

Background: Therapeutic inhibition of circulating PCSK9 reduces LDL-c levels.
Results: A synthetic PCSK9-binding peptide, which restores cellular LDL receptors, was identified.
Conclusion: Pep2-8 is the smallest PCSK9 inhibitor with a defined inhibitory mechanism described to date and structurally mimics the EGF(A) domain of the receptor.
Significance: This work demonstrates the feasibility of developing a peptide-based inhibitor of PCSK9.
Keywords: Crystal Structure, Peptides, Phage Display, Protease, Protein-Protein Interactions, LDL Receptor, PCSK9
Abstract
PCSK9 (proprotein convertase subtilisin/kexin type 9) is a negative regulator of the hepatic LDL receptor, and clinical studies with PCSK9-inhibiting antibodies have demonstrated strong LDL-c-lowering effects. Here we screened phage-displayed peptide libraries and identified the 13-amino acid linear peptide Pep2-8 as the smallest PCSK9 inhibitor with a clearly defined mechanism of inhibition that has been described. Pep2-8 bound to PCSK9 with a KD of 0.7 μm but did not bind to other proprotein convertases. It fully restored LDL receptor surface levels and LDL particle uptake in PCSK9-treated HepG2 cells. The crystal structure of Pep2-8 bound to C-terminally truncated PCSK9 at 1.85 Å resolution showed that the peptide adopted a strand-turn-helix conformation, which is remarkably similar to its solution structure determined by NMR. Consistent with the functional binding site identified by an Ala scan of PCSK9, the structural Pep2-8 contact region of about 400 Å2 largely overlapped with that contacted by the EGF(A) domain of the LDL receptor, suggesting a competitive inhibition mechanism. Consistent with this, Pep2-8 inhibited LDL receptor and EGF(A) domain binding to PCSK9 with IC50 values of 0.8 and 0.4 μm, respectively. Remarkably, Pep2-8 mimicked secondary structural elements of the EGF(A) domain that interact with PCSK9, notably the β-strand and a discontinuous short α-helix, and it engaged in the same β-sheet hydrogen bonds as EGF(A) does. Although Pep2-8 itself may not be amenable to therapeutic applications, this study demonstrates the feasibility of developing peptidic inhibitors to functionally relevant sites on PCSK9.
Introduction
PCSK9 (proprotein convertase subtilisin/kexin type 9), initially identified as NARC-1 (1), regulates LDL-c2 levels by promoting degradation of the liver LDL receptor, which results in reduced LDL receptor-dependent clearance of LDL-c particles and increased plasma LDL-c levels (2–4). PCSK9 acts by binding to the EGF(A) domain of LDL receptor on the cell surface via its catalytic domain (5). The binding affinity increases in the acidic endosomal compartment through additional interactions of LDL receptor with the C-terminal Cys-rich domain (CRD) of PCSK9 (6–8). As a result, LDL receptor recycling to the cell surface is prevented, and instead, the PCSK9·LDL receptor complex is directed to lysosomes for degradation. This “sorting” of the PCSK9·LDL receptor complex seems to be mediated by APLP2, which interacts with PCSK9 at low pH in the endosomal compartment (9).
Natural PCSK9 mutations are associated with high and low cholesterol levels (10, 11). Cohen et al. (11) found that carriers of two loss-of-function mutations had lower LDL-c levels and a dramatically reduced risk for coronary heart disease. A compound heterozygous individual with no detectable PCSK9 and extremely low LDL-c levels is healthy (12), suggesting that pharmacologic PCSK9 inhibition for lipid lowering might be safe.
PCSK9 is an ideal antibody target because it circulates in blood and has only modest binding affinity to cell surface LDL receptor and its main interaction site is an exposed ∼500-Å2 slightly convex region that is readily available for antibody binding. Recent clinical studies with anti-PCSK9 antibodies have borne this out, demonstrating strong and sustained LDL-c lowering after a single dose injection (13). Whereas the clinical development of antibody-based inhibitors has advanced rapidly, the development of small molecule inhibitors appears still to be at an early stage, probably due to the challenging nature of PCSK9 as a small molecule target. As its name implies, PCSK9 bears recognizable sequence homology to proteolytic enzymes of the subtilisin family. The molecular structure of PCSK9 determined using x-ray crystallography confirms its close conformational relationship with subtilisin-like proteases (14–16). Unlike typical enzymes, whose active sites are essential for function and generally offer good binding sites for small molecules, the PCSK9 active site is non-essential for LDL receptor down-regulation (17, 18) and remains in an inactive state due to occupancy by the prodomain C terminus after autoprocessing in the endoplasmic reticulum. Therefore, aside from an approach that would block maturation by inhibiting the autocatalytic enzyme activity during biosynthesis, the PCSK9 active site is not attractive as a traditional protease target.
The ability of PCSK9 to mediate LDL receptor degradation involves protein-protein interactions between LDL receptor and the PCSK9 prodomain (19), the catalytic domain (20), and the CRD (6–9, 21–23). Protein-protein interactions are inherently challenging small molecule targets because they frequently contain large, relatively flat contact surfaces devoid of the pockets required for efficient binding of small organic chemicals. However, protein-protein interactions have been successfully inhibited by peptides, which can recapitulate key protein contacts (24). To identify a peptidic PCSK9 inhibitor, we screened both linear and disulfide-constrained phage-displayed peptide libraries with final diversities of 1.8 × 1011 and 7.8 × 1011, respectively, and identified the 13-amino acid peptide Pep2-8. Structural and biochemical characterization provided the molecular details of its inhibitory mechanism, which includes structural mimicry of the natural binding domain of LDL receptor.
EXPERIMENTAL PROCEDURES
Construction of Phage-displayed Peptide Libraries
Two groups of phage-displayed peptide libraries, the linear peptide library called Linear-lib and cysteine-restrained cyclic library called Cyclic-lib, were constructed by fusing randomized peptides to the N terminus of M13 major coat protein p8 (25). Linear-lib consisted of random peptides with 8, 10, 12, 14, or 16 amino acids, and Cyclic-lib consisted of 14-mer random peptides with varied length between two invariant cysteines. The final diversities for Linear-lib and Cyclic-Lib were 1.8 × 1011 and 7.8 × 1011, respectively.
The soft randomized library was constructed using degenerate oligonucleotides synthesized with 70:10:10:10 mixtures of nucleotide bases, in which the wild type base was in excess. This results in the wild type amino acids occurring at ∼50% frequency at the targeted position.
Selection of Peptide Ligands for PCSK9 and Affinity Maturation of Pep2
Phage pools of Linear-lib and Cyclic-lib library were cycled through rounds of binding selections using 130, 65, 26, and 26 nm biotinylated PCSK9 in a buffer of PBS, 0.5% BSA, and 0.1% Tween 20 for round 1, 2, 3 and 4, respectively (26). After four rounds of binding selection, individual phage clones were analyzed in a high throughput spot phage ELISA using plate-immobilized PCSK9 as the target (27). Nonspecific binding was defined as phage particle binding to NeutrAvidin. Clones with phage binding signal to PCSK9 over 0.5 and signal/noise ratio of >5 were considered to be positive clones and were subjected to DNA sequence analysis.
The soft randomized library was used for affinity maturation of clone 2. Phage pools were subjected to four rounds of binding selection with decreasing concentrations of biotinylated PCSK9 from round 1 to round 4 at 130, 20, 2, and 2 nm, respectively, and produced Pep2-2.
Peptide Binding Affinity for PCSK9 Measured by a Phage Competition ELISA
The sequence of Pep2-2 was fused to the N terminus of M13 major coat protein (p8) using Kunkel mutagenesis (28). The resulting construct was transformed to Escherichia coli XL1 blue, and single colonies were grown in 1 ml of 2YT supplemented with 50 μg/ml carbenicillin, 10 μg/ml tetracycline, and M13 KO7 helper phage at 37 °C for 2 h. After the addition of kanamycin (25 μg/ml) and a 6-h incubation at 37 °C, the culture was transferred to 30 ml of 2YT supplemented with 50 μg/ml carbenicillin and 25 μg/ml kanamycin and grown at 37 °C overnight. Phage were harvested and purified using the standard protocol (27). The serially diluted phage solution was added to a 384-well MaxiSorp ImmunoPlate containing biotinylated PCSK9 immobilized to NeutrAvidin-coated wells. The plate was incubated at 4 °C for 1 h and extensively washed with PBS, 0.05% Tween 20, and bound phage was quantified after the addition of anti-M13-HRP and HRP substrate on a microplate reader at A450. After subtracting background binding (i.e. phage binding to NeutrAvidin-coated plates), the data were fitted to a binding curve using KaleidaGraph, and the subsaturating phage concentration that gave 80% of maximum binding was calculated.
For the peptide affinity determinations, biotinylated PCSK9 was immobilized to NeutrAvidin-coated MaxiSorp ImmunoPlates and incubated with a mixture of serially diluted synthetic peptide (Pep2-2 or Pep2 at 0–100 μm) and the corresponding peptide-displaying phage at the earlier determined subsaturating concentration (see above). After a 1-h incubation in PBT buffer (PBS, 0.5% BSA, 0.05% Tween 20), the plate was washed extensively, and the bound phage was detected by anti-M13-HRP. The data were fitted to a four-parameter equation (KaleidaGraph) to determine the IC50 values. For IC50 measurements of the Pep2-2 derivatives (Fig. 1C) and for the truncated Pep2-8 peptides (Table 1), we used the peptides in a concentration range of 0–250 μm and the Pep2-2-displaying phage at subsaturating concentrations. The results were the average ± S.D. of three independent experiments.
FIGURE 1.
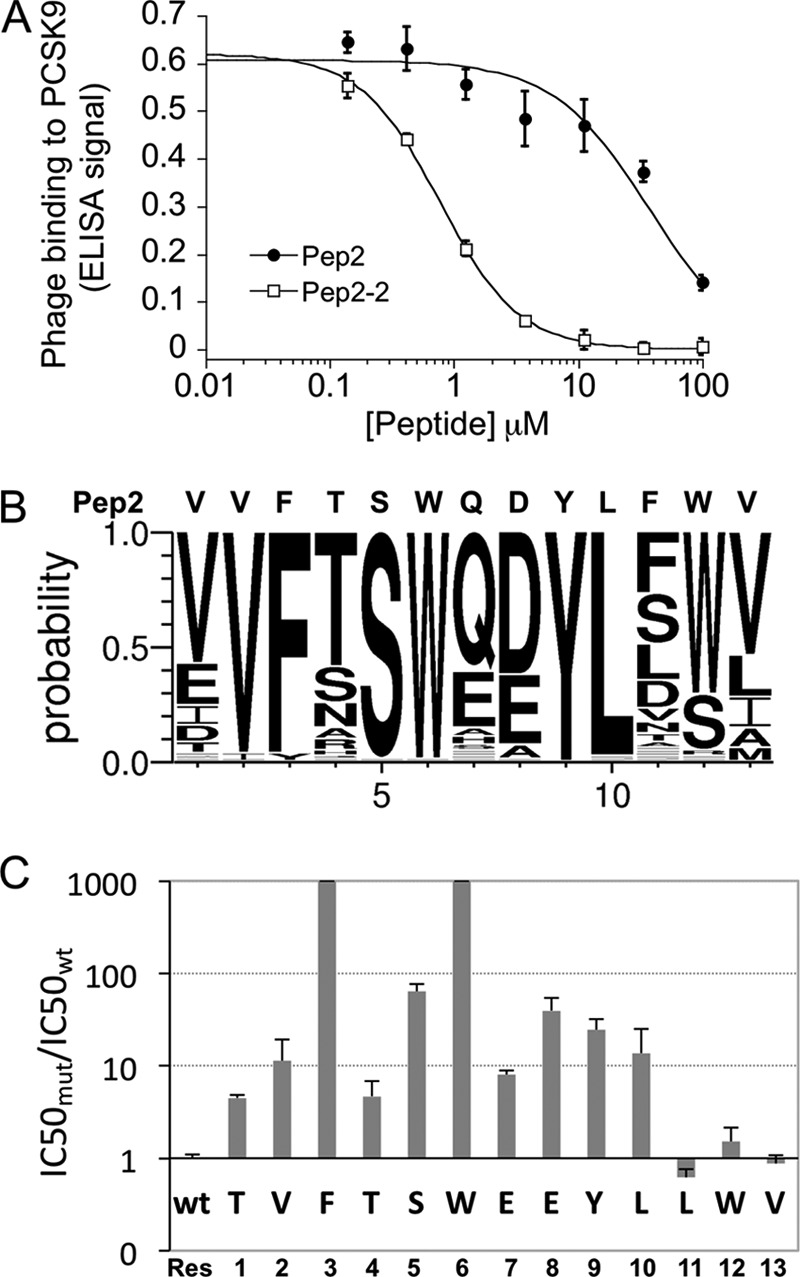
Affinity maturation of Pep2 and Ala scanning of Pep2-2. A, competition of Pep2 and affinity-matured Pep2-2 for phage binding to PCSK9. B, result of peptide randomization presented as sequence logo. C, effects of Ala substitutions on peptide activity. Error bars, S.D.
TABLE 1.
IC50 values for synthetic peptides binding to PCSK9
| Peptide ID | N- | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | -C | IC50 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μm | ||||||||||||||||
| Pep2 | Ac | V | V | F | T | S | W | Q | D | Y | L | F | W | V | Amide | 37.7 ± 1.0 |
| Pep2-2 | Ac | T | V | F | T | S | W | E | E | Y | L | L | W | V | Amide | 0.76 ± 0.01 |
| Pep2-2ΔT1 | Ac | V | F | T | S | W | E | E | Y | L | L | W | V | Amide | 4.0 ± 0.4 | |
| Pep2-2ΔV13 | Ac | T | V | F | T | S | W | E | E | Y | L | L | W | Amide | 6.2 ± 0.5 | |
| Pep2-2ΔW12, V13 | Ac | T | V | F | T | S | W | E | E | Y | L | L | Amide | 10.0 ± 0.6 | ||
| Pep2-2ΔL11-V13 | Ac | T | V | F | T | S | W | E | E | Y | L | Amide | 17.6 ± 3.7 | |||
| Pep2-2ΔL10-V13 | Ac | T | V | F | T | S | W | E | E | Y | Amide | Undetectable | ||||
| Pep2-8 | Ac | T | V | F | T | S | W | E | E | Y | L | D | W | V | Amide | 1.4 ± 0.2 |
| Pep-ctrl | Ac | T | V | A | T | S | A | E | E | Y | L | L | W | V | Amide | Undetectable |
Construction, Expression, and Purification of Wild Type and Mutant PCSK9 Proteins
Human PCSK9 cDNA containing a His8 C-terminal tag was cloned into the mammalian expression vector pRK5. Using this expression construct as a template, the PCSK9 mutants S153A, P155A, L158A, R194A, D238A, D367A, I369A, D374A, F379A, V380A, S381A, and Q382A were constructed by ACTG Inc. (Wheeling, IL) using site-directed mutagenesis. Mutations were confirmed by DNA sequencing and, in the case of S153A, P155A, and L158A, by additional N-terminal amino acid sequencing of the purified proteins. The recombinant PCSK9 proteins (wild type and mutants) were transiently expressed in Chinese hamster ovary (CHO) cells and purified from conditioned media by affinity chromatography using a nickel-nitrilotriacetic acid-agarose column (Qiagen, Germantown, MD) followed by gel filtration on a Sephacryl S 200 column (GE Healthcare) equilibrated with 50 mm Tris, pH 8.0, 500 mm NaCl, 10% glycerol. Proteins were aliquoted and stored at −80 ºC.
Production of PCSK9 Lacking the CRD (PCSK9-ΔCRD)
PCSK9-ΔCRD (Gln31–Gly452) was cloned by PCR amplification from the full-length PCSK9 cDNA using GC Advantage Polymerase (Clontech). The PCR product contained the sequence coding for PCSK9-ΔCRD and the restriction endonuclease sites BamHI and EcoRI at the 5′- and 3′-end, respectively. The PCR product was ligated into a modified pAcGP67 baculovirus transfer vector containing the DNA sequence coding for a hexahistidine affinity tag. The inserted PCR product was verified by DNA sequencing. Protein was produced in Trichoplusia ni insect cells (Expression Systems LLC, Woodland, CA) grown for 2 days after infection with baculovirus. Cells were removed from the production medium by centrifugation, and the supernatant was loaded onto a nickel-nitrilotriacetic acid resin (Qiagen) equilibrated with 50 mm Tris, pH 8.0, 500 mm NaCl and eluted with 50 mm Tris, pH 8.0, 500 mm NaCl, 250 mm imidazole. Pooled fractions were further purified on an S75 size exclusion column (GE Healthcare) equilibrated with 40 mm Tris, pH 8.0, 200 mm NaCl, 5% glycerol, 0.1 mm CaCl2. By use of a spin concentrator, the pooled fractions were concentrated to 8 mg in 0.9 ml.
Expression and Purification of Pep2-8-Fc, Pep-ctrl-Fc, and LDL Receptor-Fc Fusion Proteins
The DNA sequences encoding Pep2-8 (TVFTSWEEYLDWV) or Pep-ctrl (TVATSAEEYLFWV) plus a linker with a sequence of GGGSGAAQVTNKTHT followed by the Fc domain of human IgG1 (Cys222–Lys443) were cloned into pRK5 vector and designated as Pep2-8-Fc-pRK5 or Pep-ctrl-Fc-pRK5. The DNA sequences encoding LDL receptor ectodomain (Met1–Val787) plus a linker with a sequence of GRAQVTDKTHT followed by the Fc domain of human IgG1 (Cys222–Lys443) were cloned into pRK5 vector.
The fusion proteins were transiently expressed in CHO and purified on a Protein A resin followed by size exclusion chromatography. The identities of the proteins were confirmed by mass spectrometry, N-terminal sequencing, and SDS-PAGE. Unexpectedly, the purified LDL receptor-Fc lacked the first four LDL receptor repeats due to cleavage by an unknown protease at Gly192–Asp193 in the linker region between LDL receptor repeats 4 and 5. The binding affinity of PCSK9 to this LDL receptor-Fc construct determined by biolayer interferometry at neutral pH was 106.7 ± 5.8 nm (average ± S.D. of three experiments), suggesting that the N-terminal truncation of LDL receptor did not impair PCSK9 binding.
Binding Affinity and Specificity Determinations by Biolayer Interferometry
The binding affinities of Pep2-8_Fc and Pep-ctrl_Fc to PCSK9 were measured by biolayer interferometry on an OctetRed 384 (ForteBio, Menlo Park, CA). Anti-hIgG-Fc capture biosensors (ForteBio) were loaded with Pep2-8_Fc or Pep-ctrl_Fc in 50 mm Tris, pH 7.5, 300 mm NaCl, 1 mm CaCl2, 1 mg/ml BSA, 0.05% Tween 20, washed in the same buffer, and transferred to wells containing PCSK9 at concentrations ranging from 8 to 6000 nm in the same buffer. The kon and koff values were obtained by fitting the association and dissociation data to a 1:1 model algorithm using Octet software. The KD derived from kinetic fitting was calculated as koff/kon.
To assess Pep2-8 specificity, the binding to other members of the proprotein convertase family, including furin, PC1, PC2, PC7 (R&D Systems, Minneapolis, MN), and PC4 (Abnova, Taipei City, Taiwan), was measured. Pep2-8_Fc and Pep-ctrl_Fc were captured on biosensors as described above. The binding to each different proprotein convertase was then measured in optimized assay buffer as suggested by the manufacturer: 50 mm Tris, pH 7.5, 300 mm NaCl, 1 mm CaCl2, 1 mg/ml BSA, 0.05% Tween 20 for PC1 and PC7 (and PCSK9); 50 mm Tris, pH 8.0, 100 mm NaCl, 1 mm CaCl2, 1 mg/ml BSA, 0.04% Brij-35 for furin; 50 mm Tris, pH 8.0, 100 mm NaCl, 5 mm reduced glutathione, 1 mm CaCl2, 1 mg/ml BSA, 0.05% Tween 20 for PC4; and 50 mm NaOAc, pH 5.0, 100 mm NaCl, 1 mm CaCl2 for PC2. The loaded biosensors were washed in the respective assay buffers and transferred to pairs of wells, containing the tested proprotein convertase member and PCSK9 at the same concentration (700 nm, 750 nm, 1 μm, 500 nm, and 500 nm for furin, PC7, PC2, PC4, and PC1, respectively). The association responses over 899 s for each pair were obtained, and the binding response relative to that of PCSK9 (“relative response”) was calculated.
ELISA for Mapping the Functional Pep2-8 Binding Site on PCSK9
Biotinylated Pep2-8 was captured by NeutrAvidin-coated 384-well MaxiSorp ImmunoPlates (Nalge Nunc International, Rochester, NY). Serially diluted wild type and mutants of PCSK9 (starting at 10 μm) in PBS, 0.5% BSA, 0.05% Tween 20 were added to the wells and incubated at 4 °C for 1 h. After washing with PBS, 0.05% Tween 20, bound PCSK9 was detected by the sequential addition of anti-His HRP conjugate (1:5000 dilution; Qiagen, Hilden, Germany) and substrate 3,3′,5,5′-tetramethylbenzidine (TMBE-1000, Moss (Pasadena, MD)). The data were fitted to a four-parameter equation (KaleidaGraph) to determine the concentrations for half-maximal binding (EC50 values). The relative binding was calculated as the ratio of EC50 for wild type PCSK9 over PCSK9 variants.
Peptide Synthesis
Synthetic peptides were prepared on an automated Protein Technologies Inc. synthesizer. Peptides were assembled using standard Fmoc chemistry protocols on an Fmoc Rink amide linker attached to aminomethyl resin, and peptides were acetylated on the N terminus. Upon completion of the linear chains, peptides were cleaved off the solid support with trifluoroacetic acid/triisopropylsilane/water (95:2.5:2.5) for 1 h at room temperature. Trifluoroacetic acid was evaporated, and peptides were precipitated with ethyl ether; extracted with acetic acid, acetonitrile, and water; and lyophilized. Crude peptides were solubilized in dimethyl sulfoxide and purified by reverse phase chromatography on a C18 column using acetonitrile/water buffers. Purified fractions were analyzed by liquid chromatography mass spectrometry (PE-Sciex), pooled, and lyophilized.
ELISA for Measuring Inhibition of PCSK9 Binding to LDL Receptor and EGF(A) Domain
LDL receptor-Fc (2 μg/ml) or EGF(A)-Fc (1 μg/ml) in DPBS (with calcium) (Invitrogen) was added to 96-well plates (Nalge Nunc International) and incubated at 4 °C overnight. After washing the plates with PBS + 0.05% Tween 20, His-tagged PCSK9 (2 μg/ml for LDL receptor-Fc and 4 μg/ml for EGF(A)-Fc experiments) preincubated for 30 min with serially diluted Pep2-8 or Pep-ctrl in 25 mm HEPES, 300 mm NaCl, 2 mm CaCl2, 0.1% BSA, 0.05% Tween 20 containing 0.15% DMSO (final concentration) was added to the wells. After a 2-h incubation, plates were washed with PBS + 0.05% Tween 20, and bound PCSK9 was detected with anti-His-HRP antibody (Roche Applied Science) plus substrate TMB (KPL, Gaithersburg, MD), and A450 was measured on a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA).
HepG2 Assay for Measuring Cell Surface LDL Receptor and LDL Uptake
The effects of Pep2-8 and Pep-ctrl on PCSK9-mediated LDL receptor degradation were measured in a HepG2 cell assay essentially as described (26). Peptides at various concentrations were preincubated with 15 μg/ml PCSK9 in the presence of 0.5% DMSO for 30 min prior to the addition to cells. After a 4-h incubation, the cells were processed for measurements of LDL receptor surface levels by flow cytometry as described (26).
In the LDL uptake assay, HepG2 cells were seeded at a density of 3 × 104 cells/well in a 96-well plate. After 24 h, the medium was changed to 10% lipoprotein-deficient serum (Intracel) in DMEM (Invitrogen). After 24 h, Pep2-8 or Pep-ctrl at various concentrations was preincubated with 15 μg/ml PCSK9 in 0.5% DMSO for 30 min prior to the addition to the cells. After 1.5 h, 10 μg/ml BODIPY-LDL (Invitrogen) was added and incubated for 3.5 h. Cells were washed three times with PBS, and fluorescence was measured on a Wallac VICTOR3 plate reader (PerkinElmer Life Sciences). For both assays, the results were the average ± S.D. of three independent experiments.
Formation and Crystallization of the Complex
A 10-fold molar excess of Pep2-8 (0.5 mg) was added to 200 μl of PCSK9-ΔCRD (8.9 mg/ml) and incubated for 14 h at 4 °C. Crystallization trials were performed using commercially available screening reagents in 0.4-μl sitting drops made from protein and reservoir in a 1:1 ratio. A drop made using reservoir containing 0.2 m LiCl and 20% PEG 6000 in 0.1 m Tris, pH 8.0, produced crystals that were optimized by growth in larger drops. A crystal was harvested and passed briefly through a solution of reservoir augmented with 30% glycerol and preserved for data collection by immersion in liquid nitrogen.
Structure Determination of PCSK9-ΔCRD·Pep2-8 by X-ray Crystallography
Diffraction data extending to 1.85 Å resolution were collected at synchrotron beamline SSRL 9-2 and integrated and scaled (29, 30) in space group C2. Approximate phases were obtained by the method of molecular replacement (31), using a truncated version of a previously reported structure of PCSK9 (15) (Protein Data Bank code 2QTW). Electron density maps and intermediate molecular models were manipulated using Coot (32), and refinement was performed using Phenix.refine (33) and Refmac5 (32).
Structure Determination of Pep2-8 in Solution Using NMR
Nuclear magnetic resonance experiments were carried out on a Bruker DRX-600 spectrometer equipped with a triple-resonance cryoprobe. Samples for NMR studies were prepared by dissolving Pep2-8 in PBS (5% D2O) to a final concentration of 1.2 mm. The NMR spectra were collected at 298 K and internally referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid. Complete assignment of the proton resonances was achieved using a combination of two-dimensional total correlation spectroscopy, two-dimensional double-quantum-filtered COSY, and two-dimensional NOESY experiments. The mixing times for the total correlation spectroscopy and NOESY spectra were 98 and 150 ms, respectively. For the two-dimensional experiments, 2,048 and 512 data points were used in F2 and F1, respectively. NMR data were processed using the NMRPipe/NMRDraw package (34) and analyzed with NMRView (35). The interproton distances were estimated from the intensities of the cross-peaks observed in the NOESY experiment and classified as strong (1.8–2.8 Å), medium (1.8–3.4 Å), and weak (1.8–5.0 Å).
Structure calculations were performed with the program CNS, starting from an extended structure and using a combination of torsion angle dynamics and cartesian dynamics. A total of 127 NOE-derived interproton distances were used as experimental restraints to calculate a set of 30 structures, all of them satisfying the experimental constraints, with no NOE violations greater than 0.2 Å. From this set, the 20 structures with the lowest energies were selected for further analysis. The quality of the structures was assessed using Procheck-NMR (36). The figures representing the structures were generated using the programs MOLMOL and PyMOL.
RESULTS
Selection of PCSK9-binding Peptide by Peptide Phage Display
Two types of phage-displayed naive peptide libraries, Linear-lib and Cyclic-lib (25), were used to select for PCSK9 binding peptides by solution sorting against biotinylated PCSK9 in calcium-free buffer (26). After four rounds of panning, several clones gave modest binding signals. The peptides derived from these clones were synthesized and tested in the HepG2 LDL receptor degradation assay. We found that Pep2 (VVFTSWQDYLFWV) derived from clone 2 was able to inhibit PCSK9-mediated LDL receptor degradation with low potency (data not shown). A competition phage ELISA showed that Pep2 was a relatively weak PCSK9 binder (IC50 = 37.7 ± 1.0 μm) (Fig. 1A). To improve binding affinity, a soft-randomized library based on the Pep2 parent sequence was constructed. In this experiment, each position retained the parent amino acids at a 50% frequency. After four rounds of solution sorting, 80 phage clones showed improved binding to PCSK9 compared with the parent clone as measured by phage ELISA (data not shown). The sequence alignment, summarized in the sequence logo (Fig. 1B) of the 80 peptides, showed that Val2, Phe3, Ser5, Trp6, Tyr9, and Leu10 were highly conserved, suggesting that these residues are important for PCSK9 binding. Of note, many of the clones with high ELISA signals also replaced the parent residues Gln7 and Asp8 with a glutamate residue, indicating that the glutamate preference may account for the affinity improvements. Therefore, for further experiments, we chose Pep2-2, which gave the strongest ELISA signal and had glutamate residues in positions 7 and 8 (Table 1). The synthesized Pep2-2 bound to PCSK9 with 50-fold greater affinity than the parent peptide Pep2 in a phage competition ELISA (IC50 = 0.76 ± 0.01 μm) (Fig. 1A).
Structure-Activity Relationship of Pep2-2 and Design of Its Derivative Pep2-8
A panel of Pep2-2-based peptides in which each residue was individually replaced by alanine was synthesized and tested for PCSK9 binding in a competition phage ELISA (Fig. 1C). Alanine replacements of the three C-terminal residues Leu11, Trp12, and Val13 did not affect binding. In contrast, a dramatic binding loss (>1,000-fold) was observed for alanine substitutions of Phe3 and Trp6, suggesting that these side chains are necessary either for maintaining peptide structure or providing critical PCSK9 contacts or both. Strong effects (>20-fold loss) were also found for substitutions at Ser5, Glu8, and Tyr9.
The peptide alanine scanning results suggested that the three C-terminal residues may not be critical for the binding interaction. Therefore, we measured binding of C-terminally truncated versions of Pep2-2. Surprisingly, the sequential deletion of the four C-terminal residues resulted in decreasing binding levels, and binding became undetectable when all four residues were deleted (Table 1). Deletion of the N-terminal Thr1 resulted in a 6-fold binding loss, consistent with the reduced binding in the alanine scanning experiment. Therefore, Pep2-2 cannot be shortened without compromising binding affinity, indicating that all residues are necessary for optimal interaction with PCSK9.
The low solubility of Pep2-2 limited its further characterization in cell-based systems. Therefore, we changed Leu11 to Asp11, because the sequence logo indicated that aspartate is relatively preferred at this position (Fig. 1B). This new peptide, designated Pep2-8 (TVFTSWEEYLDWV), was synthesized and showed better solubility than Pep2-2 while maintaining a binding affinity that was comparable with that of Pep2-2 (Table 1). Pep2-8 was used for all subsequent studies.
Binding Affinity and Specificity of Pep2-8
For binding affinity measurements, we constructed a Pep2-8 Fc fusion protein (Pep2-8_Fc) that could be immobilized on Octet® anti-Fc-coated sensor tips for biolayer interferometry experiments. As a control, we made a peptide Fc fusion protein (Pep-ctrl_Fc) based on a Pep2-2 double mutant having the two important binding residues Phe3 and Trp6 replaced by alanine (Table 1). PCSK9 bound to Pep2-8_Fc with a KD value of 0.66 ± 0.11 μm (Fig. 2A), whereas the control Pep-ctrl_Fc did not give any binding signal up to 6 μm (data not shown). Furthermore, the same system was used to assess the specificity of Pep2-8 by measuring the binding to the related proprotein convertases furin, PC1, PC2, PC4, and PC7. Whereas PCSK9 gave a strong and specific binding response, the tested proprotein convertases showed only small and nonspecific binding signals (Fig. 2B), indicating that Pep2-8 specifically recognizes PCSK9.
FIGURE 2.
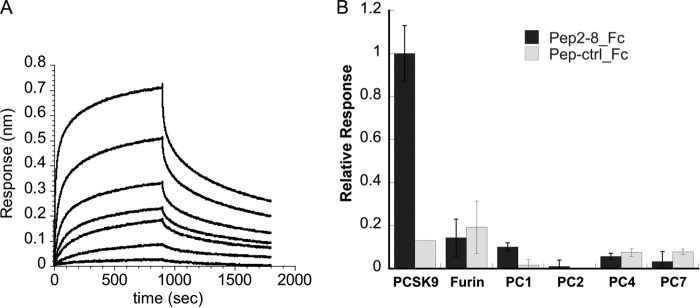
Binding affinity and specificity of Pep2-8. A, binding kinetics of Pep2-2. The binding of serially diluted PCSK9 to Pep2-8-Fc protein immobilized on sensor tip was measured by biolayer interferometry. The determined KD for Pep2-8 was 0.66 ± 0.11 μm (average ± S.D. (error bars) of three experiments). B, binding specificity of Pep2-8. The binding of sensor-captured Pep2-8-Fc and Pep-ctrl-Fc to PCSK9 and other proprotein convertases was measured by biolayer interferometry. The association responses were normalized to the PCSK9 response measured in parallel. The results are the average ± S.D. of three experiments.
Pep2-8 Restores LDL Receptor Function in HepG2 Cells
The addition of PCSK9 to HepG2 cells reduced surface LDL receptor levels by about 75%, as determined by FACS. Co-incubation with Pep2-8 restored LDL receptor surface levels in a concentration-dependent manner (Fig. 3A). The control peptide showed no effect. Moreover, Pep2-8 restored LDL uptake of PCSK9-treated HepG2 cells to about 90% of control activity at a concentration of 50 μm, whereas the control peptide showed no effect (Fig. 3B). The half-maximal effect (EC50) for LDL uptake was 6–12.5 μm, which was similar to the EC50 value of ∼12.5 μm for surface LDL receptor restoration. The results indicated that Pep2-8 was able to neutralize PCSK9 activity, resulting in the functional recovery of cellular LDL receptor.
FIGURE 3.
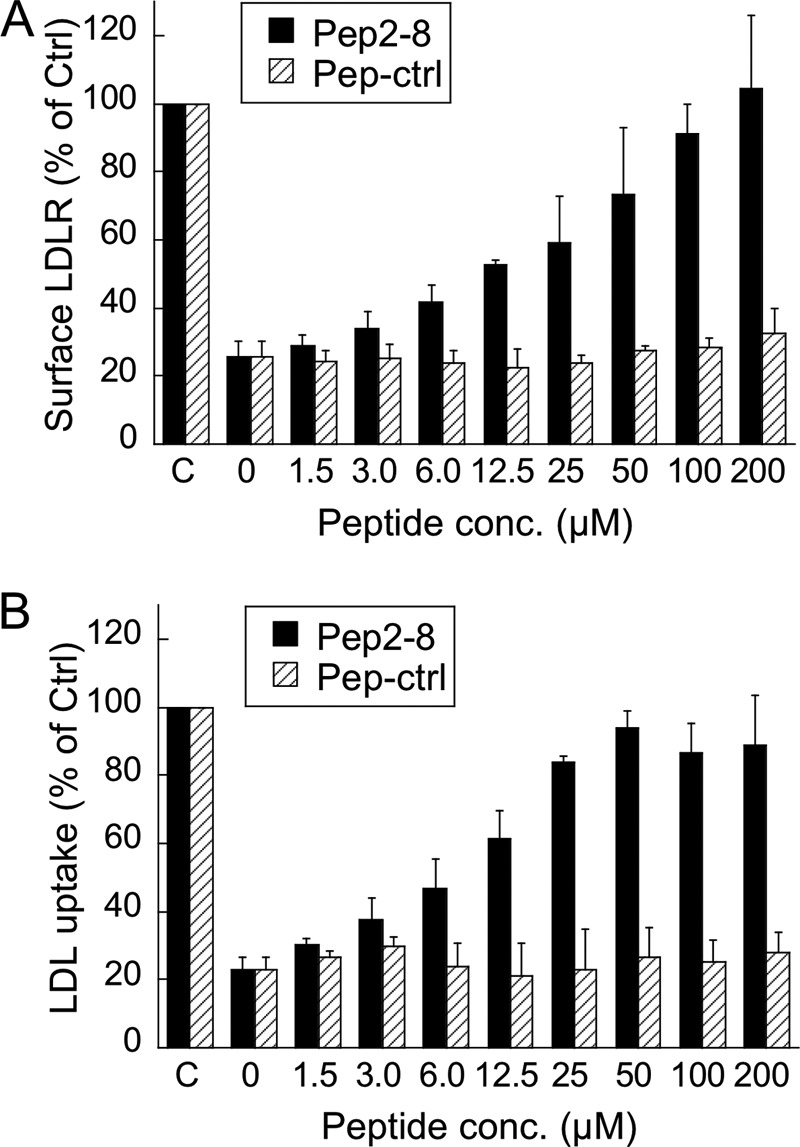
Inhibition of PCSK9 activity by Pep2-8 in HepG2 cell assays. A, cell surface LDL receptor levels in HepG2 cells. HepG2 cells were treated for 4 h with 15 μg/ml PCSK9 and increasing concentrations of Pep2-8 (black bars) or a control peptide (shaded bars), and LDL receptor surface levels were quantified by flow cytometry. B, LDL uptake by HepG2 cells. HepG2 cells were treated with 15 μg/ml of PCSK9 and peptides (as in A) for 1.5 h, after which fluorescently labeled LDL was added. After 3.5 h, the LDL uptake was quantified by fluorescence measurements. LDLR, LDL receptor. Error bars, S.D.
Pep2-8 Interferes with LDL Receptor Binding to PCSK9
PCSK9-mediated LDL receptor degradation involves all PCSK9 domains (5, 19, 20, 23). To understand how Pep2-8 interfered with LDL receptor degradation, we sought to identify the peptide-binding site on PCSK9. We found that Pep2-8 bound to PCSK9 lacking the CRD (PCSK9-ΔCRD), suggesting that the CRD does not harbor the peptide-binding site (data not shown). Furthermore, in a competition binding ELISA, Pep2-8 inhibited binding of PCSK9 to LDL receptor-Fc with an IC50 value of 0.81 ± 0.08 μm, whereas Pep-ctrl had no effect (Fig. 4A). A similar assay was used to examine whether Pep2-8 interfered with PCSK9 binding to the EGF(A) domain of LDL receptor. Pep2-8 completely inhibited this interaction with an IC50 value of 0.44 ± 0.04 μm, whereas Pep-ctrl showed no inhibition (Fig. 4B). The Pep2-8 binding site was further elucidated by measuring binding of Pep2-8 to a panel of PCSK9 mutants with alanine changes within or close to the known EGF(A) binding region on the PCSK9 catalytic domain, including Phe379, which is at the center of the EGF(A) binding site (20, 37). Analysis of each purified mutant by SDS-PAGE and by N-terminal sequencing indicated that the introduced mutations did not affect pro-PCSK9 processing at Gln152–Ser153 during biosynthesis, yielding mature PCSK9 proteins (data not shown). The binding ELISA showed that many of the mutant proteins, including F379A, had >50% reduced binding to PCSK9 (Fig. 4C), suggesting that the Pep2-8 binding site overlaps with that of EGF(A).
FIGURE 4.
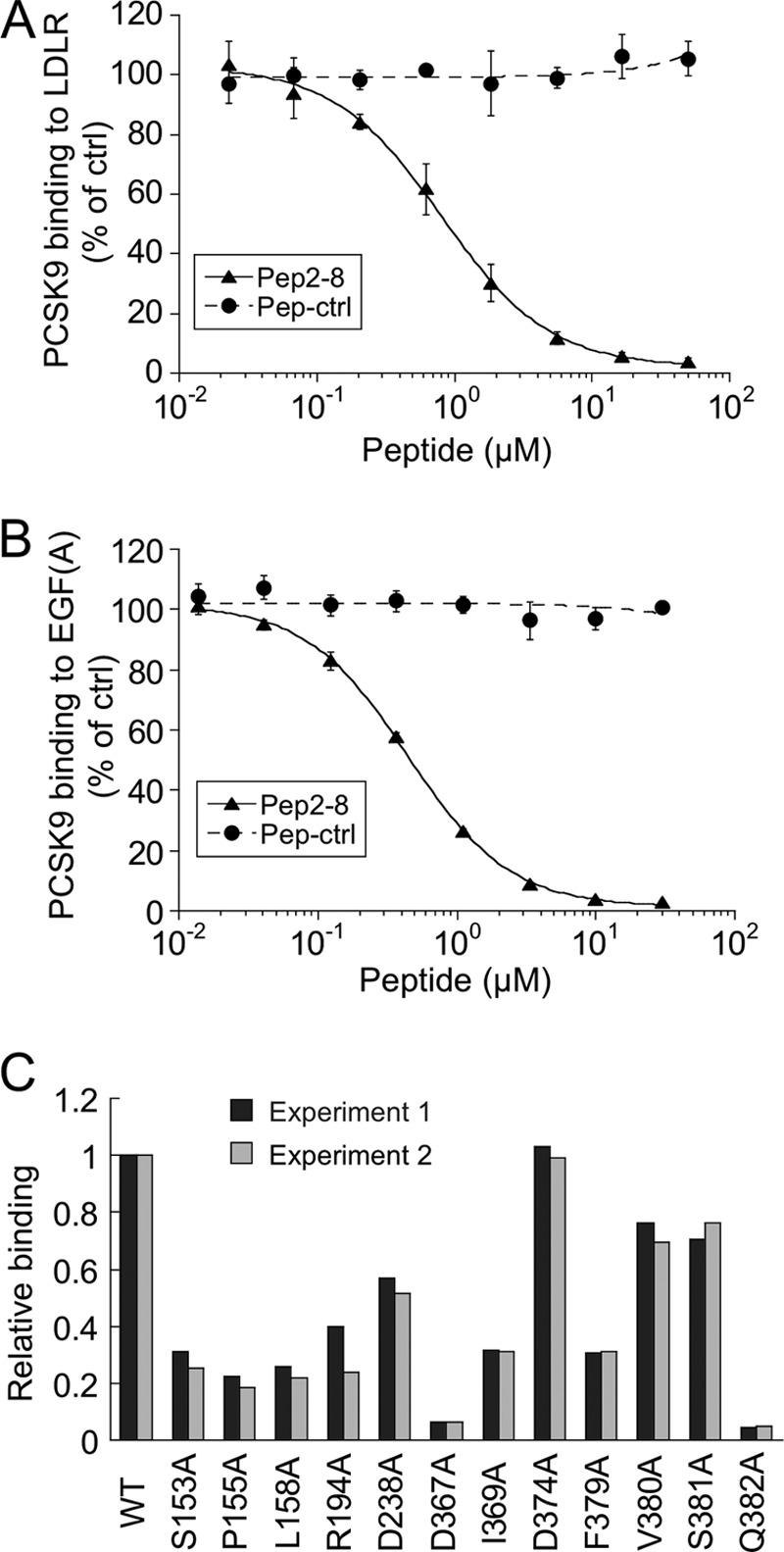
Pep2-8 competes with LDL receptor binding to PCSK9. A, inhibition of PCSK9 binding to LDL receptor. The binding of PCSK9 to plates coated with LDL receptor-Fc protein was measured in the presence of increasing concentrations of Pep2-8 (filled triangles) or a control peptide (filled circles; Pep-ctrl). The IC50 calculated for Pep2-8 was 0.81 ± 0.08 μm (average ± S.D. of three independent experiments). Error bars, S.D. B, inhibition of PCSK9 binding to EGF(A)-Fc. The binding of PCSK9 to plates coated with EGF(A)-Fc protein was measured in the presence of increasing concentrations of Pep2-8 (filled triangles) or a control peptide (filled circles; Pep-ctrl). The IC50 calculated for Pep2-8 was 0.44 ± 0.04 μm (average ± S.D. of three independent experiments). Error bars, S.D. C, mapping of Pep2-8 binding site by Ala scan. NeutrAvidin-coated 384-well Maxisorp ImmunoPlates were coated with biotinylated Pep2-8, and the binding of serially diluted wild type and PCSK9 mutant proteins (all with C-terminal His tag) was quantified by using HRP-conjugated anti-His antibody. The PCSK9 concentrations at 50% binding saturation (EC50) were determined and normalized to the wild type value (EC50 wild type/EC50 mutant; Relative binding). Shown are the results of two independent experiments.
Crystal Structure of the PCSK9-ΔCRD·Pep2-8 Complex and Solution Structure of Pep2-8 by NMR
In order to obtain a more detailed insight into the binding site and the inhibitory mechanism of Pep2-8, we determined the crystal structure of Pep2-8 bound to PCSK9-ΔCRD at 1.85 Å resolution (Table 2). Despite numerous close intermolecular contacts characterizing our crystals (Fig. 5), the overall similarity to previously reported PCSK9 structures (14, 37) is very high, with r.m.s. deviations from superposed Cα atoms, ignoring a few outliers, of less than 0.5 Å. The entire Pep2-8 is resolved from residue acetyl-Thr1 to amidyl-Val13 and interacts with PCSK9 residues that are in the catalytic domain and that are also directly contacted by the EGF(A) domain of LDL receptor (20) (Fig. 6, A and B). The surface of PCSK9 made solvent-inaccessible by Pep2-8 is smaller than that by EGF(A) (420 versus 520 Å2), but there is a strong overlap in the Pep2-8 and EGF(A) binding surfaces on PCSK9 (Fig. 6, C and D). Among the PCSK9 residues central to both interfaces are Ile369 and Phe379, which also showed effects on Pep2-8 binding in the alanine changes (Fig. 4C). Pep2-8 residues Thr1–Ser5 adopt an extended main chain conformation, with Phe3 main chain torsion angles appropriate for a β-strand. Starting with the carbonyl of Ser5 and until the end at Val13, the peptide is an α-helix (Fig. 6C). Residues Glu7, Glu8, and Asp11 are the only peptide residues with a formal charge at neutral pH, and thus an overall negative electrostatic potential is concentrated on one side of the helix. The opposite side of the helix contains the side chains from residues Trp6, Tyr9, Leu10, and Trp12. In addition, the side chain of Phe3 has a small van der Waals contact with the side chains of Tyr9 and Trp12, and the Val2 side chain is adjacent, so these residues thereby contribute to the hydrophobic surface on this side of the peptide and form the major interaction site with PCSK9 as described in more detail below.
TABLE 2.
X-ray data collection and refinement for PCSK9ΔCRD/pep2-8
| Data collection (SSRL 9-2) | |
| Space group | C2 |
| Unit cell | a = 95.10 Å, b = 70.76 Å, c = 70.41 Å, β = 96.07° |
| VM (Å3/dalton) | 2.4 |
| Resolution (Å) | 50-1.85 (1.92-1.85) |
| Rsyma,b | 0.099 (0.506) |
| No. of observations | 150,624 |
| Unique reflections | 39,638 |
| Completeness (%)b | 99.9 (99.5) |
| I/σIb | 11 (2.2) |
| Wilson B (Å2) | 15 |
| Refinement | |
| Resolution (Å) | 50–1.85 |
| Reflections (F > 0σ(F)) | 38618 |
| Final Rc, RFREE | 0.187, 0.228 |
| Molecules/asymmetric unit | 1 |
| Protein residues | 381 |
| Solvent molecules | 378 |
| Atomsd | 3348 (110) |
| Mean B-factor (Å2) | 10 |
| r.m.s. deviation | |
| Bonds (Å) | 0.010 |
| Angles (degrees) | 1.1 |
| Bonded atom B-factors (Å2) | 4.0 |
| No. of TLS groups | 8 |
| Ramachandran (%) | 96/3/1 |
a Rsym = Σ‖I| − |〈I〉‖/Σ |〈I〉|, where I is the intensity of a single observation and 〈I〈 is the average intensity for symmetry-equivalent observations.
b Values for the highest resolution shell are shown in parenthesis.
c r = Σ |Fo − Fc|/Σ|Fo|, where Fo and Fc are observed and calculated structure factor amplitudes, respectively. RFREE is calculated as R for reflections sequestered from refinement.
d The number of atoms assigned less than unit occupancy is shown in parenthesis.
FIGURE 5.
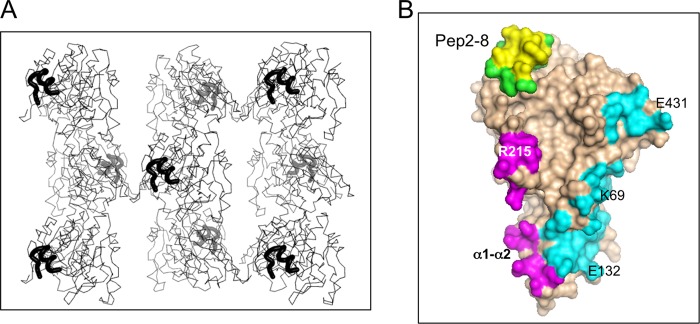
X-ray structure of Pep2-8·PCSK9-ΔCRD; crystal packing contacts. A, Cα traces of Pep2-8·PCSK9-ΔCRD are viewed along the crystal c axis (also the longer axis of the molecule), with Pep2-8 shown as a thick black line. Viewed this way, opposite ends (Pep2-8 binding site and prodomain) alternate foreground/background across and down the figure. B, the 4-Å contact zones from crystal neighbors are shown colored according to different neighboring molecules. Pep2-8 provides an intermolecular contact (yellow) but is otherwise colored green. The neighboring molecule providing the contact colored yellow is omitted from A for clarity.
FIGURE 6.

Crystal structure of the Pep2-8·PCSK9-ΔCRD complex. A and B, Pep2-8 binds the catalytic domain of PCSK9-ΔCRD at a region also used by the LDL receptor for binding to PCSK9. A, Pep2-8 (green) is a 13-amino acid peptide composed of a β-strand followed by an α-helix that presents mostly aromatic side chains to the PCSK9 surface (beige). The orange surface is the prodomain of PCSK9. B, the LDL receptor from Protein Data Bank entry 3P5C (gray) is shown with the Pep2-8·PCSK9-ΔCRD complex, after using the catalytic domains for superpositioning. Other colors are as for A, and Pep2-8 is shown as a green mesh. Note that elements of the LDL receptor are inside the Pep2-8 mesh. The yellow spheres represent Ca+2 ions associated with LDL receptor. C and D, close correspondence between PCSK9 surfaces contacted by Pep2-8 and the EGF(A) domain of LDL receptor. C, colored red is the PCSK9 surface (beige) within 4 Å of Pep2-8 (green). Pep2-8 side chains within 4 Å of PCSK9 are included as sticks and are labeled (large type). PCSK9 side chains are labeled in small type. D, colored red is the PCSK9 surface (beige) within 4 Å of EGF(A) (gray), from Protein Data Bank entry 3BPS. The yellow sphere represents a Ca+2 ion as part of the EGF(A) structure. The PCSK9 molecules are displayed in the same orientation after superposition, and selected PCSK9 residues are labeled.
The chemical shift dispersion of the NMR resonances and the presence of a substantial number of interresidue NOE contacts indicated that Pep2-8 does adopt a preferred fold when free in solution (Table 3). The solution conformation of Pep2-8 determined from the NMR data shows a remarkable correspondence to the Pep2-8 structure bound to PCSK9 (Fig. 7, A and B). The lowest energy solution structure determined using NMR, after superposition with the peptide from the x-ray structure using 13 Cα atoms, has an r.m.s. deviation of only 0.78 Å. The largest deviations between pairs of Cα atoms occur for Thr1 (1.3 Å) and Glu8 (1.2 Å). Superpositioning of only the α-helical residues yields an r.m.s. deviation of 0.31 Å, and superpositioning of residues 1–5 yields an r.m.s. deviation of only 0.27 Å. Most of the main chain torsion angles (ϕ,ψ) in the x-ray peptide coordinates are very close to those in the NMR coordinates, the two outliers being Thr4 ϕ (Δ = 36°) and Trp6 ϕ (Δ = 32°). Thus, the conformation of Pep2-8 when free or when bound to PCSK9 is a short extended N-terminal segment followed by an α-helix. Similar strand-loop-helix conformations have been described previously for small phage-derived peptides (38–40), although these examples were stabilized by a disulfide bond to tether the N terminus to the center of the helix. Pep2-8 lacks such a covalent stabilizing element, and in lieu of a disulfide bond, van der Waals contacts from Phe3 to Tyr9 and Trp12 serve to tether the N-terminal region and probably stabilize the helix.
TABLE 3.
Summary of the input restraint, statistics of the resulting structures, and restraint violations for Pep2-8 in solution resolved with NMR
| Restraints used for the structure calculations | |
| Total no. of NOE distance restraints | 127 |
| Short range (intraresidue) | 36 |
| Medium range (|i − j| ≤ 4) | 82 |
| Long range (|i − j| > 4) | 9 |
| Structural statistics | |
| r.m.s. deviations from idealized geometry | |
| Bonds (Å) | 0.0008 ± 0.0001 |
| Angles (degrees) | 0.204 ± 0.002 |
| Impropers (degrees) | 0.081 ± 0.001 |
| r.m.s. deviations from distance restraints (Å) | 0.007 ± 0.002 |
| Ramachandran statistics (%)a | |
| Residues in most favored regions | 74.1 |
| Residues in additional allowed regions | 25.5 |
| Residues in generously allowed regions | 0.5 |
| Residues in disallowed regions | 0.0 |
| Atomic pairwise r.m.s. deviation (Å) | |
| For backbone atoms (C′, Cα, N)b | 0.36 ± 0.13 |
| For all heavy atomsb | 1.1 ± 0.2 |
a Based on PROCHECK-NMR analysis.
b Residues Thr1 and Val2 at the N terminus were not included in the calculation.
FIGURE 7.
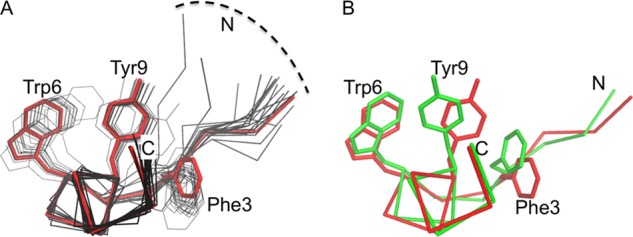
Pep2-8 solution structure by NMR and from the Pep2-8·PCSK9-ΔCRD complex x-ray structure. Peptide backbone shown with Cα atoms only. The aromatic amino acid side chains Phe3, Trp6, and Tyr9 are shown. Side chain atoms of Trp12 are omitted for clarity. N, N terminus; C, C terminus. A, the 20 lowest energy NMR structures describe a consistent peptide conformation (pairwise root mean square deviation for backbone atoms 0.36 ± 0.13 Å (Table 3)). The lowest energy NMR structure is in red, and the next 19 lowest energy models from NMR are shown in black. B, the Pep2-8 structure from the Pep2-8·PCSK9-ΔCRD x-ray structure (green) is essentially the same as the lowest energy solution structure from NMR (red).
Pep2-8 Interaction with PCSK9 and Its Structural Mimicry of the EGF(A) Domain
The hydrophobic face of Pep2-8 contacts PCSK9, being within 4 Å of residues in PCSK9 β-strands β13 (Ile368–Ala371) and β14 (Phe379–Gln382) and the loop between them, the loop between α4 (Ser225–Ser235) and β7 (Ser246–Arg251), and Pro155 near the start of α5 (Fig. 8). The Pep2-8 Trp6 side chain is within 4 Å of carbon atoms of Asp238, Ala239, and Phe379. The Pep2-8 Tyr9 side chain is within 4 Å of Ile369. Of the remaining peptide side chains comprising the hydrophobic side of the peptide, Thr1 is 3.7 Å from the Cβ atom of Cys378, and Phe3 is 4.1 Å from Ile369, but Trp12 and Val13 do not contact PCSK9. There are only three hydrogen bonds directly between Pep2-8 and PCSK9; two reciprocal β-sheet hydrogen bonds between Pep2-8 Phe3 and PCSK9 Phe379, and one between the main chain oxygen atom of Pep2-8 Thr4 and the side chain of PCSK9 Thr377. There is a single instance of one water molecule bridging Pep2-8 and PCSK9 (Thr1 and Ser381, respectively) and also several multiwater bridges (e.g. that involving the Pep2-8 Tyr9 side chain hydroxyl).
FIGURE 8.

Open book views of Pep2-8 and EGF(A) interaction with PCSK9. There is a close correspondence between PCSK9 surfaces contacted by Pep2-8 and the EGF(A) domain of LDL receptor. A, an open book presentation of the PCSK9 (beige)/Pep2-8 (green) interface. Molecular surfaces are augmented with dots for atoms where the partner protein is within 3.5 Å (red) or 4.0 Å (yellow). Black amino acids are those from the partner protein and within 4 Å. The Pep2-8 residue Trp6 is labeled in the right-hand image to aid in orienting the reader. B, an open book presentation of the PCSK9 (beige)/EGF(A) (gray) interface, with colors analogous to A. Note the very close correspondence of PCSK9 residues presented in black on the right-hand images.
The binding site identified by a PCSK9 alanine scan is imperfectly consistent with the structural contact region seen in the x-ray structure. For instance, the two largest hits, Asp367 and Gln382, are not contact residues. Rather they are at the start and the end, respectively, of β-stands β13 and β14, which together with the loop between them provide several contact residues to Pep2-8. However, Pro155 is a contact residue for both Trp6 and Tyr9, and it seems likely that the effects of alanine substitutions for Ser153 or Leu158 are best understood in terms of an indirect effect on Pro155, inasmuch as the native side chains stabilize and/or orient the catalytic domain N terminus, which is also important for LDL receptor binding (20, 37).
Of the 10 PCSK9 residues within 4 Å of Pep2-8, nine are also among the 13 residues within 4 Å of EGF(A) (i.e. Pro155, Asp238, Ile369, Ser372, Asp374, Thr377, Cys378, Phe379, and Val380 (Fig. 8). Pep2-8 and EGF(A) use the same combination of secondary structure elements, a strand and α-helix, to establish complementary surfaces capable of binding strongly overlapping surfaces on PCSK9 (Fig. 9A). Structural mimicry between Pep2-8 and EGF(A) is strongest where they each form β-sheet hydrogen bonds with PCSK9 β-strand β14 (Fig. 9, B and C). The fidelity of this correspondence is quite high, as it must be if both involve interactions characteristic of a β-sheet, and accordingly the main chains, rather than side chains, dominate contacts. Additional interactions for both Pep2-8 and EGF(A) come from helical segments via their respective side chains, which project toward PCSK9. Here the correspondence is far from perfect; the Pep2-8 helix is longer, and the helical axes diverge by about 45° from parallel (Fig. 9A). The best side chain correspondence is seen between Pep2-8 Trp6 and EGF(A) Leu298, which project into a closely similar region of PCSK9 (Fig. 9A).
FIGURE 9.

Structural mimicry of the EGF(A) domain by Pep2-8. EGF(A) and Pep2-8 employ related collections of secondary structure elements to establish a complementary surface. A, Pep2-8 (green) provides a short β-strand, a turn, and short α-helix. The analogous segments of EGF(A) (gray) are a short β-strand and a short helix from discontinuous segments of the domain. B, there is high correspondence between the ways Pep2-8 and EGF(A) interact with PCSK9 β-strand β14. Pep2-8 (green) has β-sheet type hydrogen bonds (black dashes) with PCSK9 (beige) and an additional hydrogen bond with the side chain of PCSK9 Thr377. The same segment of PCSK9 is shown with a β-strand from EGF(A) (gray), where almost identical interactions are apparent (red dashes). EGF(A) has one additional hydrogen bond not present in the Pep2-8 complex. Some atoms are omitted for clarity. C, Pep2-8 (green) main chain mimics the analogous segment of EGF(A) (gray).
DISCUSSION
PCSK9 has emerged as an important regulator of LDL-c metabolism, and current clinical studies with injectable PCSK9 inhibitors, such as antibodies, RNAi, and locked nucleic acids, hold great promise for the treatment of coronary heart disease (reviewed in Refs. 13 and 41). Undoubtedly, an orally available small molecule inhibitor of PCSK9, due to lower cost and ease of administration, would be a highly desirable alternative therapeutic agent. Pharmacologic approaches to inhibit PCSK9 with a small molecule appear challenging, because PCSK9 function is governed by protein-protein interactions. Phage display has been used successfully to identify peptides that disrupt protein-protein interactions (25, 38–40, 42, 43), and for peptides targeting the antiapoptotic Bcl-2 family members, it established a structural basis to synthesize organic small molecules for therapeutic applications (44, 45). By screening linear and constrained phage-displayed peptide libraries against PCSK9, we were able to identify Pep2-8, which neutralized PCSK9 activity and restored LDL receptor function in cellular assays.
Structural and biochemical studies revealed the molecular details of how Pep2-8 inhibits PCSK9. The peptide binding site largely overlapped with that of the EGF(A) domain; therefore, Pep2-8 acts a competitive inhibitor of LDL receptor binding. This is akin to the inhibition mechanism of anti-PCSK9 monoclonal antibodies, which also disrupt the interaction of the LDL receptor-EGF(A) domain with PCSK9 (46–49). However, unlike the extended epitope region of the antibodies, the Pep2-8 contact area is merely 420 Å2, which is even smaller than that of the EGF(A) domain. Therefore, the 13-amino acid peptide is, to our knowledge, the smallest PCSK9 inhibitor with a defined mechanism of inhibition. Other described polypeptide inhibitors include the EGF(A) domain and variants thereof (26, 37, 50, 51), the PCSK9 C-terminal domain (52), and annexin A2 (23). Peptides derived from the annexin A2-R1 domain directly inhibited PCSK9 binding to LDL receptor, but they required a length of 73 amino acids for binding interactions in the submicromolar concentration range (23). Another report showed activity of PCSK9 prodomain-derived linear peptides but without any insight into the molecular mechanisms (53).
Despite the small size of the Pep2-8 peptide, it has a well defined structure free in solution, comprising an N-terminal extended strand and turn followed by a two-turn helix. Moreover, the peptide fold is very similar in the absence or presence of PCSK9. Adopting the bound conformation in the absence of target protein allows this particular peptide to minimize the entropic losses ordinarily associated with transferring a ligand from a conformationally diverse free state to a restricted conformation when bound. The phage selection method seems particularly adept at selecting such conformationally constrained peptides, with the N-terminal strand/turn followed by a C-terminal helix being a particularly common motif (38–40). In three reported cases (38–40), the peptides also have well defined conformations in the absence of their binding partner. When comparing these strand-turn-helix peptides, a number of common structural features are found that probably contribute to their structural stability: (i) tethering the N-terminal strand to the helix to position the helix capping motif; (ii) helix capping motifs to help initiate the helical conformation; and (iii) predominantly hydrophobic residues along one face of the helix (Fig. 10). The recurrence of this motif suggests that it is one of the few ways in which a stable fold can be obtained in a peptide of ∼15 residues.
FIGURE 10.

Comparison of the Pep-2-8 fold with other strand-helix structures derived from phage display. Shown are structures of the strand-turn-helix peptides that bind to PCSK9 (Pep2-8; A), IGFBP1 (B) (39), IGF1 (C) (40), and FVIIa (D) (38). In each case, the N terminus is on the left, and the helix is on the right. The top panels show the side view of the helix, and the bottom panels show the “end on.” Side chains are depicted for those residues involved in tethering the N-terminal strand to the helix, participating in helix-cap motifs, or that form the hydrophobic surface on one face of the helix; other side chains are omitted for clarity. Helix-cap hydrogen bonds are shown in magenta, whereas helical-hydrogen bonds are in yellow. Side chains in the N-terminal strand involved in hydrophobic contacts with the helix are in green. These structures reveal a number of common features that probably contribute to the stability of the peptide fold: (i) tethering the N-terminal strand to the helix to position the helix capping motif (in the literature examples, this is achieved via a disulfide bond, whereas in Pep2-8, the hydrophobic interaction of Phe3 and hydrogen bonds from Thr4 to the side chain of Glu8 achieve the same result); (ii) helix capping motifs to help initiate the helical conformation (e.g. in Pep2-8, hydrogen bond from Ser5 side chain to the backbone amide of Glu8 and the presence of the Glu7 side chain to hydrogen-bond to its own backbone amide NH and to provide a favorable interaction with the helix dipole); (iii) predominantly hydrophobic residues along one face of the helix; the Van der Waals contacts between residues on adjacent turns help to propagate the helix and also strengthen the i, i − 4 hydrogen bonds by sequestering them from solvent. The arrangement of strand-loop-helix provides a scaffold from which to present a variety of relatively flat hydrophobic surfaces (bottom panels) for interacting with partner proteins.
In the three cases described above (38–40), the peptides bind at a protein site ordinarily used for protein-protein interactions. Given the often flat and hydrophobic nature of such surfaces, the strand-turn-helix peptide motif seems to be particularly adept at complementing these regions, with the surface formed by the strand and the hydrophobic face of the helix providing a relatively flat surface with which to interact with the target protein. The N-terminal strand size and composition and the particular residues on the helix provide sufficient diversity for such peptides to bind selectively to a variety of protein targets. In the case of Pep2-8, the strand-turn-helix conformation positions the side chains of three key interacting residues, Phe3, Trp6, and Tyr9, in close proximity to form a central hydrophobic binding area. The important contributions of these residues to PCSK9 binding are also reflected in the >1,000-fold binding loss as a result of Alanine substitutions of Phe3 and Trp6. Thus, the hydrophobic patch on one side of the Pep2-8 serves a dual role to stabilize the structure (with entropic benefit for binding) and to also form van der Waals contacts with the partner protein. The length of Pep2-8 could not be reduced without inflicting a substantial affinity loss, but alanine substitutions of the three C-terminal residues Asp11, Trp12, and Val13 had no binding effects. The crystal structure explains this paradoxical finding and shows that side chains from these peptide positions do not contact PCSK9, consistent with the Ala scan results. Nonetheless, these residues extend the α-helix and thereby stabilize it and in this way indirectly influence the preceding residues in the helix that do contact PCSK9.
The structural details revealed an unexpected mimicry between Pep2-8 and important PCSK9-interacting elements of the EGF(A) domain. Pep2-8 uses its N-terminal extended segment to recreate the anti-parallel β-sheet main chain interactions between EGF(A) residues 305–310 and PCSK9 residues 379–382 (β14). In addition, the Pep2-8 α-helix provides additional contacts to PCSK9, as does the EGF(A) helical segment running from 295 to 301, although we note that the backbone atoms of these helical segments are not well aligned structurally. The peptide geometry allows Phe3 and Tyr9 of Pep2-8 to make favorable hydrophobic contacts with Ile369 and Phe379 of PCSK9. Asn301 in the helical segment of EGF(A) is involved in two intramolecular hydrogen bonds, but the polar nature of its side chain suggests that it is not making productive contacts with PCSK9. Interestingly, replacing Asn301 by a leucine has been shown to improve the binding to PCSK9 (26). Thus, the phage selection process led to a peptide with a binding site on PCSK9 that strongly overlaps that of a biological binding partner and in doing so yielded two structural elements that mimic the key structural elements of the biological binding partner while improving some of the specific contacts that are unproductive in the native complex.
The correspondence between PCSK9 residues contacted by Pep2-8 and EGF(A) is high (Fig. 8), and the amount of PCSK9 surface buried by Pep2-8 is about 80% of that buried by EGF(A), yet Pep2-8 is only 13 amino acids long versus about 40 residues for EGF(A). Thus, Pep2-8 is a minimized version of EGF(A) with roughly equivalent binding affinity (26, 37). The breadth of solutions to in vitro discovery of EGF(A) mimics is suggested by the reported structure of a phage-derived anti-PCSK9 antibody fragment 1D05 in complex with PCSK9 (48). This antibody also contacts PCSK9 residues contacted by EGF(A) and Pep2-8 and places key structure elements in rough analogy to the Pep2-8 extended N-terminal segment (for 1D05, the β-strand following the complementarity-determining region (CDR-H3) and C-terminal α-helix (for 1D05, CDR-H2) (Fig. 11). In this case, the structural analogy between the Pep2-8 helix and CDR-H2 from 1D05 (a β-hairpin loop) is obviously quite poor, and 1D05 has many additional aspects of its recognition of PCSK9 because it buries about twice the PCSK9 surface area of Pep2-8 and has an about 150-fold better binding than Pep2-8. Nonetheless, even as large and high affinity a PCSK9-binding protein as 1D05 has some elements in common with Pep2-8 and EGF(A). In PCSK9, the presence of a hydrophobic patch (centered on Ala239, Ile369, and Phe379) adjacent to backbone NH and CO groups with unsatisfied hydrogen bonds (strand β14) clearly provides an attractive binding site for peptides, proteins and antibodies. Due to the non-directional nature of van der Waals contacts, there is considerable variety in the side chain vectors that ligands use to interact with the hydrophobic patch, whereas the specific geometric constraints required for hydrogen bond interactions dictate that the optimal interactions require a complementary β-strand on the ligand.
FIGURE 11.
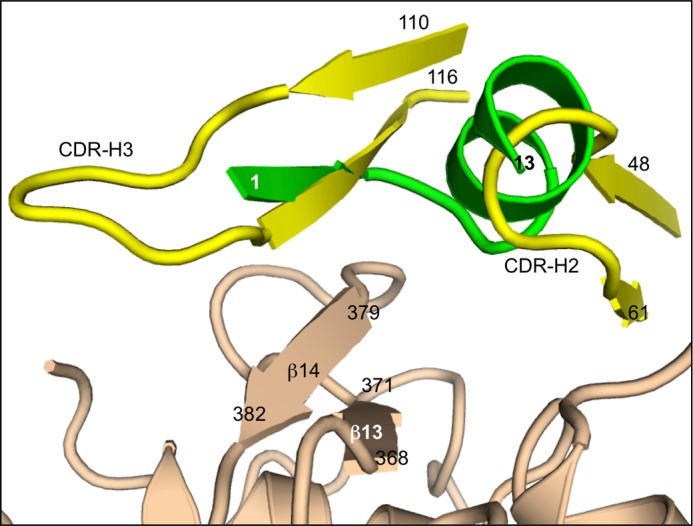
EGF(A) mimicry by antibody 1D05 in comparison with Pep2-8. Three PCSK9-binding proteins employ related collections of secondary structure elements to establish a complementary surface. Pep2-8 (green) provides a short β-strand, a short loop, and short α-helix. The analogous segments from antibody 1D05 (yellow) (48) are a β-strand leading from CDR-H3 and the initial loop of CDR-H2. PCSK9 β-strands formed by residues 368–371 and 379–382 and the loop connecting them provide important contacts.
Peptide phage display provides a robust tool to identify ligands for targets of biological interest. The libraries are economical to establish (utilizing DNA synthesis and molecular biology manipulations), selection experiments are straightforward to perform, and the use of DNA sequencing methods provides a facile route to identify binding elements within complex mixtures of up to 1011 unique members. However, phage display is limited by accessing only the diversity represented by the 20 naturally occurring amino acids. In contrast, synthetic combinatorial libraries offer a far greater scope for molecular diversity (non-natural amino acids, including d-stereochemistry; non-peptidic backbones; inclusion of moieties not present in naturally occurring biopolymers) with the caveat that they are more expensive to prepare and require more complex schemes to identify those molecules that bind to the target.
The work herein describes the identification of novel peptide ligands from phage libraries that are able to bind selectively to PCSK9 with an affinity comparable with the endogenous ligand EGF(A). The small size of the peptide (13 residues) serves to identify a focused epitope on PCSK9 and highlights key interactions that ligands must make to obtain a high binding energy. Although peptides are not usually considered as orally bioavailable drug candidates, several examples exist in the literature where peptides have served as the starting point for the successful development of small molecule drugs that exhibit oral bioavailability (44, 45, 54). Conceivably, Pep2-8 could provide such a steppingstone to go from protein to small molecule drug antagonists of the LDL receptor-PCSK9 interaction. However, the specific geometry of the EGF(A) binding site suggests that this will be challenging because the PCSK9 surface is relatively flat without concavities that are a prominent feature of potent, small molecule ligand binding sites. Indeed, significant optimization of Pep2 family members failed to identify peptides with IC50 much below 1 μm, suggesting that there may be an intrinsic limit on the energy available through interaction with this site. On the other hand, it is possible that the biostability of Pep2-8 could be improved by chemical modifications or the introduction of non-peptidic hydrocarbon backbones. Nonetheless, we remain optimistic that approaches such as the one described here may yield reagents and insights into ways of affecting PCSK9 function, possibly through interaction at allosteric sites or by binding to other domains of this large protein.
Acknowledgments
Diffraction data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL), operated for the United States Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research, National Institutes of Health, NIGMS (including Grant P41GM103393), and National Center for Research Resources Grant P41RR001209.
The atomic coordinates and structure factors (code 4NMX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LDL-c
- LDL cholesterol
- CRD
- C-terminal Cys-rich domain of PCSK9
- PCSK9-ΔCRD
- PCSK9 lacking the CRD
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- r.m.s.
- root mean square.
REFERENCES
- 1. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1). Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Costet P., Krempf M., Cariou B. (2008) PCSK9 and LDL cholesterol. Unravelling the target to design the bullet. Trends Biochem. Sci. 33, 426–434 [DOI] [PubMed] [Google Scholar]
- 3. Horton J. D., Cohen J. C., Hobbs H. H. (2009) PCSK9. A convertase that coordinates LDL catabolism. J. Lipid Res. 50, S172–S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seidah N. G. (2009) PCSK9 as a therapeutic target of dyslipidemia. Expert Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 5. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 6. Holla Ø. L., Cameron J., Tveten K., Strøm T. B., Berge K. E., Laerdahl J. K., Leren T. P. (2011) Role of the C-terminal domain of PCSK9 in degradation of the LDL receptors. J. Lipid Res. 52, 1787–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamamoto T., Lu C., Ryan R. O. (2011) A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J. Biol. Chem. 286, 5464–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tveten K., Holla Ø. L., Cameron J., Strøm T. B., Berge K. E., Laerdahl J. K., Leren T. P. (2012) Interaction between the ligand-binding domain of the LDL receptor and the C-terminal domain of PCSK9 is required for PCSK9 to remain bound to the LDL receptor during endosomal acidification. Hum. Mol. Genet. 21, 1402–1409 [DOI] [PubMed] [Google Scholar]
- 9. DeVay R. M., Shelton D. L., Liang H. (2013) Characterization of PCSK9 trafficking reveals a novel lysosomal targeting mechanism via APLP2. J. Biol. Chem. 288, 10805–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 11. Cohen J. C., Boerwinkle E., Mosley T. H., Jr., Hobbs H. H. (2006) Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272 [DOI] [PubMed] [Google Scholar]
- 12. Zhao Z., Tuakli-Wosornu Y., Lagace T. A., Kinch L., Grishin N. V., Horton J. D., Cohen J. C., Hobbs H. H. (2006) Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 79, 514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stein E. A., Swergold G. D. (2013) Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr. Atheroscler. Rep. 15, 310. [DOI] [PubMed] [Google Scholar]
- 14. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 15. Hampton E. N., Knuth M. W., Li J., Harris J. L., Lesley S. A., Spraggon G. (2007) The self-inhibited structure of full-length PCSK9 at 1.9 Å reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 104, 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. (2007) The crystal structure of PCSK9. A regulator of plasma LDL-cholesterol. Structure 15, 545–552 [DOI] [PubMed] [Google Scholar]
- 17. Li J., Tumanut C., Gavigan J. A., Huang W. J., Hampton E. N., Tumanut R., Suen K. F., Trauger J. W., Spraggon G., Lesley S. A., Liau G., Yowe D., Harris J. L. (2007) Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem. J. 406, 203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNutt M. C., Lagace T. A., Horton J. D. (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 19. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mayer G., Poirier S., Seidah N. G. (2008) Annexin A2 is C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J. Biol. Chem. 283, 31791–31801 [DOI] [PubMed] [Google Scholar]
- 22. Ni Y. G., Condra J. H., Orsatti L., Shen X., Di Marco S., Pandit S., Bottomley M. J., Ruggeri L., Cummings R. T., Cubbon R. M., Santoro J. C., Ehrhardt A., Lewis D., Fisher T. S., Ha S., Njimoluh L., Wood D. D., Hammond H. A., Wisniewski D., Volpari C., Noto A., Lo Surdo P., Hubbard B., Carfí A., Sitlani A. (2010) A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J. Biol. Chem. 285, 12882–12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seidah N. G., Poirier S., Denis M., Parker R., Miao B., Mapelli C., Prat A., Wassef H., Davignon J., Hajjar K. A., Mayer G. (2012) Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PLoS One 7, e41865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ross N. T., Katt W. P., Hamilton A. D. (2010) Synthetic mimetics of protein secondary structure domains. Phil. Trans. R. Soc. A. 368, 989–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stanger K., Steffek M., Zhou L., Pozniak C. D., Quan C., Franke Y., Tom J., Tam C., Krylova I., Elliott J. M., Lewcock J. W., Zhang Y., Murray J., Hannoush R. N. (2012) Allosteric peptides bind a caspase zymogen and mediate caspase tetramerization. Nat. Chem. Biol. 8, 655–660 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y., Zhou L., Kong-Beltran M., Li W., Moran P., Wang J., Quan C., Tom J., Kolumam G., Elliott J. M., Skelton N. J., Peterson A. S., Kirchhofer D. (2012) Calcium-independent inhibition of PCSK9 by affinity-improved variants of the LDL receptor EGF(A) domain. J. Mol. Biol. 422, 685–696 [DOI] [PubMed] [Google Scholar]
- 27. Tonikian R., Zhang Y., Boone C., Sidhu S. S. (2007) Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc. 2, 1368–1386 [DOI] [PubMed] [Google Scholar]
- 28. Kunkel T. A., Roberts J. D., Zakour R. A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 29. Otwinowski Z., Minor W. (1996) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 30. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 35. Johnson B. A., Blevins R. A. (1994) NMR View. A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 36. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR. Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 37. Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo Surdo P., Calzetta A., Noto A., Baysarowich J., Mattu M., Talamo F., De Francesco R., Sparrow C. P., Sitlani A., Carfí A. (2009) Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 38. Dennis M. S., Eigenbrot C., Skelton N. J., Ultsch M. H., Santell L., Dwyer M. A., O'Connell M. P., Lazarus R. A. (2000) Peptide exosite inhibitors of factor VIIa as anticoagulants. Nature 404, 465–470 [DOI] [PubMed] [Google Scholar]
- 39. Lowman H. B., Chen Y. M., Skelton N. J., Mortensen D. L., Tomlinson E. E., Sadick M. D., Robinson I. C., Clark R. G. (1998) Molecular mimics of insulin-like growth factor 1 (IGF-1) for inhibiting IGF-1. IGF-binding protein interactions. Biochemistry 37, 8870–8878 [DOI] [PubMed] [Google Scholar]
- 40. Schaffer M. L., Deshayes K., Nakamura G., Sidhu S., Skelton N. J. (2003) Complex with a phage display-derived peptide provides insight into the function of insulin-like growth factor I. Biochemistry 42, 9324–9334 [DOI] [PubMed] [Google Scholar]
- 41. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 42. Pan B., Li B., Russell S. J., Tom J. Y., Cochran A. G., Fairbrother W. J. (2002) Solution structure of a phage-derived peptide antagonist in complex with vascular endothelial growth factor. J. Mol. Biol. 316, 769–787 [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y., Appleton B. A., Wiesmann C., Lau T., Costa M., Hannoush R. N., Sidhu S. S. (2009) Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 5, 217–219 [DOI] [PubMed] [Google Scholar]
- 44. Franklin M. C., Kadkhodayan S., Ackerly H., Alexandru D., Distefano M. D., Elliott L. O., Flygare J. A., Mausisa G., Okawa D. C., Ong D., Vucic D., Deshayes K., Fairbrother W. J. (2003) Structure and function analysis of peptide antagonists of melanoma inhibitor of apoptosis (ML-IAP). Biochemistry 42, 8223–8231 [DOI] [PubMed] [Google Scholar]
- 45. Zobel K., Wang L., Varfolomeev E., Franklin M. C., Elliott L. O., Wallweber H. J., Okawa D. C., Flygare J. A., Vucic D., Fairbrother W. J., Deshayes K. (2006) Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 1, 525–533 [DOI] [PubMed] [Google Scholar]
- 46. Chan J. C., Piper D. E., Cao Q., Liu D., King C., Wang W., Tang J., Liu Q., Higbee J., Xia Z., Di Y., Shetterly S., Arimura Z., Salomonis H., Romanow W. G., Thibault S. T., Zhang R., Cao P., Yang X. P., Yu T., Lu M., Retter M. W., Kwon G., Henne K., Pan O., Tsai M. M., Fuchslocher B., Yang E., Zhou L., Lee K. J., Daris M., Sheng J., Wang Y., Shen W. D., Yeh W. C., Emery M., Walker N. P., Shan B., Schwarz M., Jackson S. M. (2009) A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 106, 9820–9825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liang H., Chaparro-Riggers J., Strop P., Geng T., Sutton J. E., Tsai D., Bai L., Abdiche Y., Dilley J., Yu J., Wu S., Chin S. M., Lee N. A., Rossi A., Lin J. C., Rajpal A., Pons J., Shelton D. L. (2012) Proprotein convertase substilisin/kexin type 9 antagonism reduces low-density lipoprotein cholesterol in statin-treated hypercholesterolemic nonhuman primates. J. Pharmacol. Exp. Ther. 340, 228–236 [DOI] [PubMed] [Google Scholar]
- 48. Ni Y. G., Di Marco S., Condra J. H., Peterson L. B., Wang W., Wang F., Pandit S., Hammond H. A., Rosa R., Cummings R. T., Wood D. D., Liu X., Bottomley M. J., Shen X., Cubbon R. M., Wang S. P., Johns D. G., Volpari C., Hamuro L., Chin J., Huang L., Zhao J. Z., Vitelli S., Haytko P., Wisniewski D., Mitnaul L. J., Sparrow C. P., Hubbard B., Carfí A., Sitlani A. (2011) A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J. Lipid Res. 52, 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stein E. A., Mellis S., Yancopoulos G. D., Stahl N., Logan D., Smith W. B., Lisbon E., Gutierrez M., Webb C., Wu R., Du Y., Kranz T., Gasparino E., Swergold G. D. (2012) Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 366, 1108–1118 [DOI] [PubMed] [Google Scholar]
- 50. Shan L., Pang L., Zhang R., Murgolo N. J., Lan H., Hedrick J. A. (2008) PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem. Biophys. Res. Commun. 375, 69–73 [DOI] [PubMed] [Google Scholar]
- 51. McNutt M. C., Kwon H. J., Chen C., Chen J. R., Horton J. D., Lagace T. A. (2009) Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J. Biol. Chem. 284, 10561–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Du F., Hui Y., Zhang M., Linton M. F., Fazio S., Fan D. (2011) Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J. Biol. Chem. 286, 43054–43061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Palmer-Smith H., Basak A. (2010) Regulatory effects of peptides from the pro and catalytic domains of proprotein convertase subtilisin/kexin 9 (PCSK9) on low-density lipoprotein receptor (LDL-R). Curr. Med. Chem. 17, 2168–2182 [DOI] [PubMed] [Google Scholar]
- 54. Andronati S. A., Karaseva T. L., Krysko A. A. (2004) Peptidomimetics - antagonists of the fibrinogen receptors. Molecular design, structures, properties and therapeutic applications. Curr. Med. Chem. 11, 1183–1211 [DOI] [PubMed] [Google Scholar]