with 80 mm β-mercaptoethanol. Resistance to reduction of the induced disulfides in the presence of 80 mm β-mercaptoethanol in the folded state was demonstrated by inactivation of the reducing agent with 100 mm N-ethylmaleimide (Fluka) followed by SDS-PAGE under non-reducing conditions (see “Quantification of EL5 cross-linking efficiencies”).
PglB mutant with deletion of TM helices 8 and 9 (mutant 7) was purified from membranes as described above. This mutant was not desalted but was directly concentrated to 3.5 mg/ml after elution from the Ni-NTA column and further analyzed by SDS-PAGE and MALDI-MS to prove the expected molecular weight.
PglB mutant carrying a cleavable EL5 loop (mutant 8) was purified from membranes as described above, and the desalted protein was incubated in a 1:3 molar ratio of PglB/3C protease (prepared in house) for 2 h at 4 °C. The protein was concentrated and repurified by preparative size exclusion chromatography (Superdex 200, GE Healthcare) and reconcentrated for final functional analysis.
Expression and Purification of Glycoengineered GFP
Glycoengineered GFP was overexpressed from pCL67 in E. coli BL-21 (DE3) Gold cells in 5-liter baffled flasks using Terrific Broth medium supplemented with 1% (w/v) glycerol. Cells were grown to A600 of 2.0 and induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside for 4 h at 37 °C. Cells were harvested by centrifugation, and cell pellets were resuspended in a buffer containing 100 mm Tris-HCl, pH 8.0, and 100 mm NaCl at a cell mass/buffer ratio (w/v) of 1:5. Cells were disrupted by sonication on ice (3 × 3 min), and GFP was purified from the supernatant by Ni-NTA affinity chromatography and desalted into a buffer containing 100 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 0.5 mm EDTA. The desalted protein was analyzed by size exclusion chromatography (Superdex 200, GE Healthcare), protein concentration was determined by absorbance at 280 nm, and the protein was stored at 4 °C.
Expression and Purification of HmuT-Q147C
HmuT-Q147C periplasmic substrate-binding protein was expressed as described previously (20). Cells were resuspended in a buffer containing 50 mm Tris-HCl, pH 8.0, and 150 mm NaCl at a cell mass/buffer ratio (w/v) of 1:5, and cells were disrupted by sonication on ice (3 × 3 min). HmuT-Q147C was purified from the supernatant by Ni-NTA affinity chromatography and desalted into a buffer containing 10 mm Mes-NaOH, pH 6.5, 100 mm NaCl, 0.5 mm EDTA, and 3% (v/v) glycerol. The protein was supplemented with 10 mm tris(2-carboxyethyl)phosphine and stored at −20 °C.
Quantification of EL5 Cross-linking Efficiencies
The cross-linking efficiency of PglB mutants Pos2 (F311C/F579C) and Pos3 (F308C/K528C) could be determined from a mobility shift to increased molecular weight after SDS-PAGE in the presence of non-reducing sample buffer. Samples were treated with an excess of N-ethylmaleimide (Fluka) prior to the addition of SDS sample buffer. Band intensities of oxidized and reduced species were determined from scanned gels (ImageJ). Mutant Pos1 (N321C/L374C) did not reveal a mobility shift upon cross-linking, and the residual non-cross-linked fraction was quantified by labeling of the free cysteines. The sample was incubated with a 3-fold molar excess of fluorescein 5-maleimide (Thermo Scientific) over cysteines for 10 min at room temperature, and the reaction was quenched with a molar excess of N-ethylmaleimide. The protein HmuT-Q147C carrying a single, surface-exposed cysteine (20, 21) was used as an internal standard. HmuT-Q147C labeling was performed after preparative size exclusion chromatography (to remove tris(2-carboxyethyl)phosphine) according to the procedure for PglB labeling. The labeled proteins were mixed in an equimolar ratio and resolved by SDS-PAGE, and fluorescent bands were visualized by using a Typhoon Trio Plus imager (GE Healthcare) with excitation at 488 nm and a 526-nm SP emission filter. Band intensities were determined from fluorescence gel scans (ImageJ). Fluorescence bands were additionally correlated to Immunoblot signals of the same gel. Immunodetection of PglB and HmuT-Q147C was performed using an anti-His5 antibody (Qiagen).
Synthesis of DQNAT Acceptor Peptide Labeled with 5-Carboxyfluorescein
Peptide synthesis was performed as described previously (13).
Fluorescence Anisotropy Measurements
Measurements of acceptor peptide binding by fluorescence anisotropy were performed as described previously (13).
Extraction of LLOs
Isolation of LLOs containing the C. jejuni heptasaccharide was performed as described before (17). LLOs with the truncated disaccharide glycan structure (GalNAc-α1,3-diNAcBac-α1-PP-Und) were extracted from E. coli SCM6 cells carrying plasmid DC3 (containing an inactivated pglB gene) by a mixture of chloroform/MeOH of 2:1. Extracts were dried in a rotary evaporator and reconstituted in a buffer containing 10 mm Mes, pH 6.5, 100 mm NaCl, and 1% (w/v) Triton X-100. The concentration of reconstituted LLOs was determined by titrating various amounts of LLO against a constant amount of fluorescently labeled acceptor peptide in an in vitro glycosylation assay (see below).
In Vitro Glycosylation Assay
In vitro glycosylation assays were performed as described previously (13) using fluorescently labeled acceptor peptide or glycoengineered GFP (10 μm each) as acceptor substrate and C. jejuni LLO (heptasaccharide) or the disaccharide LLO (35 μm each) as donor substrate. For turnover rate determination, a total of 5 samples were taken in 6-min intervals, and the concentration of PglB mutants was adjusted (1 nm to 50 μm) so that the reaction was in a linear range. Data were fitted by linear regression to determine turnover rates.
For EL5 peptide titration experiments, 15 μm PglB mutant 5 (Tyr-plug mutant) was incubated with various amounts of undecapeptides EL5 (acetyl-VLYQLKFYVFK-NH2) and EL5_mut (acetyl-VLYQLKFAVFK-NH2) (GenScript), respectively. Five samples were taken at appropriate time intervals (3–10 min) so that the reaction was in a linear range.
For analysis of GFP glycosylation, Triton-PAGE was used. Reactions were stopped by the addition of 4× native sample buffer (375 mm Tris-HCl, pH 6.8, 250 mm EDTA, 16.5% (w/v) sucrose, 0.1% (w/v) Triton X-100). Samples were diluted 7-fold prior to analysis by Triton-PAGE in minigels (8 × 8 cm) consisting of an 8.5% resolving gel (0.5 m Tris-HCl, pH 8.8, 7% sucrose, 0.1% Triton X-100) and a 5% stacking gel (125 mm Tris-HCl, pH 6.8, 0.1% Triton X-100). Gels were run at 4 °C in a running buffer containing 50 mm Tris and 384 mm glycine. Fluorescent bands for glycosylated and non-glycosylated GFP were visualized by using a Typhoon Trio Plus imager (GE Healthcare) with excitation at 488 nm and a 610-nm emission filter. The amount of formed glyco-GFP was determined analogously to the amount of formed glycopeptides (13).
Mass Spectrometry
Mass spectra were acquired using a commercial MALDI-TOF/TOFTM mass spectrometer as described previously (22). The measurement was performed in the linear positive ion mode, and ionization was achieved with an Nd:YAG laser (355 nm). The mass spectrum was the average of 1,000 laser shots acquired at random sample positions. The sample (2 μm) was directly mixed with the matrix solution (sinapinic acid, 20 mg/ml in water/acetonitrile/trifluoroacetic acid, 49.95:49.95:0.1) in a 1:1 (v/v) ratio before spotting. A mixture of cytochrome c and bovine serum albumin was applied for external calibration. Before data processing, the spectrum was background-subtracted and smoothed using a Savitzky-Golay algorithm (Igor Pro 6.2., Wavemetrics).
NMR Spectroscopy
NMR spectra were recorded at 283 K on Avance III 600-, 700-, or 750-MHz Bruker spectrometers equipped with a QCI, TCI, or TXI probe, respectively. The undecapeptide acetyl-VLYQLKFYVFK-NH2 (GenScript) was dissolved in either D2O or 95% H2O, 5% D2O at a concentration of 4 mm. Standard two-dimensional spectra were recorded for resonance assignment of the peptide: 1H-1H TOCSY (mixing times of 13 and 80 ms), 13C-1H HSQC, long range 13C-1H HSQC, 13C-1H HMQC-COSY, and 1H-1H NOESY spectra (mixing time of 150 ms). All spectra are referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid, whereas 13C chemical shifts are indirectly referenced using a scaling factor Ξ of 0.251449530 (23). All spectra were processed with Topspin 3.1 (Bruker) and analyzed by Sparky 3 (T. D. Goddard and D. G. Kneller, University of California, San Francisco). Chemical shift assignments are shown in supplemental Figs. S5 and S6.
RESULTS
Cross-Linking EL5 to PglB Core near the Peptide Binding Site Abolishes Catalysis
We first mutated the three cysteines in the sequence of C. lari PglB by mutagenesis (C17A, C30A, and C350S) and tested the in vitro activity and the sequon binding affinity of the resulting Cys-less enzyme (Table 1 and Fig. 1C). Cys-less PglB bound the bacterial glycosylation sequon DQNAT with an unchanged affinity compared with wild type enzyme (Kd of 1.18 ± 0.02 μm compared with 1.02 ± 0.06 μm for wild type PglB (13)) but showed a 2.2-fold reduction in glycosylation rate (0.69 ± 0.02 peptides/s compared with 1.50 ± 0.04 peptides/s for wild type PglB (13)). We then designed covalent cross-links to restrict the motion of EL5 (Gly282–Gln330) by engineering double cysteine mutants at three distinct locations of PglB. The first cross-link we engineered was the double mutant N321C/L374C, attaching EL5 to TM helix 12, close to the peptide binding region (Fig. 1A, Pos1). Oxidative disulfide bond formation at Pos1 was determined to be 96–98% efficient by calibrated fluorescein labeling of unreacted cysteines, and the disulfide bond could be completely reduced upon the addition of 80 mm β-mercaptoethanol (Table 1 and supplemental Fig. S2A). This cross-link strongly affected peptide binding because we observed a 45-fold reduction in affinity for the oxidized compared with the reduced enzyme (Table 1 and Fig. 1B). Because the cross-linking efficiency was not 100%, our data suggested that the cross-linked species is unable to bind the peptide (or binds it with an unmeasurably low affinity) and that the remaining, non-cross-linked PglB (which has a Kd of 2.6 ± 0.2 μm) accounts for the determined Kd of 117 ± 11 μm in the oxidized sample. This interpretation is consistent with the observed 40-fold increase in peptide glycosylation rate upon reduction of the disulfide cross-link at Pos1 (Table 1). We could exclude nonspecific effects by β-mercaptoethanol in the reduced sample because no significant effects on peptide binding or glycosylation were observed for the Cys-less mutant (Table 1 and Fig. 1C). In addition, the presence of 80 mm β-mercaptoethanol in the reaction did not affect PglB stability (supplemental Fig. S2B).
TABLE 1.
Acceptor peptide binding and glycosylation turnover rates of PglB variants containing EL5 disulfide cross-links
Conditions specified as oxidized refer to copper-induced disulfide cross-linking, and those specified as reduced indicate the presence of 80 mm β-mercaptoethanol. Dissociation constants (Kd) were derived from curves shown in Fig. 1, B and C, and turnover rates were derived from three independent experiments. The indicated errors represent the S.E. of the fit for each turnover rate and dissociation constant determination.
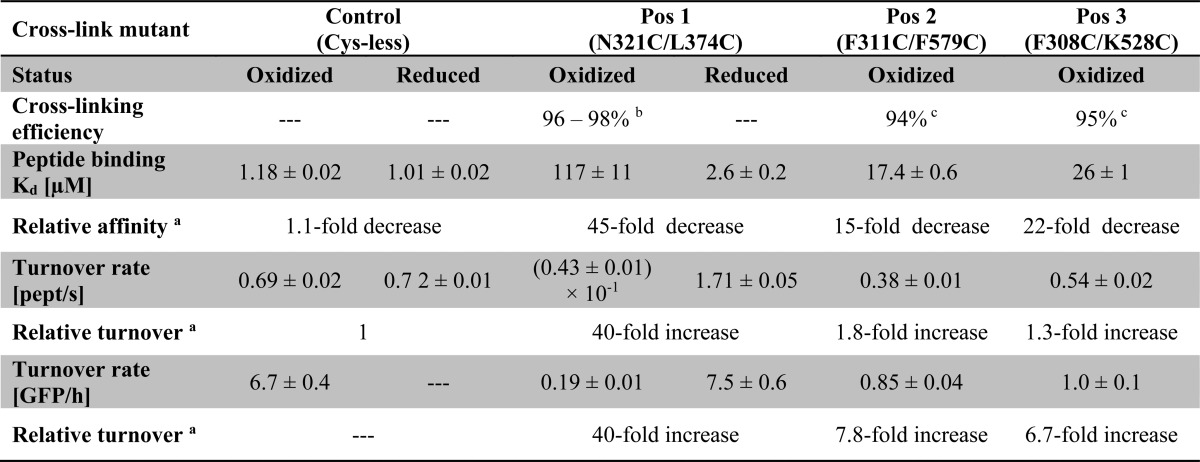
a Values for relative affinity and relative turnover refer to the ratio between the reduced and the oxidized condition of the same mutant. In the case of mutants for Pos2 and Pos3 cross-linking, they refer to the ratio between the oxidized sample of the Cys-less mutant (control) and the corresponding oxidized sample.
b Cross-linking efficiencies were determined by labeling of free cysteines with fluorescein 5-maleimide and subsequent quantification of fluorescence band intensities from scanned gels (supplemental Fig. S2A).
c Cross-linking resulted in a size shift after SDS-PAGE, and cross-linking efficiencies were determined from band intensities of Coomassie-stained gels (supplemental Fig. S2, C and D).
FIGURE 1.

Disulfide cross-linking of EL5 to core PglB. A, schematic representation of C. lari PglB (PDB code 3RCE) with the TM domain in blue, the periplasmic domain in green, and bound acceptor peptide in yellow. EL5 connecting TM9 and TM10 is colored red, with the structurally disordered segment (25 residues) depicted by a red dashed line. Residues selected for cross-linking are shown in a ball and stick representation and colored cyan. The pairwise mutation to cysteines at three distinct positions is illustrated by orange dashed circles, and corresponding residues are indicated. B and C, peptide binding of different cross-linking mutants quantified by fluorescence anisotropy. Purified PglB was titrated into a solution containing 1 μm fluorescently labeled peptide variant (containing the sequon DQNAT) and 10 mm MnCl2. Data points reflect the mean of 20 measurements of the same sample. Error bars, S.D. Curves labeled “oxidized” indicate samples that were cross-linked with CuCl2 during the purification process, whereas “reduced” indicates samples that contained 80 mm β-mercaptoethanol.
To evaluate the effect of substrate peptide size on the activity of our cross-linked PglB variants, we investigated the in vitro turnover of a GFP construct that contained the bacterial glycosylation sequon DQNAT flanked by flexible GGS sequences in a surface-exposed loop (Fig. 2A). Engineered GFP versions have previously been used both as a markers to monitor the N-glycosylation status of eukaryotic cell lines and as in vitro substrates of C. jejuni PglB (17, 24). Our in vitro glycosylation of GFP resulted in a mobility shift to increased molecular weight in native Triton-PAGE, and the educt/product ratio of the reaction could be directly quantified from fluorescence band intensities (Fig. 2B). We found Cys-less PglB to have a turnover rate of 6.7 ± 0.4 GFP molecules/h (Table 1). The EL5 cross-link at Pos1 resulted in a virtually unchanged activity in the presence of reducing agents (7.5 ± 0.6 GFP molecules/h) but a 40-fold decrease upon disulfide cross-linking (Table 1). Considering that our engineered GFP protein probably has a lower affinity for PglB than the chemically synthesized, fluorescently labeled substrate peptide, the ∼40-fold increase in activity both in peptide and GFP glycosylation strongly suggested that the cross-linked PglB species (Pos1) is indeed completely inactive and that the remaining, non-cross-linked species (∼2.5%) accounts for the observed basal activity in the oxidized sample. Therefore, we concluded that cross-linking EL5 at Pos1 completely inactivated the enzyme.
FIGURE 2.
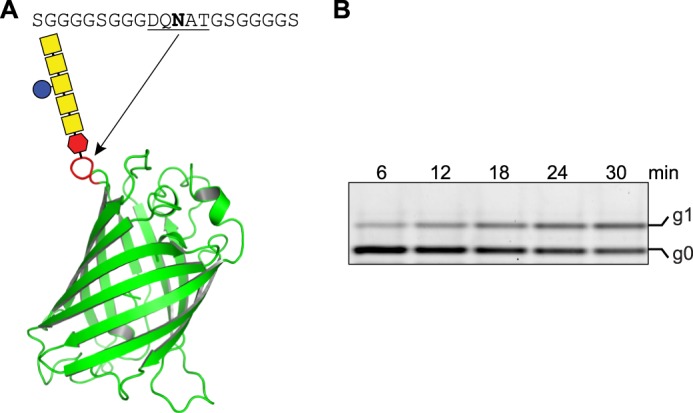
In vitro glycosylation of glycoengineered GFP. A, schematic representation of glycoengineered GFP (PDB code 1EMA) where a bacterial glycosylation site was introduced after Lys156 (red loop). The introduced site (DQNAT sequon) flanked by a GGGGS spacer is indicated above the image. B, determination of in vitro glycosylation turnover rate of glycoengineered GFP by Cys-less PglB. Glycosylated GFP (g1) is separated from non-glycosylated GFP (g0) after Triton-PAGE. Bands were visualized by a fluorescence gel scan at 488-nm excitation and 610-nm emission. The concentration of substrate GFP was 10 μm, and PglB concentration was 1.3 μm. The amount of glyco-GFP formed over time was determined from band intensities of the fluorescence gel scan, where the sum of the signals for glycosylated and non-glycosylated GFP for each lane was defined as 100%.
Cross-Linking EL5 Distant from the Peptide Binding Site Decreases Glycosylation Rate
We next investigated two cross-links that attached EL5 to the periplasmic domain, more remote from the sequon binding site than Pos1 (Fig. 1A). Disulfide bond formation efficiencies at Pos2 and Pos3 were determined to be 94 and 95%, respectively, but both cross-links could only be completely reduced under denaturing conditions upon the addition of SDS (supplemental Fig. S2, C and D). We therefore compared the activities of PglB cross-linked at Pos2 and Pos3 with those of Cys-less PglB. We found that cross-linking at Pos2 or Pos3 reduced the peptide affinities 15- and 22-fold, respectively. In analogy to the cross-link at Pos1, the observed binding curves could be interpreted as the cross-linked species being unable to bind peptide and the remaining, small fraction of non-cross-linked PglB being fully active. However, the determined glycosylation rates suggest a distinct conclusion; the cross-link at Pos2 showed a turnover rate of 0.38 ± 0.01 peptides/s, which is only 1.8-fold slower than that of Cys-less PglB, indicating that even the cross-linked species must be catalytically active and therefore able to bind peptide substrate. This observation is even more pronounced in the cross-link at Pos3, because the turnover rate for the substrate peptide was only 1.3-fold lower compared with Cys-less PglB. We also determined GFP substrate turnover rates for cross-links at Pos2 and Pos3. In both cases we found a more pronounced reduction in GFP than in peptide glycosylation (7.8-fold lower for Pos2 and 6.7-fold lower for Pos3; Table 1). We concluded that cross-linking EL5 at some distance from the peptide binding site has a strong effect both on sequon binding and on glycosylation of protein substrates but only a moderate effect on glycosylation of small peptide substrates.
A Short Motif in EL5, the Tyr-plug, Is Essential for PglB Function
In the C. lari PglB structure with bound acceptor peptide (12), the first 24 residues of EL5 (Leu283–Ala306) were disordered and not visible in the electron density (supplemental Fig. S1). Considering their location, it is tempting to speculate that these residues might interact with the glycan of bound LLO. A sequence alignment revealed that EL5 from W. succinogenes has the lowest similarity among different bacterial PglB homologs (Fig. 3B), and because the W. succinogenes glycan exhibits structural differences compared with that of C. lari (25–27), we replaced the EL5 sequence of C. lari PglB with the corresponding sequence of W. succinogenes (mutant 1). When analyzing the resulting in vivo activity, we found only a marginal reduction in acceptor substrate glycosylation compared with wild type C. lari PglB, suggesting that the EL5 sequence does not directly account for glycan specificity (Fig. 3C). We next created several EL5 versions where we inserted, replaced, and deleted residues in the disordered region (Fig. 3, A and B). An insertion of 8 amino acids at the beginning or end of the disordered region (mutants 6 and 2, respectively) did not impair PglB activity. Unexpectedly, neither did a deletion of transmembrane helices 8 and 9 (mutant 7), which alters the location of the first residue of EL5 (Fig. 3, A–C). It is notable that despite a reduction in mass by 5.2 kDa (determined by analyzing the purified PglB variant by MALDI-MS (22), mutant 7 and WT PglB have similar electrophoretic mobilities in SDS-PAGE (supplemental Fig. S3).
FIGURE 3.

Functional analysis of the disordered part of EL5. A, surface representation of C. lari PglB (PDB code 3RCE) with the TM domain in blue, the periplasmic domain in green, bound acceptor peptide in yellow, and EL5 in red. The disordered part of EL5 is indicated by a red dashed line. Cyan lines indicate regions of replacements, black lines indicate sequence deletions, and brackets indicate insertions. The white dashed line visualizes the removal of TM helices 8 and 9. Numbers correspond to the sequences shown in B and describe mutations introduced into EL5 from the C to the N terminus, successively away from the active site. B, sequence alignment of EL5 in bacterial PglB homologs. The sequence corresponding to the disordered part of EL5 is indicated by a red dashed line. Conservation of the catalytically important residue Glu319 is highlighted in black, and conservation of aromatic amino acids in the disordered part of EL5 is highlighted in magenta. Other conserved residues in the disordered region are shown in gray. For mutant 1, EL5 from C. lari PglB (Gly282–Gln330) was replaced by EL5 from W. succinogenes EL5 (Ala283–Ile329). Lines, brackets, and numbers are as in A. C–E, in vivo glycosylation assay in E. coli to investigate the activity of different EL5 mutants. Shown are immunoblots, detecting acceptor protein 3D5 (top), bacterial N-glycans (middle), and PglB (bottom). PglB mutants are indicated above the lanes. Glycosylation results in a mobility shift from the non-glycosylated (g0) to the glycosylated form of the acceptor protein (g1). Functional PglB is partially autoglycosylated at Asn535 and Asn556, resulting in two additional bands (g1 and g2). All experiments were performed at least in triplicate, and representative samples are shown. C, numbers above the lanes correspond to the numbers shown in A and B.
Sequence alignments further suggested that four aromatic residues in EL5 are conserved in bacterial homologs of PglB, whereas the middle section of EL5 varies in length and sequence (Fig. 3B). Replacement of the non-conserved region of EL5 by a flexible (GGS)3 sequence (mutant 3) did not affect the in vivo activity of PglB, and deletion of this region (mutant 4) only slightly reduced catalysis. However, we found that when a stretch of 8 amino acids, including the four aromatic residues, was replaced with a 3C cleavage site, PglB activity was completely abolished (mutant 5; Fig. 3, A–C). We termed this short stretch the “Tyr-plug” because of the contained tyrosines. When we analyzed the Tyr-plug mutant (mutant 5) in greater detail by previously established in vitro assays, we unexpectedly found that peptide substrate binding was only slightly affected (1.6-fold weaker binding than WT PglB), whereas glycosylation turnover was reduced 14,000-fold (Table 2 and Fig. 4A).
TABLE 2.
Peptide binding affinities and turnover rates for different EL5 mutants
Dissociation constants (Kd) were derived from curves shown in Fig. 4. The errors represent the S.E. of the fit. The relative turnover refers to the activity of an individual mutant compared with WT PglB. Activities were determined using fluorescently labeled acceptor peptide, and individual turnover rates were derived from three independent experiments.
| Treatment | EL5 Tyr-plug (mutant 5) | EL5 cleave (mutant 8) |
|
|---|---|---|---|
| Uncut | Cut | ||
| Kd (μm) | 1.66 ± 0.04 | 0.88 ± 0.03 | 2.32 ± 0.05 |
| Relative turnover (reduction) | 14,000-fold | 1.3-fold | 1.7-fold |
FIGURE 4.
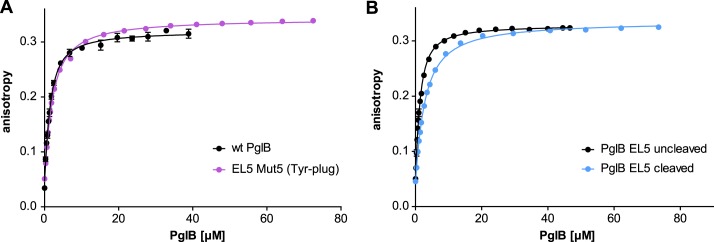
Peptide binding of different EL5 mutants quantified by fluorescence anisotropy. Purified PglB was titrated into a solution containing 1 μm fluorescently labeled peptide variant (containing the sequon DQNAT) and 10 mm MnCl2. Data points reflect the mean of 20 measurements of the same sample. Error bars, S.D. A, binding curve of PglB with replacement of the essential EL5 Tyr-plug sequence element (mutant 5, violet curve) compared with WT PglB (black curve, as presented in Ref. 13). B, binding curve of PglB with a cleavable EL5 (mutant 8 as presented in Fig. 6). The black curve represents PglB with an intact EL5 (uncleaved), whereas the blue curve results from PglB with cleaved EL5.
Tyr293 Has a Key Role in the Function of the Tyr-plug
In mutant 5, all four aromatic residues of the Tyr-plug (Tyr288, Phe292, Tyr293, and Phe295) were replaced. These residues are generally conserved among bacterial PglB homologs (Fig. 3B), and because aromatic residues are frequently involved in sugar interactions via π-stacking (28–30), we wished to clarify if the aromatic side chains were responsible for the observed drop in PglB reactivity. We thus created various alanine double mutations and tested their activities in vivo. We found that all double mutants that contained the Y293A mutation showed impaired function (Fig. 3D). A subsequent analysis of single mutants confirmed that Y293A leads to almost complete loss of activity, whereas the other mutations showed very little to no effect (Fig. 3D). Although tyrosine showed the strongest activity, the aromatic residues tryptophan, phenylalanine, and even histidine were tolerated at position 293 (Fig. 3E).
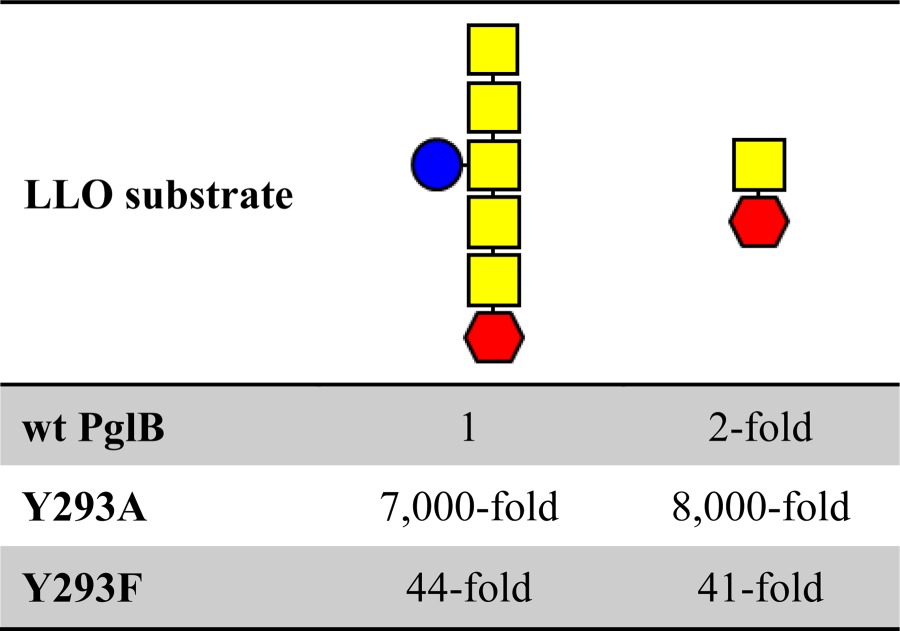
Because an aromatic side chain at position 293 was essential for PglB function, we initially assumed that Tyr293 forms a stacking interaction with one of the distal GalNAc residues of the LLO glycan, particularly because Gal-configured units represent ideal stacking partners for aromatic residues (30). To study the effect of a truncated glycan structure, we took advantage of an engineered version of the C. jejuni pgl gene cluster, yielding a LLO donor with a disaccharide structure (GalNAc-α1,3-diNAcBac-α1-PP-Und) (16). We used extracts of this short LLO variant along with WT C. jejuni heptasaccharide LLO at equal concentrations in an in vitro glycosylation assay and determined glycosylation turnover rates for different Tyr293 mutants. Using full-length C. jejuni LLO, we found reductions in turnover of 44-fold and 7,000-fold when Tyr293 was mutated to Phe and Ala, respectively (Table 3), confirming our in vivo findings that Tyr293 predominantly accounts for the effect of the Tyr-plug mutant (mutant 5) and that replacing Tyr293 by other aromatic amino acids affects PglB activity much less than removing the aromatic ring. Using the disaccharide LLO, we found a 2-fold reduction for WT PglB, which we interpret to be a result of slightly different biophysical properties of the truncated LLO. To our surprise, the turnover rates of mutants Y293F and Y293A were 41-fold and 8,000-fold reduced with the disaccharide LLO (Table 3). This excluded an interaction of Tyr293 with one of the distal GalNAc residues of the LLO.
TABLE 3.
Relative glycosylation turnover rates of different LLO donor substrates and PglB Tyr293 mutants
The relative turnover (-fold reduction) refers to the activity of an individual reaction compared with the reaction of WT PglB and LLO with the heptasaccharide structure. Activities were determined using fluorescently labeled acceptor peptide, and individual turnover rates were derived from three independent experiments. Red hexagon, di-N-acetylbacillosamine (diNAcBac); yellow square, N-acetylgalactosamine; blue circle, glucose.
A Synthetic Peptide Containing the Tyr-plug Confers Functionality
Our in vivo glycosylation studies suggested that the exact location of the Tyr-plug (including Tyr293) within EL5 is not important for catalysis because insertions before and after the motif were tolerated. This finding strongly suggested that the Tyr-plug had a defined three-dimensional conformation during catalysis. We therefore performed solution NMR analyses of a synthetic undecapeptide containing the Tyr-plug sequence (peptide EL5-11, VLYQLKFYVFK). The assignment of a corresponding two-dimensional NOESY spectrum of peptide EL5-11 did not reveal any long range NOEs, indicating the absence of a secondary structure (supplemental Fig. S4). The lack of a specific conformation of peptide EL5-11 was further confirmed by the assignment of a 15N-1H HSQC spectrum showing a small dispersion of obtained signals (supplemental Fig. S5). Nevertheless, the synthetic EL5-11 peptide did retain certain properties needed to support catalysis, because it could, to some extent, rescue the impaired function of a Tyr-plug mutant in vitro. When we determined glycosylation rates of the Tyr-plug mutant, we found that supplementing the reaction with peptide EL5-11 increased the glycosylation activity up to 3.6-fold at 100 μm peptide concentration. The activity decreased at higher peptide concentrations, which may be caused by limited peptide solubility or by nonspecific interactions with the detergent micelle (Fig. 5B). Considering the 14,000-fold reduction in activity for the Tyr-plug mutant (Table 2), the rescuing effect of the EL5-11 peptide might appear marginal. However, it is a highly specific effect depending on the presence of Tyr293, because a control peptide containing the Y293A mutation did not affect the activity of the Tyr-plug mutant (Fig. 5B). A limited local concentration of peptide EL5-11 caused by diffusion and by competition against the existing, mutated EL5 (Fig. 5A) might explain this small effect.
FIGURE 5.
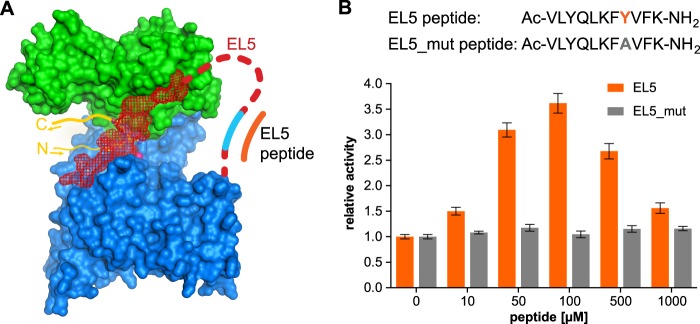
Complementation of impaired EL5 function by a synthetic EL5 peptide. A, surface representation of PglB, as shown in Fig. 3A, with replacement of the essential Tyr-plug indicated in cyan. B, the turnover rate of the Tyr-plug mutant (mutant 5) was determined at various concentrations of synthetic EL5 peptide. Data points reflect single turnover rate determinations. Error bars, S.E. of each fit. The sequence of used EL5 peptides is shown above the diagram.
The Two Functions of EL5 Are Independent
To investigate if the two functions of EL5 in catalysis are independent, we introduced a proteolytic cleavage site into EL5. The eight amino acid insertions in mutants 2 and 6 (and the sequence replacement of mutant 5) all represent 3C protease cleavage sites, but neither construct was found to be efficiently cleaved by 3C protease, possibly due to steric effects. We therefore designed a mutant where 10 non-conserved EL5 residues were replaced by a 3C cleavage site flanked by (GGS)2 spacer sequences on both sides (mutant 8; Fig. 6A). This mutant was active in vivo, could be cleaved efficiently with 3C protease in vitro, and showed good monodispersity before and after protease treatment (Fig. 6, B–D). Upon cleavage, peptide binding was reduced 2.6-fold relative to the uncut variant (Table 2 and Fig. 4B). In addition, cleavage only resulted in a marginal effect on glycosylation turnover, because the cut version revealed a 1.3-fold lower turnover rate than the uncut protein (Table 2). We conclude that the catalytically essential Tyr-plug in EL5 does not have to be covalently connected to the structurally ordered, peptide-binding segment of EL5 to fulfill its function.
FIGURE 6.

Design and analysis of PglB with a cleavable EL5. A, amino acid sequence of cleavable EL5 (mutant 8). The non-conserved region of EL5 (boldface type) was replaced by a 3C proteolytic cleavage site (red) flanked by a (GGS)2 element (blue) on both sides. The underlined residues are still ordered in the PglB structure. B, in vivo glycosylation assay in E. coli to investigate the activity of PglB with a cleavable EL5. Shown are immunoblots detecting acceptor protein 3D5 (top), bacterial N-glycans (middle), and PglB (bottom). PglB mutants are indicated above the lanes. Glycosylation results in a mobility shift from the non-glycosylated (g0) to the glycosylated form of the acceptor protein (g1). Functional PglB is partially autoglycosylated at Asn535 and Asn556, resulting in two additional bands (g1 and g2). C, SDS-PAGE of uncleaved and cleaved PglB. Fragments resulting from proteolytic cleavage are indicated with arrows. D, size exclusion chromatograms of purified and concentrated PglB EL5 cleave samples after peptide binding (fluorescence anisotropy) measurements. Black profile, PglB with intact EL5; red profile, sample with cleaved EL5. Note that the superimposed chromatograms are not normalized for the amount of protein loaded onto the gel filtration column. The analysis is used exclusively to assess monodispersity and integrity of this PglB mutant.
DISCUSSION
The long periplasmic loop EL5 was a remarkable yet insufficiently understood feature of the PglB structure. The C-terminal half of EL5 is involved in generating a tight binding site for acceptor peptide, suggesting that in a peptide-free state, PglB might adopt a distinct conformation where EL5 was disengaged from the core enzyme, as depicted in a three-step reaction cycle (supplemental Fig. S1). We tested this hypothesis using cysteine cross-linking experiments to restrict the flexibility of EL5. We indeed found that a cross-link at the peptide binding site (Pos1) abolished sequon binding and catalysis. In this PglB variant, EL5 may be unable to achieve an open conformation that is required for peptide binding, or the sequon binding site might have been distorted by the cross-link. The other two cross-links (positions 2 and 3), which are not overlapping with the peptide binding site, showed a reduced but clearly measurable activity. Our data allow the following conclusions. 1) Sequon binding can occur even when EL5 is only partially disengaged from the enzyme. 2) Sequon binding is not the rate-limiting step during catalysis in vitro because the strong reduction of peptide binding affinity (22-fold for Pos3) only leads to a slight reduction (1.3-fold for Pos3) in turnover. This interpretation is in agreement with previous observations that a replacement of the +2 Thr in glycosylation sequons by Ser, which reduced peptide binding 4-fold, only lowered turnover 1.2-fold (13). 3) The larger the peptide substrate, the stronger the effect of EL5 disengagement (for a comparison of peptide versus GFP glycosylation, see Table 1). 4) Partial disengagement of EL5 is sufficient for the release of glyco-products from the enzyme. Notably, product dissociation can only occur if either the polypeptide or the sugar moiety passes through the opening provided by EL5. Our observation that glycosylated GFP can be released even in the presence of a disulfide cross-link at Pos3 suggests that the N-glycan moiety of glyco-products is pulled through the provided opening, because GFP would be unable to fit.
X-ray structures of the archaeal Stt3 homolog AglB from Archeoglobus fulgidus in two crystal forms have recently been reported (31). Neither AglB structure contained bound acceptor peptide or LLO substrate. The structures revealed a transmembrane fold that is similar to that of our C. lari PglB structure. The entire loop EL5 is disordered in the AglB structure with bound zinc and sulfate ions, in line with our finding that EL5 is disengaged from the enzyme core in the apo-state of PglB. In the other AglB structure, the entire loop EL5 is ordered, including the N-terminal half, which is located at a distance of ∼15–20 Å from the active site of the enzyme, suggesting that a considerable conformational change would be required for interaction with the reducing end sugar. In addition to large scale conformational rearrangements of EL5, conformational fluctuations within the extracellular domain of the archaeal Stt3 homolog from Pyrococcus furiosus (AglB) have been reported to be important for function (32), suggesting that the extracellular/periplasmic domain probably adjusts its conformation upon substrate binding and EL5 engagement.
In contrast to the C-terminal half, any role of the N-terminal half of EL5 in catalysis was completely unknown, and the corresponding segment was disordered in the x-ray structure of sequon-bound C. lari PglB (12). Through a combination of systematic mutagenesis and in vivo activity analysis, we now identified a previously unrecognized motif that proved essential for catalytic activity. Unexpectedly, mutations in the newly found Tyr-plug had stronger effects on PglB activity than many of the previously described mutations at the active site, even of catalytically essential residues (13, 14). However, replacing the Tyr-plug sequence did not affect substrate peptide binding. This suggested a role in the interaction with the LLO molecule, the glycan donor of the reaction. Within the Tyr-plug, Tyr293 has a critical effect, but it was dependent on the immediate sequence context. The observation that Tyr293 could be replaced by other aromatic residues supported the notion that residue 293 might be involved in an interaction with a sugar moiety of the LLO substrate, because aromatic residues are frequently involved in π-stacking with carbohydrates (28–30). However, our experiments excluded a direct interaction of Tyr293 with one of the distal sugar residues of the C. jejuni LLO, which agrees both with the observation that C. jejuni PglB can transfer diverse carbohydrate structures in vivo (33, 34) and with in vitro studies of eukaryotic OST that identified the dolichol-linked monosaccharide Dol-PP-GlcNAc as the minimal glycosylation unit (35).
On the one hand, the exact position of the Tyr-plug within EL5 seemed to be irrelevant, because insertions, replacements, and even deletions in its vicinity were tolerated. On the other hand, the essential function of Tyr293 could not be taken over by any of the other aromatic residues within the Tyr-plug (Tyr288, Phe292, or Phe295). The inescapable conclusion was that the Tyr-plug adopts a defined three-dimensional conformation, at least during one of the rate-limiting steps in catalysis. We could indeed rescue (albeit to a limited extent) the effect of a Tyr-plug replacement in PglB by adding a synthetic peptide containing the Tyr-plug motif. However, this peptide did not adopt a defined structure on its own, as we could show by solution NMR analysis. This suggests that the Tyr-plug only adopts a defined conformation in the context of the enzyme and in the presence of bound LLO (Fig. 7A).
FIGURE 7.

Proposed function of the Tyr-plug in PglB. A, surface representation of PglB colored as in Fig. 3A. Binding of LLO donor substrate results in an interaction between the reducing end sugar and the Tyr-plug (thick red line). Red hexagon, diNAcBac; yellow square, N-acetylgalactosamine; blue circle, glucose; circled P, phosphate group; gray line, undecaprenyl (Und). B, mechanistic hypothesis of Tyr-plug function. Yellow dashed line indicates the sequon backbone. Hydrogen bonds between PglB residues Asp56 and Glu319 and the acceptor Asn contribute to the activation of the carboxamide group for the nucleophilic attack on the anomeric carbon (orange circle). The C-2 acetamido group of the reducing end sugar can be accommodated in a cavity of conserved residues in the active site of PglB (blue dashed line). The Tyr-plug is proposed to interact with the reducing end sugar and with the PglB surface. This is expected to restrict the rotation of the O-glycosidic bond and the first O–P bond (red arrows), increasing catalytic efficiency. R, oligosaccharyl; Und, undecaprenyl.
What might the essential catalytic role of the Tyr-plug be? Fig. 7 illustrates our mechanistic hypothesis. Once LLO substrate is bound to PglB, its pyrophosphate moiety is bound by interactions with the conserved residue Arg375 and the divalent metal cation, both of which are essential for catalysis (Fig. 7B) (12, 13). Although the pyrophosphate is now fixed, the reducing end sugar is not, because a rotation around the O-glycosidic bond and around the O–P bond of the pyrophosphate moiety would be allowed (red arrows in Fig. 7B), both of which would prevent proper placement of the glycan for a nucleophilic attack by the activated carboxamide group of the Asn side chain of the sequon. We therefore propose that the function of the Tyr-plug is to engage in contacts both with the active site of PglB and with the reducing end sugar of LLO, forcing the reducing end sugar into a reactive conformation and thus increasing the rate of the transfer reaction. The aromatic side chain of Tyr293 is essential and might directly stack against the reducing end sugar diNAcBac (Fig. 7B). In addition, Tyr293 might interact with the C-4 acetamido group of diNAcBac, because methyl groups of acetamido moieties are often recognized by aromatic residues in different lectins (30, 36–39). A tentative modeling of the LLO with the diNAcBac properly aligned for a nucleophilic attack places the C2-acetamido group into a PglB cavity that contains residues conserved in all Stt3 homologs. This cavity might also be involved in restricting sugar rotation, in particular rotation around the anomeric bond (Fig. 7B). The C2-acetamido group of the reducing end sugar is indeed essential both for bacterial and eukaryotic LLO substrates (34, 40).
Does the Tyr-plug also exist in archaeal and eukaryotic Stt3? Although the chemical reaction and the basic mechanism are conserved among OST homologs, it is currently unclear if eukaryotic and archaeal enzymes use a similar Tyr-plug to restrict the motion of the reducing end sugar during the catalytic step. Sequence alignments would support this hypothesis, because Stt3 homologs also contain aromatic residues (including Tyr and Phe) in EL5 at a similar distance from the catalytically essential Glu319 residue, which is also conserved. However, it will be very challenging to study these effects in vivo or in vitro. Most eukaryotic OSTs contain seven additional subunits (41) that might contribute to binding of the LLO and the correct positioning of the reducing end sugar moiety. Additionally, the glycan structure transferred by eukaryotic OST (GlcNAc2Man9Glc3) is different and much bigger than its bacterial counterpart, which could imply that steric effects restricting sugar motions play an additional role. Furthermore, the reducing end sugar is a GlcNAc in eukaryotic LLOs and even a hexose in some archaeal LLOs (4, 31, 42, 43), suggesting mechanistic diversity.
In summary, our study shows that EL5 of PglB has two distinct functions. Whereas the N-terminal half of EL5 is supposed to form a critical interaction with the LLO during catalysis, the C-terminal part contributes to peptide binding and contributes the conserved active site residue Glu319 that binds both the divalent metal and the acceptor Asn of the sequon. Intriguingly, the two functions of EL5 require no covalent link between the N- and C-terminal halves of the loop, as shown by our ability to specifically cleave EL5 with only minor effects on activity. Although the importance of the Tyr-plug for catalysis is now established, insight into the mechanistic details will require structural evidence, in particular a LLO-bound co-crystal structure of OST.
Acknowledgments
We thank E. Weber-Ban and R. Glockshuber for access to the fluorometer and M. Aebi for helpful discussions. We are grateful to the D-BIOL NMR Platform for access to NMR spectrometers. We thank A. Geerlof for providing the protocol for 3C protease expression and purification and A. Zeltina for providing HmuT-Q147C cells.
This work was supported by a grant from NCCR Structural Biology Zurich (to K. P. L.) and Swiss National Science Foundation Grants SNF 200020-125020 (to J. L. R.), SNF 200020-124663 (to R. Z.), and SNF 31003A-131075/1 and SNF 31003A-146191/1 (to K. P. L.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S6.
- LLO
- lipid-linked oligosaccharide
- EL1 and EL5
- external loop 1 and 5, respectively
- Ni-NTA
- nickel-nitrilotriacetic acid
- OST
- oligosaccharyltransferase
- TM
- transmembrane
- HSQC
- heteronuclear single-quantum correlation
- HMQC
- heteronuclear multiple-quantum correlation.
REFERENCES
- 1. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 2. Kornfeld R., Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 3. Varki A. (1993) Biological roles of oligosaccharides. All of the theories are correct. Glycobiology 3, 97–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eichler J. (2013) Extreme sweetness. Protein glycosylation in archaea. Nat. Rev. Microbiol. 11, 151–156 [DOI] [PubMed] [Google Scholar]
- 5. Nothaft H., Szymanski C. M. (2010) Protein glycosylation in bacteria. Sweeter than ever. Nat. Rev. Microbiol. 8, 765–778 [DOI] [PubMed] [Google Scholar]
- 6. Schwarz F., Aebi M. (2011) Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582 [DOI] [PubMed] [Google Scholar]
- 7. Nilsson I., Kelleher D. J., Miao Y., Shao Y., Kreibich G., Gilmore R., von Heijne G., Johnson A. E. (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J. Cell Biol. 161, 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan Q., Lennarz W. J. (2002) Studies on the function of oligosaccharyl transferase subunits. Stt3p is directly involved in the glycosylation process. J. Biol. Chem. 277, 47692–47700 [DOI] [PubMed] [Google Scholar]
- 9. Hese K., Otto C., Routier F. H., Lehle L. (2009) The yeast oligosaccharyltransferase complex can be replaced by STT3 from Leishmania major. Glycobiology 19, 160–171 [DOI] [PubMed] [Google Scholar]
- 10. Nasab F. P., Schulz B. L., Gamarro F., Parodi A. J., Aebi M. (2008) All in one. Leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wacker M., Linton D., Hitchen P. G., Nita-Lazar M., Haslam S. M., North S. J., Panico M., Morris H. R., Dell A., Wren B. W., Aebi M. (2002) N-Linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–1793 [DOI] [PubMed] [Google Scholar]
- 12. Lizak C., Gerber S., Numao S., Aebi M., Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 13. Gerber S., Lizak C., Michaud G., Bucher M., Darbre T., Aebi M., Reymond J. L., Locher K. P. (2013) Mechanism of bacterial oligosaccharyltransferase. In vitro quantification of sequon binding and catalysis. J. Biol. Chem. 288, 8849–8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lizak C., Gerber S., Michaud G., Schubert M., Fan Y. Y., Bucher M., Darbre T., Aebi M., Reymond J. L., Locher K. P. (2013) Unexpected reactivity and mechanism of carboxamide activation in bacterial N-linked protein glycosylation. Nat. Commun. 4, 2627. [DOI] [PubMed] [Google Scholar]
- 15. Lefebre M. D., Valvano M. A. (2002) Construction and evaluation of plasmid vectors optimized for constitutive and regulated gene expression in Burkholderia cepacia complex isolates. Appl. Environ. Microbiol. 68, 5956–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Linton D., Dorrell N., Hitchen P. G., Amber S., Karlyshev A. V., Morris H. R., Dell A., Valvano M. A., Aebi M., Wren B. W. (2005) Functional analysis of the Campylobacter jejuni N-linked protein glycosylation pathway. Mol. Microbiol. 55, 1695–1703 [DOI] [PubMed] [Google Scholar]
- 17. Kowarik M., Numao S., Feldman M. F., Schulz B. L., Callewaert N., Kiermaier E., Catrein I., Aebi M. (2006) N-Linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science 314, 1148–1150 [DOI] [PubMed] [Google Scholar]
- 18. Baar C., Eppinger M., Raddatz G., Simon J., Lanz C., Klimmek O., Nandakumar R., Gross R., Rosinus A., Keller H., Jagtap P., Linke B., Meyer F., Lederer H., Schuster S. C. (2003) Complete genome sequence and analysis of Wolinella succinogenes. Proc. Natl. Acad. Sci. U.S.A. 100, 11690–11695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lizak C., Fan Y. Y., Weber T. C., Aebi M. (2011) N-Linked glycosylation of antibody fragments in Escherichia coli. Bioconjug. Chem. 22, 488–496 [DOI] [PubMed] [Google Scholar]
- 20. Mattle D., Zeltina A., Woo J. S., Goetz B. A., Locher K. P. (2010) Two stacked heme molecules in the binding pocket of the periplasmic heme-binding protein HmuT from Yersinia pestis. J. Mol. Biol. 404, 220–231 [DOI] [PubMed] [Google Scholar]
- 21. Woo J. S., Zeltina A., Goetz B. A., Locher K. P. (2012) X-ray structure of the Yersinia pestis heme transporter HmuUV. Nat. Struct. Mol. Biol. 19, 1310–1315 [DOI] [PubMed] [Google Scholar]
- 22. Chen F., Gerber S., Heuser K., Korkhov V. M., Lizak C., Mireku S., Locher K. P., Zenobi R. (2013) High-mass matrix-assisted laser desorption ionization-mass spectrometry of integral membrane proteins and their complexes. Anal. Chem. 85, 3483–3488 [DOI] [PubMed] [Google Scholar]
- 23. Markley J. L., Bax A., Arata Y., Hilbers C. W., Kaptein R., Sykes B. D., Wright P. E., Wuthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids (IUPAC Recommendations 1998). Pure Appl. Chem. 70, 117–142 [DOI] [PubMed] [Google Scholar]
- 24. Losfeld M. E., Soncin F., Ng B. G., Singec I., Freeze H. H. (2012) A sensitive green fluorescent protein biomarker of N-glycosylation site occupancy. FASEB J. 26, 4210–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dell A., Galadari A., Sastre F., Hitchen P. (2010) Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010, 148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nothaft H., Szymanski C. M. (2013) Bacterial protein N-glycosylation. New perspectives and applications. J. Biol. Chem. 288, 6912–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jervis A. J., Butler J. A., Lawson A. J., Langdon R., Wren B. W., Linton D. (2012) Characterization of the structurally diverse N-linked glycans of Campylobacter species. J Bacteriol. 194, 2355–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morales J. C., Reina J. J., Díaz I., Aviñó A., Nieto P. M., Eritja R. (2008) Experimental measurement of carbohydrate-aromatic stacking in water by using a dangling-ended DNA model system. Chemistry 14, 7828–7835 [DOI] [PubMed] [Google Scholar]
- 29. Oldham M. L., Khare D., Quiocho F. A., Davidson A. L., Chen J. (2007) Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450, 515–521 [DOI] [PubMed] [Google Scholar]
- 30. Weis W. I., Drickamer K. (1996) Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 65, 441–473 [DOI] [PubMed] [Google Scholar]
- 31. Matsumoto S., Shimada A., Nyirenda J., Igura M., Kawano Y., Kohda D. (2013) Crystal structures of an archaeal oligosaccharyltransferase provide insights into the catalytic cycle of N-linked protein glycosylation. Proc. Natl. Acad. Sci. U.S.A. 110, 17868–17873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nyirenda J., Matsumoto S., Saitoh T., Maita N., Noda N. N., Inagaki F., Kohda D. (2013) Crystallographic and NMR evidence for flexibility in oligosaccharyltransferases and its catalytic significance. Structure 21, 32–41 [DOI] [PubMed] [Google Scholar]
- 33. Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., Aebi M. (2005) Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wacker M., Feldman M. F., Callewaert N., Kowarik M., Clarke B. R., Pohl N. L., Hernandez M., Vines E. D., Valvano M. A., Whitfield C., Aebi M. (2006) Substrate specificity of bacterial oligosaccharyltransferase suggests a common transfer mechanism for the bacterial and eukaryotic systems. Proc. Natl. Acad. Sci. U.S.A. 103, 7088–7093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tai V. W., Imperiali B. (2001) Substrate specificity of the glycosyl donor for oligosaccharyl transferase. J. Org. Chem. 66, 6217–6228 [DOI] [PubMed] [Google Scholar]
- 36. Weis W., Brown J. H., Cusack S., Paulson J. C., Skehel J. J., Wiley D. C. (1988) Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333, 426–431 [DOI] [PubMed] [Google Scholar]
- 37. Wright C. S. (1984) Structural comparison of the two distinct sugar binding sites in wheat germ agglutinin isolectin II. J. Mol. Biol. 178, 91–104 [DOI] [PubMed] [Google Scholar]
- 38. Wright C. S. (1990) 2.2 Å resolution structure analysis of two refined N-acetylneuraminyl-lactose–wheat germ agglutinin isolectin complexes. J. Mol. Biol. 215, 635–651 [DOI] [PubMed] [Google Scholar]
- 39. Schubert M., Bleuler-Martinez S., Butschi A., Wälti M. A., Egloff P., Stutz K., Yan S., Collot M., Mallet J. M., Wilson I. B., Hengartner M. O., Aebi M., Allain F. H., Künzler M. (2012) Plasticity of the β-trefoil protein fold in the recognition and control of invertebrate predators and parasites by a fungal defence system. PLoS Pathog. 8, e1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tai V. W., O'Reilly M. K., Imperiali B. (2001) Substrate specificity of N-Acetylglucosaminyl(diphosphodolichol) N-acetylglucosaminyl transferase, a key enzyme in the dolichol pathway. Bioorg. Med. Chem. 9, 1133–1140 [DOI] [PubMed] [Google Scholar]
- 41. Kelleher D. J., Gilmore R. (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R [DOI] [PubMed] [Google Scholar]
- 42. Calo D., Kaminski L., Eichler J. (2010) Protein glycosylation in Archaea. Sweet and extreme. Glycobiology 20, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 43. Abu-Qarn M., Yurist-Doutsch S., Giordano A., Trauner A., Morris H. R., Hitchen P., Medalia O., Dell A., Eichler J. (2007) Haloferax volcanii AglB and AglD are involved in N-glycosylation of the S-layer glycoprotein and proper assembly of the surface layer. J. Mol. Biol. 374, 1224–1236 [DOI] [PubMed] [Google Scholar]