

Background: IL-4 protects vascular endothelial cells (ECs) from complement-mediated junctional injury and cytotoxicity.
Results: IL-4 induces up-regulated expression of junctional claudin-5 through STAT6 and FoxO1. Claudin-5 up-regulation contributes to, but is not sufficient, for protection.
Conclusion: IL-4-induced claudin-5 is an important component in protection of ECs from complement.
Significance: Regulation of claudin-5 expression may play a role controlling EC inflammatory injury.
Keywords: Cell Junctions, Complement, Endothelium, Foxo, Vascular Biology, IL-4, Claudin-5
Abstract
Injury to endothelial cells (ECs) often results in cell retraction and gap formation. When caused by antigen aggregation or complement, this injury can be prevented by pretreatment of the ECs with IL-4, suggesting that IL-4 modifies the intercellular junction. Therefore, we investigated the effects of IL-4 on expression of intercellular junction proteins and whether such effects are required for IL-4-induced resistance of ECs against complement-mediated injury. We found that IL-4 induces up-regulation of the junction protein claudin-5 in porcine ECs through activation of Jak/STAT6 and phosphorylation and translocation of FoxO1 from the nucleus to the cytoplasm. Increased claudin-5 expression resulted in increased transmembrane electrical resistance of the endothelial monolayer and participated in IL-4-induced protection of the ECs from complement injury. Down-regulation of FoxO1 using siRNA by itself caused up-regulation of claudin-5 expression and partial protection from cytotoxicity. This protection was enhanced by stimulation with IL-4. We previously reported that increased phospholipid synthesis and mitochondrial protection were required for IL-4-induced resistance of ECs against complement injury and now we demonstrate a contribution of claudin-5 expression in IL-4-induced protection.
Introduction
The vascular endothelium has critical functions that are regulated by multiple mechanisms. When exposed to injurious agents such as inflammatory mediators endothelial activation plays a central role in the development of atherosclerosis, ischemia-reperfusion injury, and the vasculopathy of graft rejection. In this regard various cytokines are known to act on endothelial cells (ECs),2 with effects that can be protective or deleterious. We have previously established that IL-4 and IL-13 are able to induce protection in porcine ECs against apoptosis caused by TNF-α (1, 2). IL-4 induces activation of Jak3/STAT6 and phosphorylation of Bad, ultimately resulting in effective protection of the ECs from apoptosis (2). A form of cell injury distinct from apoptosis is the cytotoxicity caused by the membrane attack complex of complement (3, 4). Our previous studies have established that IL-4 and IL-13 are also able to induce resistance of ECs against cytotoxicity mediated by complement (1, 5, 6). This resistance is not due to reduced binding of complement to the cell membrane but is intrinsic to the cells and requires activation of Akt/SREBP-1 and phospholipid synthesis and is associated with mitochondrial protection (6, 7).
We have also previously shown that pretreatment with IL-4 prevents the actin rearrangement, cell retraction, and formation of intercellular gaps in ECs that are caused by sublytic complement (5) or antigen aggregation.3 These findings suggested to us that, for full effectiveness, the effects of IL-4 that result in protection from complement may also include proteins that are part of the intercellular junction or the subjacent cytoskeleton. Because the effects of IL-4 on the EC intercellular junction are unknown, in our present work, we first asked whether pretreatment of ECs with IL-4 would cause changes in the expression of the main proteins that form the intercellular junction and the cytoskeletal protein β-actin. There are two groups of intercellular junction proteins in ECs: the tight junction proteins claudin-5, zona occludens protein 1 (ZO-1), and occludin, with claudin-5 being the most important, and the adherens junction proteins vascular endothelial (VE)-cadherin and associated catenins (8, 9). Other adhesion molecules, similar to PECAM-1, are part of the intercellular junction but also participate in other cell-to-cell contacts (10). These proteins provide junctional adhesion among cells of the monolayer, control paracellular permeability, and are active players in cell signaling.
Here, we report that IL-4 induces up-regulation of claudin-5 expression in ECs through activation of Jak/STAT6 and phosphorylation and translocation of FoxO1 from the nucleus to the cytoplasm. We also report that these changes are part of the mechanism by which IL-4 induces resistance of the ECs against complement-mediated injury.
EXPERIMENTAL PROCEDURES
Treatment with Cytokines and Inhibitors
ECs were explanted from pig aortae and cultured and identified as described previously (11). Experiments were performed with EC monolayers from five different preparations in passages 3 to 7, 2, to 3 days post-confluence in gelatin-coated plates (Corning). All incubations were carried out at 37 °C in 5% CO2, 95% air. ECs were incubated with pig rIL-4 (R & D Systems) at 10 ng/ml or 20 ng/ml in DMEM containing 1% FBS (1). For experiments with inhibitors, ECs were made quiescent by incubation with 1% FBS-DMEM overnight. ECs were incubated with Jak inhibitor 1 (EMD Bioscience) or PF-956980 (Sigma) for 1 h before adding IL-4 and continuing the incubation as indicated. After washing, the ECs were tested for viability (12) and used for experiments.
siRNA Transfection of ECs
21-nucleotide siRNAs to target the mRNA of porcine genes for claudin-5 (GenBankTM accession no. FJ873112), VE-cadherin (GenBankTM accession no. AB046120), FoxO1 (GenBankTM accession no. NM214014), and β-actin (GenBankTM accession no. DQ845171) were designed and synthesized by Applied Biosystems. One day before transfection, the ECs were plated at 12,500 cells/cm2 in 10% FBS/DMEM. When the cells were 30–50% confluent, the medium was replaced with antibiotic-free 10% FBS/DMEM and the siRNAs were transfected into the cells with Lipofectamine RNAiMax reagent (Invitrogen) following the protocol provided by the manufacturer. After 4 h, the transfection mixtures were replaced with 10% FBS/DMEM containing antibiotics and incubated for 3 days to confluence before incubation with IL-4; this procedure reduces possible EC activation by siRNA. Cell viability was >90% as assessed using the vital dye neutral red (Sigma). Knockdown for the desired protein was evaluated by immunoblotting.
Cytotoxicity Assay
Pretreated ECs in 48-well plates were incubated for 2 h with 150 μl of a human serum pool at 15 to 30% in RPMI 1640 as a source of anti-pig Abs and complement (12). In each experiment, a corresponding concentration of the serum pool that was incubated previously at 60 °C for 30 min to inactivate complement was used as control. After washing, EC viability was measured using neutral red and the percentage of specific killing was calculated as described previously (1, 12). Values are given as mean ± S.E. of at least three independent experiments performed in triplicate. We previously established the validity of this assay to measure necrotic cell death caused by human complement at the serum concentrations and incubation times used in our current study (1).
Western Blotting
For immunoblot analysis of junction proteins in total cell extracts, the pretreated ECs in six-well plates were lysed in 1× NuPAGE sample buffer (Invitrogen) containing protease inhibitor mixture (Complete, Roche Applied Science). For immunoblot analysis of phosphoprotein, ECs in six-well plates were made quiescent by incubation with 1% FBS-DMEM overnight and treated with IL-4. The cells were then lysed in radioimmune precipitation assay buffer containing 50 mm TBS, pH 6.8, 1% Triton X-100, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mm sodium orthovanadate, and protease inhibitor mixture. For nuclear and cytoplasmic fractions, ECs in 15-cm dishes were also made quiescent and treated with IL-4. The cells were then processed using the NE-PER kit from Pierce Biotechnology according to the manufacturer's instructions. Cell extracts (10–20 μg protein) were electrophoresed under reducing conditions in 4–12% NuPAGE gels (Invitrogen) and transferred onto nitrocellulose membranes (Millipore). After blocking, the membranes were incubated with a primary Ab, and following incubation with horseradish peroxidase-conjugated secondary Ab, bands were visualized by chemiluminescence using Lumi-glo (Cell Signaling). Abs cross-reacting with the following pig proteins were used: STAT6 (Cell Signaling); ZO-1 (Invitrogen); β-catenin (Novus Biologicals); and β-actin (Abcam). Abs against claudin-5 (H-52), VE-cadherin (C-19), PECAM-1 (M-20), FoxO1 (FKHR, H-130), phospho-Thr-24-FoxO1, phospho-Ser-319-FoxO1, and horseradish peroxidase-conjugated donkey secondary Abs were from Santa Cruz Biotechnology. Anti-pig GAPDH and anti-TATA-binding protein Abs were from Pierce. Blots were stripped with ReBlot PLUS (Millipore) and retested as required. The amount of protein signal was determined from densitometry using Molecular Analyst (Bio-Rad).
Immunocytochemistry
Pretreated cells that were grown in chamber slides (Nunc) were fixed in 4% paraformaldehyde in PBS for 10 min, washed three times with PBS, then permeabilized with 0.1% Triton X-100 in PBS for 10 min, and washed again with PBS. For single label experiments, the ECs were then blocked with PBS containing 3% bovine serum albumin. They were subsequently incubated for 2 h at room temperature with primary Abs, washed, and incubated for 1 h at room temperature with Alexa Fluor 488-conjugated donkey secondary Abs (Jackson ImmunoResearch Laboratories). For actin visualization, ECs in chamber slides were fixed, permeabilized, and labeled with phalloidin-tetramethylrhodamine isothiocyanate (Sigma), as described previously (13). Samples were examined with an Olympus IX70 fluorescence imaging system. For FoxO1 translocation experiments, EC monolayers in chamber slides were made quiescent, treated with IL-4, and fixed and permeabilized as above. After blocking with PBS containing 3% donkey serum, the cells were incubated with a mixture of a rabbit mAb to FoxO1 (Cell Signaling) and goat Ab to VE-cadherin for 2 h at room temperature. They were washed with PBS and incubated for 1 h at room temperature with anti-goat IgG-Alexa Fluor 488 (green) and anti-rabbit IgG-Alexa Fluor 594 (red) (Jackson ImmunoResearch Laboratories). The nuclei were stained with DAPI, and slides were mounted with Vectashield (Vector Laboratories), and the EC monolayers were examined using a laser-scanning confocal microscope (Olympus FluoView 1000 BX2). Images were processed with Adobe Photoshop. The mean fluorescence intensity of FoxO1 in the nucleus, and cytoplasm was determined in unenhanced images using the histogram in Photoshop. The nuclei were outlined with the lasso tool, and the mean red channel intensity of the nuclei was recorded. The nuclei were then deleted, and the mean red channel intensity of the cytoplasm was recorded. The values shown are the averages of the mean fluorescence intensity in four separate images from two experiments containing 15–22 cells per image.
Membrane Barrier Function
We assessed EC membrane barrier function by measurement of transendothelial electrical resistance (TER) (14). ECs were cultured on gold electrodes using ECIS array slides (type 8W10E+, Applied Biophysics) coated with gelatin. When the monolayers were at confluence, 1% FBS-DMEM was added, and after incubation for 2 h, the arrays were placed in the ECIS instrument. (ZΘ-ECIS, Applied Biophysics) and, after an additional 15 min, IL-4 or medium was added, and real-time changes of resistance were monitored at 4 kHz. For experiments using siRNA, the arrays were placed in the instrument when the cells were at 30–50% confluence, and the cells were transfected with siRNA, as above.
Statistical Analysis
p values were obtained using a two-sample t test assuming equal variance. A p value = or < 0.05 was considered statistically significant.
RESULTS
IL-4 Induces Up-regulation of Claudin-5 Expression
We have previously reported that formation of intercellular gaps in EC monolayers caused by sublytic amounts of human complement could be fully prevented by pretreatment of the ECs with IL-4 (5). Given the location of this injury caused by complement and its abrogation by IL-4, we asked whether protection could be due to IL-4 causing changes in expression of junction proteins or β-actin. Using immunofluorescence microscopy we found that control cells exposed to medium alone expressed claudin-5 in very small amounts, while ECs incubated with IL-4 for 40 h exhibited strong claudin-5 expression at the intercellular junction (Fig. 1A). In contrast, VE-cadherin, PECAM-1 and β-catenin were strongly expressed in cells treated or not with IL-4, with minor or no changes caused by IL-4. Using Western blotting, no increment in expression of any of the examined proteins was found after 1 h of treatment with IL-4. We found that the increment in claudin-5 expression was demonstrable at 6 h and was very strong at 24 h (Fig. 1, B and C). In contrast, no increment in expression of VE-cadherin, PECAM-1, β-catenin, ZO-1, and β-actin was found after 6 or 24 h of treatment with IL-4.
FIGURE 1.

IL-4 induces up-regulation of claudin-5 expression. A, ECs were incubated with medium or 10 ng/ml IL-4 for 40 h, processed for detection of junction proteins claudin-5, VE-catherin, PECAM-1, and β-catenin, and examined by fluorescence microscopy. Results are representative of three independent experiments. Original magnification, ×40. Scale bars, 20 μm. B and C, time-dependent induction of claudin-5 (CL) in comparison with VE-catherin (VE), PECAM-1 (PE), β-catenin (Cat), ZO-1, and β-actin (Act). ECs were incubated with 10 ng/ml IL-4 for 1, 6, or 24 h. Cell extracts were assessed for protein expression using immunoblotting. Bars represent ratios of band density of indicated protein to that of GAPDH loading control relative to time 0 and are means ± S.E. of three experiments. IL-4-treated versus medium alone (*, p = 0.05 and **, p < 0.001). Immunoblots are representative of three independent experiments.
Induction of Protection from Complement Cytotoxicity by IL-4 in ECs Is in Part Dependent on Up-regulated Expression of Claudin-5
We next asked whether up-regulation of claudin-5 expression was required for the protection induced by IL-4 against complement-mediated cytotoxicity, the ultimate injury caused by the membrane attack complex of complement on ECs. As shown in Fig. 2A, base line expression of claudin-5 with no IL-4 treatment was very low in control ECs and even lower in ECs that received siRNA for claudin-5 and no IL-4, and both had similar levels of cytotoxicity, suggesting that base-line expression of claudin-5 does not have a role in protection from complement killing. These ECs with silenced claudin-5 protein expression were partially blocked from developing protection from complement killing after IL-4 treatment, in contrast to ECs that were pretreated with medium alone which showed greater protection. These results were obtained using three different sequences for claudin-5. The mean % reversal of protection were as follows: for all experiments in Fig. 2A, 60.0% (nine experiments); for siRNA-1, 71.3% (four experiments); for siRNA-2, 67.9% (three experiments); and for siRNA-3, 40.3% (two experiments). These findings suggest that a component of the mechanism by which IL-4 induces protection from complement consists of the IL-4-mediated up-regulation of claudin-5 expression. However, down-regulation of β-actin or VE-cadherin with siRNA (Fig. 2, A and B) did not decrease claudin-5 expression and did not interfere with IL-4-induced protection. Down-regulation of β-actin or VE-cadherin with siRNA without IL-4 treatment caused a reduction in cytotoxicity, and the IL-4-induced protection was proportionally more complete than in medium controls (Fig. 2B). Although we have not explored this further for VE-cadherin, all additional experiments with β-actin caused no changes in degree of protection by IL-4 compared with controls, as shown in Figs. 2A, 3D, and 6C. These experiments suggest that β-actin and VE-cadherin may contribute to protection when down-regulated and that they may not be involved in the additional protection induced by IL-4.
FIGURE 2.

Induction of protection from complement by IL-4 requires up-regulated expression of claudin-5. ECs were transfected with 2.5 nm siRNA targeting claudin-5 (three different sequences) (A), VE-cadherin (B), or β-actin (A and B), and then incubated with 10 ng/ml IL-4 for 48 h. The ECs were used to prepare cell extracts for immunoblotting or, after washing, tested for sensitivity to complement by incubation for 2 h with 20–30% human serum. Bar graphs are means ± S.E. of results from nine experiments in A and four experiments in B. *, p < 0.001, degree of cytotoxicity in IL-4-treated samples versus medium alone; **, p < 0.05, IL-4 control versus IL-4 siRNA-treated; #, p < 0.001, medium alone versus medium siRNA. The immunoblots are representative of nine independent experiments (A) and four independent experiments (B).
FIGURE 3.

IL-4 induces up-regulation of claudin-5 and protection from complement through Jak/STAT6 activation. A, Jak inhibitor 1 (Jak inh 1) and the specific Jak3 inhibitor PF-956980 (Jak3 inh) reduce claudin-5 protein expression induced by IL-4. ECs were incubated with the inhibitor for 1 h before adding 10 ng/ml IL-4 for 24 h and analyzed by immunoblotting (representative of three independent experiments). B, knockdown of STAT6 protein expression abrogates IL-4-induced up-regulation of claudin-5. ECs were transfected with siRNA targeting STAT6 and then incubated with 10 ng/ml IL-4 for 24 h and analyzed by immunoblotting (representative of five independent experiments). C, abrogation of IL-4-induced protection from complement using Jak inhibitor 1 or Jak3 inhibitor. ECs were incubated for 1 h with inhibitor at 1 μm before adding 10 ng/ml IL-4 for 48 h and then were tested for sensitivity to complement. D, abrogation of IL-4-induced protection from complement using siRNA for STAT6. ECs were transfected with 2.5 nm siRNA targeting STAT6 or β-actin as control and then incubated with 10 ng/ml IL-4 for 48 h. The ECs were then tested for sensitivity to complement. Bar graphs are means ± S.E. of results from four independent experiments. *, p < 0.001, degree of cytotoxicity in IL-4-treated samples versus medium alone.
FIGURE 6.
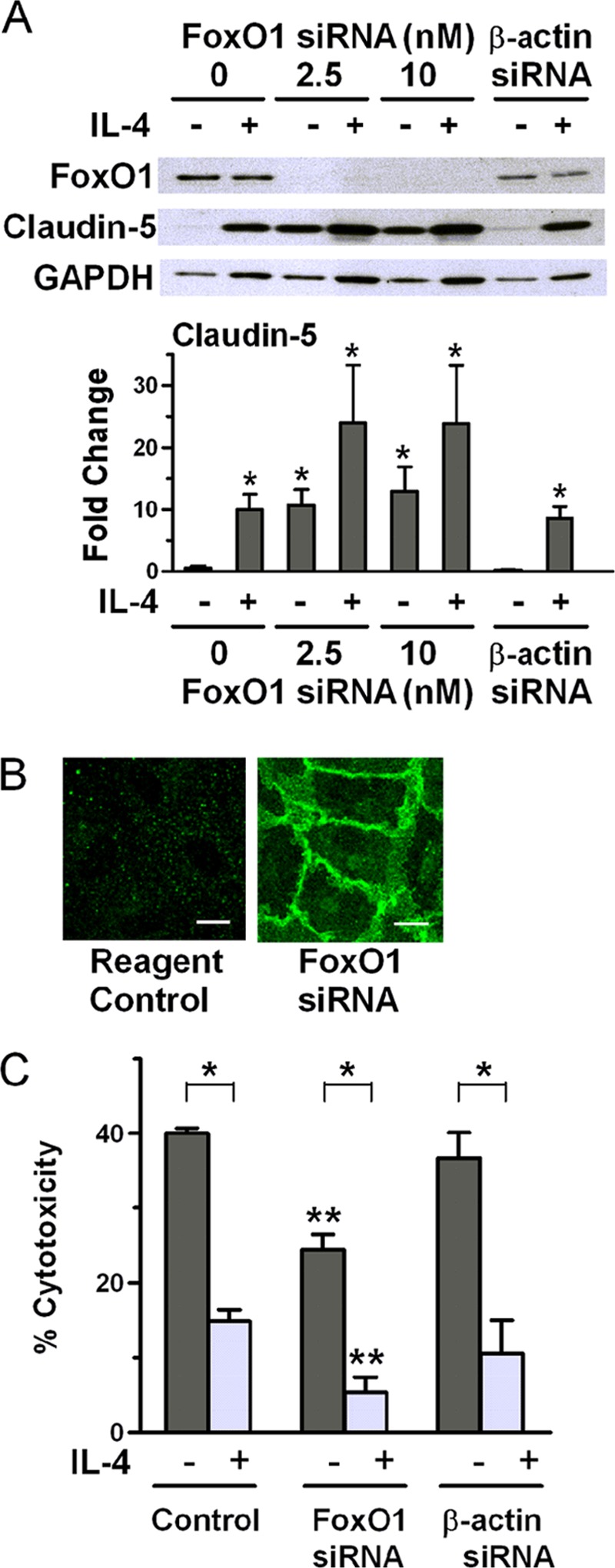
Down-regulation of FoxO1 without stimulation with IL-4 up-regulates claudin-5 expression and causes protection from complement. A, ECs were transfected with siRNA targeting FoxO1 or β-actin as control and then incubated with medium or 10 ng/ml IL-4 for 24 h and analyzed by immunoblotting (representative of three independent experiments). Bars are ratios of band density of claudin-5 to that of GAPDH loading control and are means ± S.E. of three experiments. *, p < 0.02, ECs without any treatment versus all other conditions. B, ECs were treated with Lipofectamine reagent in medium or transfected with 2.5 nm siRNA targeting FoxO1, and at confluence, the cells were examined by immunofluorescence for claudin-5. Scale bars, 10 μm. C, ECs were transfected with 10 nm siRNA targeting FoxO1 (two different sequences) or β-actin as control and then incubated with medium or 10 ng/ml IL-4 for 48 h. The ECs were then tested for sensitivity to complement. Bar graphs are means ± S.E. of results from seven independent experiments. *, p < 0.001, medium versus IL-4-treated; **, p < 0.001, medium control versus medium siRNA and IL-4 control versus IL-4 siRNA.
IL-4 Induces Up-regulation of Claudin-5 and Protection from Complement Cytotoxicity through Jak/STAT6 Activation
Because Jak/STAT6 is the classical signaling mechanisms triggered by IL-4 stimulation (15, 16), we asked whether this pathway was required for IL-4-induced up-regulation of claudin-5 in porcine ECs. We used “Jak inhibitor 1′” to inhibit Jak1, Jak2, Jak3 (17), and the Jak3-specific inhibitor PF-956980 (18). As shown in Fig. 3A, both inhibitors reduced the IL-4-induced up-regulation of claudin-5 to a similar extent. Reduction of claudin-5 expression by the specific inhibitor of Jak3 suggests that Jak3 is involved in STAT6 activation by IL-4 in ECs, but the participation of Jak1 and Jak2 cannot be excluded. Neither Jak inhibitor had any effect on the expression of VE-cadherin. We investigated the role of STAT6 in IL-4-induced up-regulation of claudin-5 using siRNA to down-regulate expression of STAT6. ECs treated with 10 nm STAT6 siRNA expressed no detectable STAT6 and exhibited no claudin-5 up-regulation upon incubation with IL-4, whereas expression of VE-cadherin was unaffected (Fig. 3B). These results demonstrate that IL-4 up-regulates claudin-5 expression in ECs through activation of the Jak/STAT6 pathway. We then showed that Jak/STAT6 activation was required for IL-4 induction of protection from complement because both Jak inhibition and STAT6 down-regulation interfered with induction of protection (Fig. 3, C and D). In contrast, siRNA for β-actin that was used as control did not interfere with protection. We also found that inhibitors of PI3K or Akt had no effect on the up-regulation of claudin-5 by IL-4, using Western blot analysis (results not shown).
IL-4 Induces Phosphorylation of FoxO1 and Changes Its Cellular Location
In a mouse EC line, it has been previously shown that claudin-5 expression is regulated by FoxO1, which binds to a regulatory element of the claudin-5 gene and suppresses its expression (19). Upon phosphorylation, FoxO1 is released and moves to the cytosol, allowing claudin-5 gene expression. Given our finding that IL-4 induces strong up-regulation of claudin-5, we asked whether IL-4 causes phosphorylation of FoxO1 at Thr-24 and Ser-319, known targets of its post-translational modification (20). As shown in Fig. 4, A and B, we found that during the first hour of incubation of ECs with IL-4, there was phosphorylation of FoxO1 at both Thr-24 and Ser-319. Because phosphorylated FoxO1 is known to dissociate from the regulatory element on the claudin-5 gene and to move to the cytoplasm where it is degraded (19), we investigated the effect of EC stimulation with IL-4 on the distribution of FoxO1 in the nucleus and cytoplasm. We found that both phosphorylated and unphosphorylated FoxO1 decreased in the nucleus, whereas in the cytoplasm, both first remained near the initial levels and decreased somewhat by 2 or 4 h of incubation (Fig. 4C). We attribute these reductions to the known rapid degradation of phosphorylated FoxO1 that takes place in the proteasomes (21, 22). This IL-4-induced redistribution and rapid loss of FoxO1 was confirmed using confocal immunofluorescence microscopy for assessment of protein localization using florescence intensity measurements (Fig. 5). To clearly visualize the nuclei and residual FoxO1, we used co-localization with DAPI. We found that in control cells, FoxO1 was localized predominantly in the nucleus, but after 2 h of incubation with IL-4, about half of the FoxO1 was lost from the nucleus. In the cytoplasm, 2 h of IL-4 treatment reduced FoxO1 staining, which likely was caused by rapid degradation (21, 22). These results suggest that IL-4 stimulation of ECs causes transfer of FoxO1 from the nucleus to the cytoplasm, where it is rapidly degraded.
FIGURE 4.

IL-4 induces phosphorylation of FoxO1 and changes its cellular location. A and B, ECs were incubated with 10 ng/ml IL-4 for the times indicated and subjected to immunoblot analysis. Bars represent ratios of band density of pThr-24-FoxO1 (A) and pSer-319-FoxO1 (B) to that of GAPDH loading control and are means ± S.E. of four experiments in A and three experiments in B. C, IL-4 promotes nuclear exclusion of pSer-319-FoxO1 and FoxO1. ECs were incubated with 10 ng/ml IL-4 for the times indicated, and cytoplasmic and nuclear fractions were subjected to immunoblotting. Bars represent ratios of normalized densities for each fraction using the corresponding loading control, GAPDH and TATA-binding protein (TBP) for cytoplasmic and nuclear fraction, respectively, and are means ± S.E. of three experiments. *, p < 0.05, IL-4-treated versus time 0.
FIGURE 5.
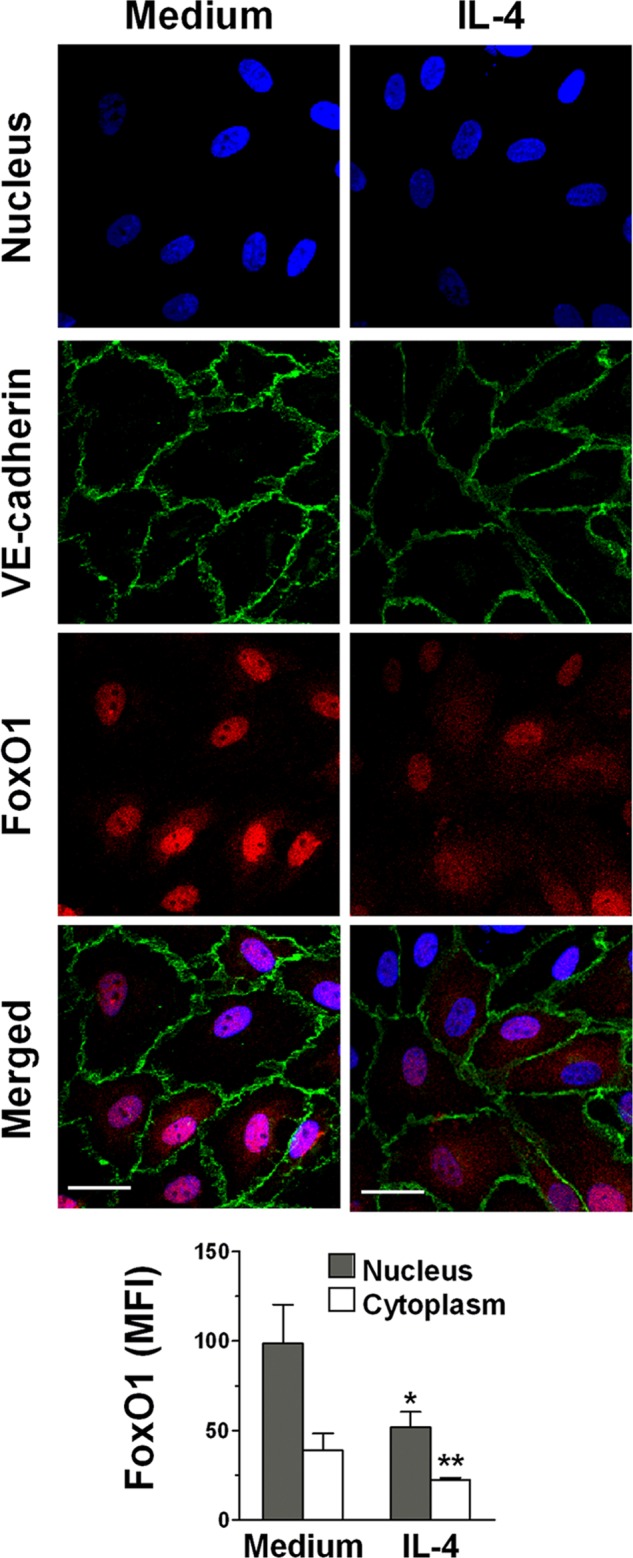
IL-4 induces translocation of FoxO1 to the cytoplasm. ECs were treated with medium or 10 ng/ml IL-4 for 2 h. The nuclei were visualized with DAPI (blue), and the cells were stained for VE-cadherin (green) and FoxO1 (red). The EC monolayers were then examined by confocal microscopy as the merged images of DAPI, VE-cadherin, and FoxO1. Photomicrographs are representative of three independent experiments. Original magnification, ×60. Scale bars, 20 μm. Bar graph represents mean fluorescence intensity (MFI) of FoxO1 in the nucleus and cytoplasm quantitated as described under “Experimental Procedures.” *, p < 0.02 and *, p < 0.05, IL-4-treated versus medium alone (nucleus and cytoplasm, respectively).
Down-regulation of FoxO1 in the Absence of Stimulation with IL-4 Up-regulates Claudin-5 Expression and Confers Partial Protection from Complement Cytotoxicity
Because loss of FoxO1 from the nucleus may ultimately be responsible for the effect of IL-4 on ECs that results in up-regulation of claudin-5, we assessed claudin-5 expression by cells with knockdown FoxO1 expression. We found that FoxO1 knockdown with siRNA by itself results in up-regulation of claudin-5 expression to a level similar to that obtained with IL-4 stimulation alone or with IL-4 stimulation and β-actin siRNA as a control (Fig. 6A). Higher levels of claudin-5 expression were found in cells that had both down-regulated FoxO1 and IL-4 treatment. Using immunofluorescence, we then showed that the increased claudin-5 resulting from down-regulation of FoxO1 migrated to the intercellular space (Fig. 6B). The ECs that exhibited increased levels of claudin-5 expression following knockdown of FoxO1 and no IL-4 treatment were partially protected from complement cytotoxicity (Fig. 6C). These results were obtained using two different sequences for FoxO1. In comparison with medium controls, no changes were seen using β-actin as a control. This result suggests that for induction of more effective protection from complement cytotoxicity by IL-4 other mechanisms are required in addition to up-regulated claudin-5 expression.
IL-4 Induces Claudin-5-dependent Increased Transendothelial Electrical Resistance of the Monolayer, Partially Preventing the Loss of Barrier Function Caused by Complement
Because claudin-5 is known to promote EC barrier function, we asked whether IL-4 caused claudin-5-dependent increase of intercellular electrical resistance, an indicator of the barrier function. We assessed TER of EC monolayers during exposure to IL-4 and found a gradual increase in TER over time, achieving the maximum level after ∼40 h of incubation, which usually persisted for ∼30 h of additional incubation (Fig. 7A). No changes were seen in monolayers that were incubated with medium alone. We next compared changes in TER caused by exposure to complement in ECs that were preincubated with IL-4 or medium alone for 70 h. We found that, initially, complement caused a drop in TER whether or not the endothelial monolayers were pretreated with IL-4. However, the monolayers that were pretreated with IL-4 before addition of complement stabilized at a higher level and remained at that level for 2.4 h of incubation (Fig. 7B). This result suggests that the protective effect of IL-4 against complement may in part be due to the strengthening of the barrier function caused by IL-4. We then used siRNA to knockdown expression of claudin-5 and found that in these monolayers, the IL-4-induced increase in electrical resistance was largely suppressed and identical to that in control cells incubated with medium and no IL-4 (Fig. 7C). Controls using Lipofectamine reagent alone or reagent plus β-actin siRNA before incubation with IL-4 did not alter the ability of IL-4 to increase TER. These results demonstrate that IL-4 induces increased TER in endothelial monolayers through up-regulation of claudin-5.
FIGURE 7.
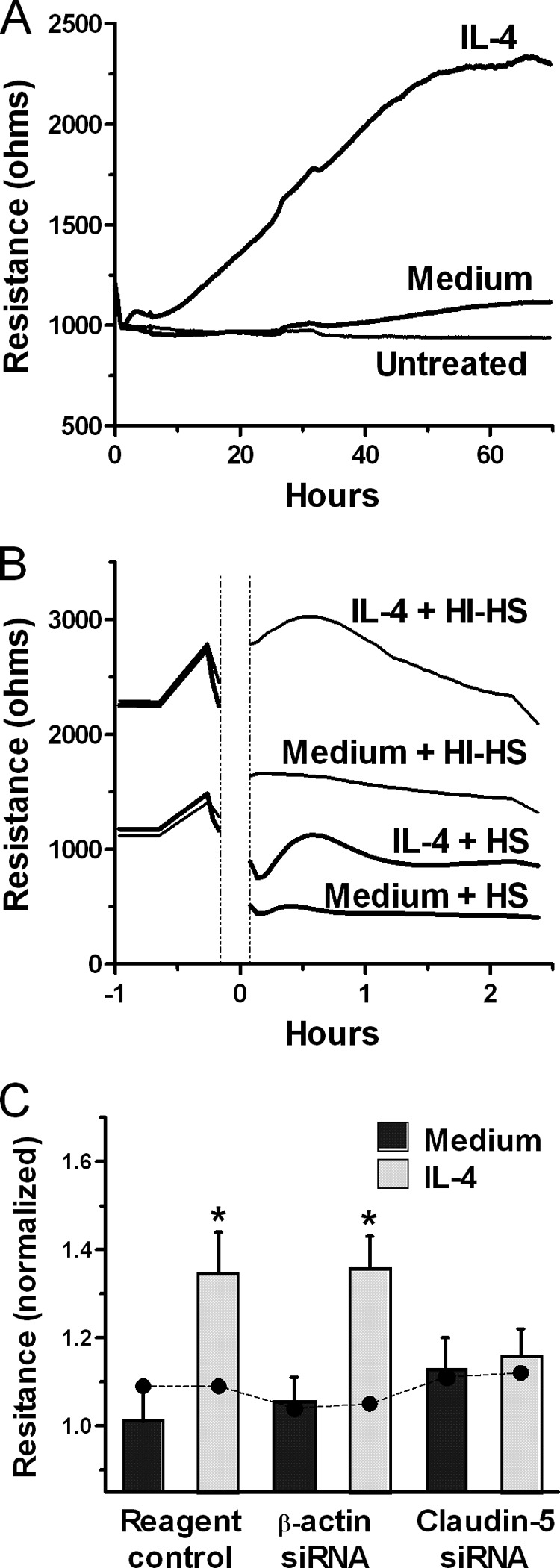
IL-4 induces claudin-5-dependent increase in electrical resistance of the EC monolayer, reducing the loss of barrier function caused by complement. A, TER measurements were performed continuously on EC monolayers in growth medium in duplicate (untreated) or with 1% FBS/DMEM alone (medium) or containing 10 ng/ml IL-4 (IL-4) in triplicate. Results are representative of three independent experiments. B, TER measurements of ECs that were pretreated with 10 ng/ml IL-4 or medium for 70 h, then washed and, at time 0, treated with 20% human serum (HS) or 20% heat-inactivated human serum (HI-HS). Then, TER measurements were continued for 2.4 h. Results are representative of two independent experiments. C, ECs were treated with the transfection reagent control or transfected with siRNA for β-actin or claudin-5, and when the cells reached confluence, medium or 10 ng/ml IL-4 was added, and incubation continued for 50 h. The bar graphs are normalized TER values at 50 h (means ± S.E. of four experiments). Filled circles are normalized TER values obtained before addition of medium or IL-4. Claudin-5 and β-actin down-regulation induced by specific siRNA was confirmed by Western blot. *, p < 0.02, normalized TER values at 50 h of IL-4-treatment versus medium.
DISCUSSION
In this study, we report the following novel findings: first, IL-4 activates porcine ECs resulting in up-regulation of the intercellular adhesion molecule claudin-5, and second, this increased claudin-5 expression plays a role in the induction by IL-4 of EC resistance against complement injury. Many substances are able to adversely modify the EC junction, including thrombin (23), antibodies and EC antigen-binding lectins (11, 13), and the membrane attack complex of complement (24). Because the injury caused by complement can be prevented by IL-4 (5), we hypothesized that IL-4 modifies one or more components of the junction so that the EC monolayer can resist the deleterious effect of complement. We found that IL-4 induces strong and selective up-regulation of the tight-junction protein claudin-5, suggesting that this change in expression might be one mechanism for IL-4-induced protection from complement. Therefore, we deemed it important to study the mechanisms involved in IL-4 stimulation resulting in claudin-5 up-regulation because this process could potentially play a physiologic role in regulating claudin-5 functions and because it may open a novel approach to induce or prevent cytoprotection proximal to claudin-5.
We found that IL-4, through activation of Jak/STAT6, induces phosphorylation of the transcription factor FoxO1, which results in transfer of FoxO1 from its predominant location in the nucleus to the cytoplasm. FoxO1, in a complex with β-catenin and T-cell factor, has been reported to suppress transcription of the claudin-5 gene, since loss of FoxO1 from the nucleus following its phosphorylation allows transcription of this gene (19). A kinase activated by Jak/STAT6 that might contribute to phosphorylate FoxO1 in ECs might be Akt because this kinase is known to phosphorylate FoxO1 (20) and is required for IL-4 protection of ECs from complement (1). However, inhibition of PI3K or Akt did not alter the IL-4-induced claudin-5 up-regulation. Therefore, it is likely that other kinases are important for this effect of IL-4, as there are many potential phosphorylation sites on FoxO1 (20). We also found that, without stimulation with IL-4, knockdown of FoxO1 causes increased expression of claudin-5, highlighting the crucial role of FoxO1 in regulation of claudin-5 expression. In addition to FoxO1 it has been reported that SOX18 (25) and ERG (26) are also involved in the transcriptional regulation of claudin-5 expression.
Claudin-5 is a member of the large family of claudin proteins, each exclusively or predominantly localized in a particular tissue. Claudin-5 is expressed only in the endothelium where it is the dominant claudin and is required for formation of confluent EC monolayers (19) and is a target for estrogen in vascular endothelium (27). Claudin-5KO mice are known to die soon after birth due to problems with the development of the blood brain barrier that results from abnormal permeability of the endothelium (28). Claudin-5 is the main adhesion protein responsible for the control of paracellular permeability, which can be assessed by measuring the electrical resistance of the EC monolayer (14, 29). In our study, we found that IL-4 causes a marked and steady increase in electrical resistance that parallels the time course of increased claudin-5 expression and reaches a plateau after ∼40 h of incubation. Addition of complement to these IL-4-treated cells resulted in partial loss of the electrical resistance, in contrast to the complete collapse of the electrical resistance of control cells, suggesting that the protective effect of IL-4 may in part be due to the increment in monolayer integrity caused by IL-4. Our study also demonstrates that the increase in electrical resistance caused by IL-4 is due to the increase in claudin-5 because we found that knockdown of claudin-5 expression suppressed the IL-4-induced increase in electrical resistance. These results demonstrate that ECs with up-regulated expression of claudin-5 have increased control of the monolayer permeability, which may provide a foundation to facilitate repair mechanisms after complement attack.
In our study, we demonstrate that up-regulation of claudin-5 expression, although a participant in protection from complement, is not sufficient to afford strong protection, as claudin-5 up-regulation achieved through knockdown of FoxO1 only conferred partial protection. This observation suggests that, in addition to claudin-5 up-regulation, activation of other mechanisms is necessary for protection. A contributing role might be played by other intercellular adhesion proteins, especially occludin, because this protein is an important tight junction protein. Moreover, nonspecific effects due to siRNA-induced EC activation may have played a role in our results. Finally, we emphasize our previously reported findings that IL-4-induced resistance of ECs against complement-mediated cytotoxicity requires the participation of PI3K and Akt, activation of SREBP-1, and increased phospholipid synthesis (5–7, 13). The requirement of increased phospholipid synthesis for protection may, at least in part, take place through increased cardiolipin synthesis and mitochondrial protection (7). Although we have not yet investigated the mechanisms that account for the requirement of phospholipid synthesis in conjunction with increased claudin-5 expression to achieve protection, our study shows that the two activation effects of IL-4 may be needed. Thus, we reported previously that the critical protection afforded by IL-4 from the severe injury of mitochondria structure and function that follows complement attack is associated with IL-4-mediated increase of phospholipid synthesis (7). This mitochondrial protection may be needed together with the increased claudin-5 expression.
Our current findings and our previous studies indicate that IL-4 and IL-13 may be important as regulatory cytokines of the vascular endothelium under physiological as well as pathological situations. In vitro, with cytokine doses as small as 1 ng/ml added extrinsically to EC cultures (1) and picogram amounts of cytokines secreted by ECs that were transduced with low doses of adeno-IL-4, strong protection from complement could be achieved (5). Moreover, in vivo transfer of the IL-4 gene into a pig iliac artery resulted in protection from the complement-mediated injury of the endothelium and sub-endothelium that occurs immediately upon perfusion of the artery with human blood in control arteries (5). Although our models have used pig endothelium and human blood or serum, and as such serve mainly as models for xenotransplantation, we have shown previously that IL-4 is also capable of inducing protection against complement in human ECs (7).
Although it has been extensively documented that in many pathological conditions induction of cytoprotection may be desirable, in other situations, especially cancer, cytoprotection should be avoided and even suppressed (30–33). Therefore our studies on IL-4-induced mechanisms of protection should be interpreted as a two-way process, whereupon it should be possible to intervene to promote or to interfere with induction of cytoprotection. Moreover, the search for inducers or inhibitors of cytoprotection should indeed focus on those substances that act as proximally as possible to the final cell processes that are responsible for the cytoprotection, as shown in other models such as regulation of heme oxygenase-1 and carbon monoxide, with potential clinical applications (34, 35). The direct use of an inducer such as IL-4 would be problematic because such substances often have pleotropic effects, for example triggering proinflammatory mediators. Therefore, a main thrust of our studies on mechanisms of IL-4-induced protection was to understand the pathways that culminate in induction of protection, as summarized in Fig. 8. Additional work is needed to define potential sites for intervention such as: a) selective overexpression of mitochondrial cardiolipin, as opposed to global stimulation of phospholipid synthesis (7); b) inhibition of the DNA-binding site for the FoxO1/β-catenin/T-cell factor complex that suppresses claudin-5 transcription (19, 36); and c) selective activation of phospholipid synthesis or enzymatic activities required for proper positioning of claudin-5 at the intercellular junction (37). Being able to intervene pharmacologically at distal stages of the activation pathways stimulated by IL-4 that lead to protection could be applicable to protect the endothelium in vascular processes such as atherosclerosis, ischemia reperfusion injury, and transplant vasculopathy.
FIGURE 8.
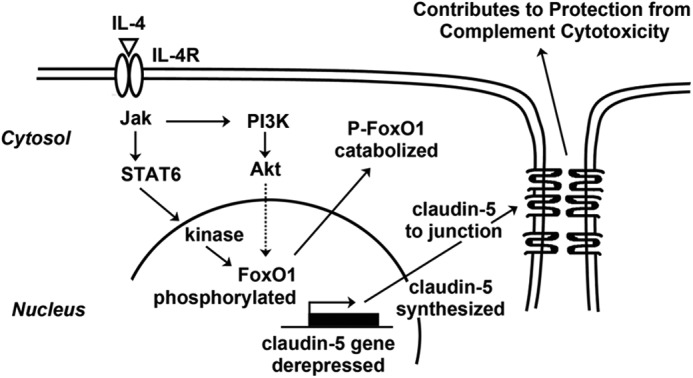
Proposed mechanism activated by IL-4 stimulation of ECs resulting in claudin-5 up-regulation that contributes to protection from complement-mediated cytotoxicity. Not indicated in the diagram is the absolute requirement for protection of a second activation process that includes PI3K-Akt-SREB1-phospholipid synthesis (1, 6, 7).
Acknowledgments
We thank Dr. Santanu Banerjee for invaluable assistance with the TER studies and Julia Nguyen for technical support with the confocal study. We acknowledge the assistance of the staff of the Imaging Centers at the University of Minnesota.
This work was supported by National Institutes of Health Grant RO1 HL062195 (to A. P. D.) and by the Division of Cardiothoracic Surgery, Department of Surgery, University of Minnesota. G. M. V. is a recipient of research funds and is a consultant for Sangart, Inc.
B. A. Benson and A. P. Dalmasso, unpublished data.
- EC
- endothelial cell
- TER
- trans-endothelial electrical resistance
- VE
- vascular endothelial
- ZO-1
- zona occludens protein 1.
REFERENCES
- 1. Grehan J. F., Levay-Young B. K., Fogelson J. L., François-Bongarçon V., Benson B. A., Dalmasso A. P. (2005) IL-4 and IL-13 induce protection of porcine endothelial cells from killing by human complement and from apoptosis through activation of a phosphatidylinositide 3-kinase/Akt pathway. J. Immunol. 175, 1903–1910 [DOI] [PubMed] [Google Scholar]
- 2. Black S. M., Benson B. A., Idossa D., Vercellotti G. M., Dalmasso A. P. (2011) Protection of porcine endothelial cells against apoptosis with interleukin-4. Xenotransplantation 18, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Müller-Eberhard H. J. (1988) Molecular organization and function of the complement system. Annu. Rev. Biochem. 57, 321–347 [DOI] [PubMed] [Google Scholar]
- 4. Dalmasso A. P. (2002) The complement barrier to xenotransplantation in Xenotransplantation (Platt J. L., ed), pp. 139–171, Humana Press, Totowa, NJ [Google Scholar]
- 5. Black S. M., Grehan J. F., Rivard A. L., Benson B. A., Wahner A. E., Koch A. E., Levay-Young B. K., Dalmasso A. P. (2006) Porcine endothelial cells and iliac arteries transduced with AdenoIL-4 are intrinsically protected, through Akt activation, against immediate injury caused by human complement. J. Immunol. 177, 7355–7363 [DOI] [PubMed] [Google Scholar]
- 6. Black S. M., Schott M. E., Benson B. A., Rutherford M. S., Young B. K., Dalmasso A. P. (2008) Interleukin-4 induces lipogenesis in porcine endothelial cells, which in turn is critical for induction of protection against complement-mediated injury. Transplant. Proc. 40, 638–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Black S. M., Schott M. E., Batdorf B. H., Benson B. A., Rutherford M. S., Levay-Young B. K., Dalmasso A. P. (2010) IL-4 induces protection of vascular endothelial cells against killing by complement and melittin through lipid biosynthesis. Eur. J. Immunol. 40, 803–812 [DOI] [PubMed] [Google Scholar]
- 8. Bazzoni G., Dejana E. (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84, 869–901 [DOI] [PubMed] [Google Scholar]
- 9. Wallez Y., Huber P. (2008) Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 1778, 794–809 [DOI] [PubMed] [Google Scholar]
- 10. Woodfin A., Voisin M. B., Nourshargh S. (2007) PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 27, 2514–2523 [DOI] [PubMed] [Google Scholar]
- 11. Dalmasso A. P., Benson B. A., Johnson J. S., Lancto C., Abrahamsen M. S. (2000) Resistance against the membrane attack complex of complement induced in porcine endothelial cells with a Gal α(1–3)Gal binding lectin: up-regulation of CD59 expression. J. Immunol. 164, 3764–3773 [DOI] [PubMed] [Google Scholar]
- 12. Dalmasso A. P., He T., Benson B. A. (1996) Human IgM xenoreactive natural antibodies can induce resistance of porcine endothelial cells to complement-mediated injury. Xenotransplantation 3, 54–62 [Google Scholar]
- 13. Grehan J. F., Levay-Young B. K., Benson B. A., Abrahamsen M. S., Dalmasso A. P. (2005) αGal ligation of pig endothelial cells induces protection from complement and apoptosis independently of NF-κB and inflammatory changes. Am. J. Transplant. 5, 712–719 [DOI] [PubMed] [Google Scholar]
- 14. Tiruppathi C., Malik A. B., Del Vecchio P. J., Keese C. R., Giaever I. (1992) Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 89, 7919–7923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelms K., Keegan A. D., Zamorano J., Ryan J. J., Paul W. E. (1999) The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17, 701–738 [DOI] [PubMed] [Google Scholar]
- 16. Hebenstreit D., Wirnsberger G., Horejs-Hoeck J., Duschl A. (2006) Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 17, 173–188 [DOI] [PubMed] [Google Scholar]
- 17. Pedranzini L., Dechow T., Berishaj M., Comenzo R., Zhou P., Azare J., Bornmann W., Bromberg J. (2006) Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 66, 9714–9721 [DOI] [PubMed] [Google Scholar]
- 18. Steele A. J., Prentice A. G., Cwynarski K., Hoffbrand A. V., Hart S. M., Lowdell M. W., Samuel E. R., Wickremasinghe R. G. (2010) The JAK3-selective inhibitor PF-956980 reverses the resistance to cytotoxic agents induced by interleukin-4 treatment of chronic lymphocytic leukemia cells: potential for reversal of cytoprotection by the microenvironment. Blood 116, 4569–4577 [DOI] [PubMed] [Google Scholar]
- 19. Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., Dejana E. (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y., Wang Y., Zhu W. G. (2011) Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell. Biol. 3, 276–282 [DOI] [PubMed] [Google Scholar]
- 21. Matsuzaki H., Daitoku H., Hatta M., Tanaka K., Fukamizu A. (2003) Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 100, 11285–11290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang H., Tindall D. J. (2011) Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 1813, 1961–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawkitinarong K., Linz-McGillem L., Birukov K. G., Garcia J. G. (2004) Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am. J. Respir. Cell Mol. Biol. 31, 517–527 [DOI] [PubMed] [Google Scholar]
- 24. Saadi S., Platt J. L. (1995) Transient perturbation of endothelial integrity induced by natural antibodies and complement. J. Exp. Med. 181, 21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fontijn R. D., Volger O. L., Fledderus J. O., Reijerkerk A., de Vries H. E., Horrevoets A. J. (2008) SOX-18 controls endothelial-specific claudin-5 gene expression and barrier function. Am. J. Physiol. Heart Circ. Physiol. 294, H891–H900 [DOI] [PubMed] [Google Scholar]
- 26. Yuan L., Le Bras A., Sacharidou A., Itagaki K., Zhan Y., Kondo M., Carman C. V., Davis G. E., Aird W. C., Oettgen P. (2012) ETS-related gene (ERG) controls endothelial cell permeability via transcriptional regulation of the claudin 5 (CLDN5) gene. J. Biol. Chem. 287, 6582–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burek M., Arias-Loza P. A., Roewer N., Förster C. Y. (2010) Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 30, 298–304 [DOI] [PubMed] [Google Scholar]
- 28. Nitta T., Hata M., Gotoh S., Seo Y., Sasaki H., Hashimoto N., Furuse M., Tsukita S. (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 161, 653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Becker P. M., Verin A. D., Booth M. A., Liu F., Birukova A., Garcia J. G. (2001) Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L1500–L1511 [DOI] [PubMed] [Google Scholar]
- 30. Balla J., Vercellotti G. M., Jeney V., Yachie A., Varga Z., Jacob H. S., Eaton J. W., Balla G. (2007) Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal. 9, 2119–2137 [DOI] [PubMed] [Google Scholar]
- 31. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 32. Kroemer G., Galluzzi L., Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163 [DOI] [PubMed] [Google Scholar]
- 33. Li Z., Chen L., Qin Z. (2009) Paradoxical roles of IL-4 in tumor immunity. Cell. Mol. Immunol. 6, 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Otterbein L. E. (2009) The evolution of carbon monoxide into medicine. Respir. Care 54, 925–932 [DOI] [PubMed] [Google Scholar]
- 35. Gozzelino R., Jeney V., Soares M. P. (2010) Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354 [DOI] [PubMed] [Google Scholar]
- 36. Dejana E., Taddei A., Randi A. M. (2007) Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim. Biophys. Acta 1775, 298–312 [DOI] [PubMed] [Google Scholar]
- 37. Van Itallie C. M., Gambling T. M., Carson J. L., Anderson J. M. (2005) Palmitoylation of claudins is required for efficient tight-junction localization. J. Cell Sci. 118, 1427–1436 [DOI] [PubMed] [Google Scholar]