

Abstract
Background:
Heat shock protein 90 (HSP90) has a key role in the maintenance of the cellular proteostasis. However, HSP90 is also involved in stabilisation of oncogenic client proteins and facilitates oncogene addiction and cancer cell survival. The development of HSP90 inhibitors for cancer treatment is an area of growing interest as such agents can affect multiple pathways that are linked to all hallmarks of cancer. This study aimed to test the hypothesis that targeting cysteine residues of HSP90 will lead to degradation of client proteins and inhibition of cancer cell proliferation.
Methods:
Combining chemical synthesis, biological evaluation, and structure–activity relationship analysis, we identified a new class of HSP90 inhibitors. Click chemistry and protease-mass spectrometry established the sites of modification of the chaperone.
Results:
The mildly electrophilic sulphoxythiocarbamate alkyne (STCA) selectively targets cysteine residues of HSP90, forming stable thiocarbamate adducts. Without interfering with the ATP-binding ability of the chaperone, STCA destabilises the client proteins RAF1, HER2, CDK1, CHK1, and mutant p53, and decreases proliferation of breast cancer cells. Addition of a phenyl or a tert-butyl group in tandem with the benzyl substituent at nitrogen increased the potency. A new compound, S-4, was identified as the most robust HSP90 inhibitor within a series of 19 derivatives.
Conclusion:
By virtue of their cysteine reactivity, sulphoxythiocarbamates target HSP90, causing destabilisation of its client oncoproteins and inhibiting cell proliferation.
Keywords: Hsp90, NRF2, cysteine, HSF1
Heat shock protein 90 (HSP90) is a ubiquitously expressed ATP-dependent molecular chaperone that is evolutionarily highly conserved (Prodromou, 2012). In an unstressed eukaryotic cell, 1–2% of the cytosolic protein pool is comprised of HSP90, making it one of the most abundant cellular proteins (Taipale et al, 2010). HSP90 and its co-chaperones, collectively known as the HSP90 complex machinery, are involved in the correct folding, stabilisation, and activation of a broad range of approximately 200 ‘client' proteins. Under normal conditions, the HSP90 complex machinery follows a dynamic chaperone cycle using the energy of ATP hydrolysis to fold proteins, which participate in trafficking, signal transduction, receptor maturation, and immunity. Furthermore, HSP90 is vital for the cellular responses to environmental changes, preserving the integrity of the proteome under conditions of stress (Taipale et al, 2010). Very recently, using C. elegans as a model organism, it was found that activation of HSP90 in response to an imbalance in proteostasis in one tissue functions in a cell-nonautonomous manner to initiate a protective response in adjacent tissues and restore the balance within the whole organism (van Oosten-Hawle et al, 2013).
In addition to its critical functions in the normal cell, HSP90 also acts as a molecular chaperone for a large number of oncogenic client proteins (Whitesell and Lindquist, 2005). One such client protein is HER2, a receptor tyrosine kinase that has an important role in cell growth and survival through regulating signal transduction pathways such as the mitogen-activated protein kinase (MAPK) pathway. Overexpression of HER2 has been linked to malignant transformation (Yoshida et al, 2011). Another client protein, the serine/threonine kinase RAF1, functions as a molecular link between the membrane-bound Ras GTPases and the MAPK signalling cascade, hence serving as a regulatory switch for cell fate decisions such as apoptosis and proliferation (Chen et al, 2011). Many of the HSP90 client oncoproteins are mutant versions and rely on the chaperone for their stability. Furthermore, compared with normal cells, cancer cells are subject to increased cellular stress such as hypoxia and acidosis, and hence are more heavily dependent on the HSP90 complex machinery to survive. Not surprisingly, cancer cells often have increased amounts of HSP90 contributing to 3–5% of the total amount of protein (Johnson et al, 2010). This highlights the importance of targeting HSP90 in cancer, and the development of HSP90 inhibitors for cancer treatment is actively being pursued (Trepel et al, 2010).
The naturally occurring benzoquinone ansamycin antibiotic geldanamycin (GA), its derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG), and several purine scaffold-based HSP90 inhibitors, target the ATP-binding pocket within the N-terminal domain of the chaperone (Figure 1A; Jhaveri et al, 2012). In contrast, the antibiotic novobiocin and its analogues, and the chemotherapeutic agent cisplatin, target the C-terminal domain of HSP90 (Itoh et al, 1999; Marcu et al, 2000; Rosenhagen et al, 2003; Zhao and Blagg, 2013). It has been speculated that, in addition to competing with ATP for binding to the N-terminal domain of HSP90, inhibitors such as GA and 17-AAG, because of the electrophilic properties of their benzoquinone moiety, may have the ability to react with cysteine residues of HSP90 and cause conformational changes that could further hinder the function of the chaperone (Beck et al, 2011). Modifications of cysteine residues of HSP90 by sulphhydryl-reactive small molecules have been shown to occur both in vitro and in vivo (Nardai et al, 2000; Carbone et al, 2005; Martinez-Ruiz et al, 2005; Shibata et al, 2011). HSP90 is among the proteins that are modified by the electrophilic lipid peroxidation product 4-hydroxy-2-nonenal (Jacobs and Marnett, 2007, 2010; Smathers et al, 2011) and by its azido- and alkynyl-tagged derivatives (Vila et al, 2008). As all cysteines of HSP90 are located within its middle domain, small molecules that react with those cysteines may represent a new class of inhibitors distinct from those that target either the N-terminal ATP-binding pocket or the C-terminal domain of the chaperone.
Figure 1.

STCA destabilises the HSP90 client proteins HER2, RAF1, and mutant p53, and stabilises WT p53. (A) Domain structure of human HSP90β. The N-terminal domain (NTD) is the site of binding of ATP and some co-chaperones. The middle domain (MD) is where many of the HSP90 client proteins and co-chaperones interact, and the C-terminal domain (CTD) contains a dimerisation motif. The white bars indicate the distribution and positions of the cysteine residues. (B–D) MCF7 cells (2.5 × 105 per well) in six-well plates were exposed to vehicle (0.1% acetonitrile) or STCA for 16 h. Cells were then lysed in RIPA buffer, proteins in aliquots from cell lysates were resolved by SDS–PAGE, transferred to immobilon-P, and probed with antibodies against HER2, HSP70 (B), RAF1(C), and glycogen synthase kinase 3 (GSK3)α/GSK3β (D). (E) MDA-MB-231 cells (5 × 105 per well) in six-well plates were treated with vehicle (0.1% acetonitrile) or STCA for 24 h. The levels of mutant p53 and HSP70 were detected by western blot analyses. (F) MCF7 cells (2.5 × 105 per well) in six-well plates were treated with vehicle (0.1% acetonitrile) or STCA for 24 h. The levels of WT p53 were detected by western blot analyses. (G) MCF7 cells were treated with vehicle (0.1% acetonitrile) or 75 μM STCA for 2, 4, 8 or 24 h. For all panels, the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were also determined as a loading control. The data represent results from three independent experiments.
It was previously shown that cysteine residues of coenzyme A (CoA)-utilising enzymes can be selectively targeted by a mildly electrophilic sulphoxythiocarbamate-CoA analogue with the formation of stable thiocarbamate adducts (Hwang et al, 2007). Subsequently, a series of sulphoxythiocarbamate compounds were synthesised based on the structure of the isothiocyanate sulphoraphane, one of the most potent naturally occurring inducers of the cytoprotective KEAP1/NRF2 pathway (Ahn et al, 2010). In addition to the KEAP1/NRF2 pathway, sulphoxythiocarbamate alkyne (STCA) also induces HSP70, a prototypic heat shock protein (Zhang et al, 2011). Induction of HSP70 by STCA is independent of NRF2 and KEAP1, but requires heat shock factor 1 (HSF1). One possible mechanism of activation of HSF1 by STCA is through binding and inhibiting HSP90 or HSP70, the major negative regulators of HSF1. Indeed, using a mass spectrometry-based proteomic approach, we identified HSP90 and HSP70 among the proteins modified when cells were exposed to STCA (Ahn et al, 2010). We further confirmed that HSP90 is a bona fide cellular target of STCA by immunoprecipitation-western blot analysis (Zhang et al, 2011), whereas under identical experimental conditions, we were unable to detect STCA-labelled HSP70. This finding suggests that STCA may inhibit the function of HSP90. In this study, we address this possibility and describe a new class of sulphoxythiocarbamate small-molecule inhibitors of HSP90 that, by virtue of their sulphhydryl reactivity, target HSP90 to cause destabilisation of its client oncoproteins and inhibit cell proliferation.
Materials and methods
Chemicals and reagents
The detailed chemical synthesis of all new compounds is described in Supplementary Online Material. All general chemicals and reagents were of analytical grade and obtained from Sigma-Aldrich (Dorset, UK and Milwaukee, WI, USA).
HSP90 expression and purification
Human HSP90β clone was kindly provided by Dr Anutosh Chakraborty and Dr Solomon H Snyder (Johns Hopkins University). DNA-encoding full-length human HSP90β (aa 1-724) was subcloned into the pGEX 6p2 vector (GE Healthcare, Piscataway, NJ, USA) using SalI and NotI restriction sites. The plasmid was transformed into E. coli BL21 (DE3) codon plus cells. Cells were grown at 37 °C in LB media until OD600 of 0.7, then induced at 16 °C with IPTG (0.5 mM) for 16 h. Cell pellets were collected by centrifugation and lysed via double-pass on French press in a lysis buffer (20 mM NaH2PO4-Na2HPO4, pH 7.4, 500 mM NaCl, 1 mM DTT, 1 mM PMSF). Cell lysates were clarified by centrifugation at 17 700 × g. The supernatant was loaded onto Glutathione Sepharose 4 FF (GE Healthcare) for affinity purification, washed with 1 × phosphate-buffered saline (PBS), eluted with cleavage buffer (50 mM Tris, pH 7.5, 50 mM GSH), and dialysed against 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT. GST was cleaved by precision-protease at 4 °C for 4 h. HSP90β was further purified on a mono-Q column (GE Healthcare) equilibrated with 50 mM Tris pH 7.5. Bound protein was eluted with a linear salt gradient over 240 ml from 0 to 1 M NaCl. Protein purity was determined by SDS–PAGE.
HSP90 trypsin digestion, avidin purification, and MALDI-mass spectrometry of peptides modified by STCA
Purified HSP90β (ca 70 μM) in 40 mM HEPES, pH 7.5, 20 mM KCl, 1 mM TCEP was incubated with STCA (1.3 mM) or vehicle (acetonitrile, 0.1%, v/v) at 25 °C for 2 h, and dialysed against 40 mM HEPES, pH 7.5, 20 mM KCl, 0.5 mM TCEP to remove the remaining STCA. The dialysate was then subjected to click reaction in a solution containing biotin azide (0.2 mM), TCEP (1 mM), TBTA (0.2 mM), CuSO4 (1 mM) at room temperature for 1.5 h. Any precipitate was pelleted by centrifugation, dissolved in 8 M urea, and recombined to the solution. The click reaction solution was extensively dialysed against 100 mM Tris HCl, pH 7.5 to remove small molecules. The dialysate containing HSP90β (120 μg) was then treated with DTT (5 mM) at 37 °C for 30 min, alkylated by iodoacetamide (15 mM) at 37 °C for 30 min, quenched by additional DTT (15 mM), and digested with trypsin (6 μg), with addition of 10% acetonitrile. The digest was diluted 10 times with 100 mM Tris HCl, and incubated with monomeric avidin-agarose (Pierce, Rockford, IL USA) at room temperature for 2 h. The beads were washed sequentially with 1 × TBST (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween), 1 × TBS, and water. Bound peptides were eluted with 0.5 M aq. acetic acid, lyophilised, and redissolved in 0.1% TFA/water. Eluted peptides were co-crystallised in α-cyano-4-hydroxycinnamic acid (10 mg ml–1 in 50% acetonitrile/0.1% TFA) and analysed by matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) on a Voyager DE STR using Voyager Instrument Control Panel (v5.1) and Data Explorer (v4.0) (Applied Biosystems, www.appliedbiosystems.com).
Cell culture
The human breast cancer cell lines MCF7 and MDA-MB-231 were generous gifts from Dr Colin J Henderson and Dr Andrew South (University of Dundee). The human cervical cancer cell line HeLa and the human osteosarcoma cell line U2OS were kind gifts from Dr Sonia Rocha (University of Dundee). The HeLa-luc cell line was developed in the laboratory of Dr Richard I Morimoto (Northwestern University; Calamini et al, 2012). All cell lines were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) heat-inactivated FBS. The medium for the MCF7 cells also contained 1% glutamine (Invitrogen), whereas the medium for the HeLa-luc cells was supplemented with 100 μg ml–1 G418 (Invitrogen, Paisley, UK). Mouse embryonic fibroblasts (MEFs), from wild-type (WT) or HSF1-knockout (HSF1-KO) mice (Xiao et al, 1999), were cultured in DMEM supplemented with 10% (v/v) heat-inactivated FBS, non-essential amino acids, and 50 U ml–1 penicillin/streptomycin. Cells were maintained at 5% CO2 in air at 37 °C.
ATP-binding assay
MCF7 cells (0.5 × 106) growing in 6-cm dishes were treated with either 0.1% (v/v) acetonitrile (vehicle for STCA), 75 μM STCA, 0.1% DMSO (vehicle for GA and celastrol), 1 μM GA, or 0.8 μM celastrol for 24 h. Cells were then scraped into 300 μl of lysis buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.25% NP40, with one protease inhibitor tablet (Roche, Welwyn Garden City, UK) per 10.0 ml of buffer), frozen, thawed, and lysed for 30 min at 4 °C. ATP-agarose beads (Jena Bioscience, Jena, Germany) were washed with the incubation buffer (10 mM Tris pH 7.5, 150 mM NaCl, 20 mM MgCl2, 0.05% NP40, 1 mM DTT), and 200 μg of protein from each sample was added to a suspension of 30 μl of beads in 1.25 ml of buffer. The samples were incubated rotating overnight at 4 °C. The beads were collected by centrifugation, washed three times with the incubation buffer, 10 μl of SDS loading buffer, and 40 μl of the incubation buffer were added, and the samples were incubated for 5 min at 100 °C. The beads were pelleted by centrifugation, and the supernatants were collected and subjected to western blot analysis.
Western blotting
Cells were washed twice with PBS and lysed in RIPA buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS, and 1 mM EDTA), containing 1 protease inhibitor cocktail tablet (Roche) per 10 ml of buffer. The lysates (300 μl or 150 μl for each 6-cm dish or well of a six-well plate, respectively) were transferred into 1.5-ml Eppendorf tubes, which were placed on a rotator at 4 °C for 30 min before centrifugation at 16 300 × g for 10 min at 4 °C. The pellet was discarded, and the supernatant was transferred to a new tube. Protein concentrations were determined by the BCA assay (Thermo, Rockford, IL, USA). Proteins were resolved by SDS–PAGE, transferred to immobilon-P membranes, and probed with specific antibodies against HSP70 (mouse monoclonal, 1 : 1000, StressMarq, York, UK), HSP90 (mouse monoclonal, 1 : 5000, BD Biosciences, Franklin Lakes, NJ, USA), HER2 (rabbit polyclonal, 1 : 500, Millipore, Temecula, CA, USA), RAF1 (rabbit polyclonal, 1 : 200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), GSK3 (mouse monoclonal, 1 : 1000, Biosource, Camarillo, CA, USA), CHK1 (mouse monoclonal, 1 : 1000, Santa Cruz Biotechnology, Camarillo, CA, USA), pS345-CHK1 (rabbit polyclonal, 1 : 1000, Cell Signaling, Danvers, MA, USA), CDK1 (rabbit polyclonal, 1 : 1000, Cell Signaling), or p53 (DO-1) (mouse monoclonal, 1 : 1000, Abcam, Cambridge, UK). Equal loading was confirmed by probing the blots with antibodies against GAPDH (rabbit polyclonal, 1 : 5000) or β-actin (mouse monoclonal, 1 : 10 000), both from Sigma, Dorset, UK. The intensity of each band was quantified by densitometry using ImageJ (developed by NIH, http://rsbweb.nih.gov/ij/), normalised against the density of the loading control, and is shown as a histogram in the Supplementary Online Material.
Quantitative RT–PCR
The primers and probes for quantifying the levels of mRNA for hsp70a1a and hsp70a1b were from Applied Biosystems (Mm01159846_s1 and Mm03038954_s1). WT MEFs (2.1 × 105 per well) in six-well plates were exposed to vehicle (0.1% acetonitrile) or increasing concentrations of each sulphoxythiocarbamate for 8 h. Cells were lysed, total RNA was extracted using RNeasy Kit (Qiagen Ltd, Manchester, UK), and 500 ng total RNA was reverse transcribed into cDNA with Omniscript Reverse Transcription Kit (Qiagen Ltd). Real-time PCR was performed on Applied Biosystems 7900HT Fast Real-Time PCR System. The data were normalised using β-actin (mouse ACTB, Applied Biosystems, Mm00607939_s1) as an internal control.
Caspase 3 detection and cell proliferation assay
Activity of caspase 3 was determined by a flourometric assay (BD Biosciences). To determine cell proliferation, MCF7 (5 × 103 per well) or MEF (104 per well) cells were seeded in white flat-bottomed 96-well plates (Nunc). Cells were treated with 0.1% (v/v) acetonitrile (vehicle control) or increasing concentrations of sulphoxythiocarbamates for 24 or 48 h. Four hours before the end of the 24- and 48-h treatments, 10 μl of Alamar Blue dye solution (AbdSerotec, Oxford, UK) was added to the media (100 μl) within each well, and the plates were returned to the incubator. After 4 h, the fluorescence of the reduced probe was determined (ex. 560 nm/em. 590 nm, SpectraMax M2, Molecular Devices, Sunnyvale, CA, USA).
Luciferase assay
HeLa-luc cells (1.3 × 105 per well) in 12-well plates were treated with 0.1% (v/v) acetonitrile (vehicle) or STCA for 24 h. Luciferase activity was determined as described (Calamini et al, 2012).
Statistical analysis
All values are means±s.d. The differences between groups were determined by Student's t-test.
Results
STCA destabilises the HSP90 client oncoproteins HER2 and RAF1
The identification of HSP90 as a target protein of STCA (Ahn et al, 2010; Zhang et al, 2011) suggested that sulphoxythiocarbamates may function as HSP90 inhibitors. To test this possibility, we evaluated the stability of two well-established HSP90 client oncoproteins, HER2 and RAF1. The levels of both HER2 (Figure 1B) and RAF1 (Figure 1C) were decreased after treatment of MCF7 breast cancer cells with STCA. The levels of HSP70 were correspondingly dose-dependently elevated (Figure 1B). In contrast to the destabilising effect of STCA on HER2 and RAF1, the levels of the kinases GSK3α and GSK3β were only modestly changed (Figure 1D). These results show that STCA inhibits HSP90 and leads to selective degradation of its client oncoproteins, in agreement with the recently described preferential targeting and affinity capture of tumour-enriched HSP90-oncoprotein complexes by the purine scaffold-based HSP90 inhibitor PU-H71 (Moulick et al, 2011).
Persistent activation of HER2 and RAF1 is associated with sustained proliferative signalling, one of the hallmarks of cancer (Hanahan and Weinberg, 2011). Evading growth suppressors—another hallmark of cancer—is often facilitated by overexpression of mutant p53, which in turn is stabilised by HSP90 (Blagosklonny et al, 1996). We therefore tested the possibility that STCA may also destabilise mutant p53 using MDA-MB-231 cells, which carry mutant (Arg280Lys) p53. The levels of immunologically detectable mutant p53 were decreased after 24 h of exposure to STCA in a dose-dependent manner (Figure 1E). A parallel experiment in MCF7 cells showed that, in contrast to its mutant counterpart, the levels of WT p53 were increased by STCA (Figure 1F). Accumulation of WT p53 was evident 4 h after exposure to STCA, and continued to increase at the 8-h time point (Figure 1G). These results are consistent with the previously reported opposing effects of HSP90 inhibition by GA, which downregulates mutant p53 and upregulates WT p53 (Lin et al, 2008).
STCA causes apoptosis and decreases cell proliferation
To determine the functional consequences of RAF1, HER2, and mutant p53 destabilisation by STCA, we evaluated the ability of the sulphoxythiocarbamate to cause apoptosis by monitoring the activity of caspase 3. As MCF-7 cells are caspase 3-deficient, we used MDA-MB-231 cells. Compared with vehicle (control) treatment, the activity of caspase 3 was elevated by 1.5-fold 24 h after exposure to STCA (Figure 2A). Next, cell proliferation was evaluated using the probe Alamar Blue (Nociari et al, 1998). Twenty-four hours after treatment with STCA, there was a significant dose-dependent reduction in proliferation (Figure 2B), in agreement with the lower cell density that was observed in STCA-treated compared with vehicle-treated cells at the same time point (Figure 2C).
Figure 2.

STCA causes apoptosis and decreases cell proliferation. (A) MDA-MB-231 cells (106 per dish) in 6-cm dishes were treated with vehicle (0.1% acetonitrile) or 75 μM STCA for 24 h and the levels of caspase 3 were determined in cell lysates. Data are from three independent experiments. P<0.05 (Student's t-test). (B) MCF7 cells (5 × 103 per well) in 96-well plates were treated with vehicle (0.1% acetonitrile, ○), 25 μM (♦), 50 μM (▴), or 75 μM STCA (◊) for 24 or 48 h. Cell proliferation was assessed using the Alamar Blue fluorometric assay. *P<0.001 for the STCA-treated relative to vehicle-treated cells. (C) Phase contrast microscope images of MCF7 cells treated with vehicle (0.1% acetonitrile) or 75 μM STCA for 24 h. Scale bar=200 μm.
STCA does not affect the ATP-binding ability of HSP90
We then addressed the mechanism of inhibition of HSP90 by STCA. To investigate whether STCA affects the ATP-binding ability of HSP90, we performed an ATP-agarose pull down assay using cell lysates from MCF7 cells that had been treated with STCA or its solvent control. We also used two other types of control treatments: one with GA, and another with celastrol, which unlike GA, has been shown to inhibit HSP90 without altering its ATP-binding ability. As expected, treatment with GA almost completely abolished the binding of HSP90 to ATP (Figure 3). In sharp contrast, neither celastrol, nor STCA had any effect on the ATP-binding ability of HSP90, strongly suggesting that STCA does not bind to the N-terminal domain of HSP90.
Figure 3.

STCA does not affect the ability of HSP90 to bind ATP. MCF7 cells (5 × 105 per dish) in 6-cm dishes were exposed to vehicle (0.1% acetonitrile) or STCA for 24 h. Control treatments were also performed with vehicle (0.1% DMSO), 1 μM GA and 0.8 μM celastrol (CL). Cells were lysed and subjected to ATP pulldown using ATP-agarose beads. For the ATP pulldown and input samples, the levels HSP90 and GAPDH were determined by western blot analyses. The data represent results from three independent experiments.
STCA modifies cysteine residues of HSP90
The electrophilic sulphoxythiocarbamate group of STCA reacts with nucleophilic cysteine residues in its protein targets. Human HSP90β contains six cysteines, which are located within its middle domain (Figure 1A). To identify which cysteines are the targets for modification by STCA, we used a protease-mass spectrometric approach by taking advantage of the alkyne group of the compound, which allows for click reaction with biotin azide. Thus, recombinant HSP90β was incubated with STCA, then dialysed and biotinylated by click reaction. The biotinylated STCA-labelled protein was subjected to exhaustive trypsin digestion and exposed to avidin-coated beads. The captured peptides were eluted with acetic acid and analysed by MALDI-mass spectrometry. We identified two prominent peaks that were not present in control samples (Figure 4 and Supplementary Figure 1). The corresponding mass of the shorter peptide was in precise agreement with the molecular weight for modified Cys564 (m/z 1524, FENLC564*K). The mass of the longer peptide (m/z 3146) corresponded exactly to the molecular weight for peptide LVSSC589*C590*IVTSTYGWTANMER, in which one cysteine was labelled with STCA, and the other with iodoacetamide. These assignments were confirmed by tandem MS analysis, which further revealed that Cys412, within the peptide C412*LELFSELAEDK, was also labelled by STCA (Supplementary Figure 2 and Supplementary Table 1). As the middle domain of HSP90 is the site of interaction with most of its client proteins (Whitesell and Lindquist, 2005), it is very likely that chemical modification of cysteine residue(s) within this domain causes a conformational change, which interferes with the function of the chaperone, causing dissociation and subsequent degradation of client proteins. Notably, in human HSP90α, modification of Cys597 (corresponding to Cys589 in HSP90β) by nitric oxide has been shown to shift the conformational equilibrium of the chaperone towards the open conformation, and it has been suggested that this increases the energy barrier necessary for conformational changes within HSP90 during the ATPase cycle, thus ultimately reducing the activity of the chaperone (Retzlaff et al, 2009). This notion is also supported by in silico-based studies (Morra et al, 2009).
Figure 4.

STCA modifies cysteine residues of HSP90. Purified recombinant HSP90β was incubated with STCA for 2 h, dialysed to remove STCA, subject to click reaction with biotin azide, and digested by trypsin. The tryptic digest was incubated with avidin-coated beads. The captured peptides were eluted by 0.5 M acetic acid and analysed by MALDI-mass spectroscopy.
STCA enhances transcription through heat shock elements (HSEs)
Because HSP90 is the major negative regulator of HSF1, activation of the transcription factor followed by HSE-dependent induction of heat shock proteins is a common consequence of inhibition of the chaperone. To test whether STCA enhances transcription through the HSE, we used the cervical cancer HeLa HSE-luciferase reporter cell line (HeLa-luc; Calamini et al, 2012). We observed a dose-dependent induction of the reporter, and the luciferase activity was elevated by 1.6-, 2-, and 2.5-fold by 25, 50, and 75 μM STCA, respectively (Figure 5A). Western blot analysis confirmed induction of the endogenous HSP70 (Figure 5B).
Figure 5.

STCA activates transcription through HSEs. HeLa-luc cells (1.3 × 105 per well) stably transfected with the luciferase gene under the control of the HSP70.1 promoter containing the HSE, were exposed to STCA for 24 h in 12-well plates. (A) Luciferase activity was determined in cell lysates. The relative luminescence units (RLU) were quantified and normalised with respect to the vehicle control treatment and are expressed as the relative luciferase activity (treated/control ratio). Cells were also treated with celastrol (1.6 μM) as a positive control. *P<0.05 relative to the vehicle treatment. (B) The levels of endogenous HSP70 were determined in a parallel experiment by western blot analysis. GAPDH served as the loading control. The data are representative from three independent experiments.
Sulphoxythiocarbamates cause the HSF1-dependent upregulation of HSP70: structure–activity relationship analysis
Transcriptional activation of the heat shock response is used as a mechanistically unbiased approach to screen chemical libraries for identification of HSP90 inhibitors (Santagata et al, 2012). In addition, there is a correlation between degradation of HSP90 client proteins and induction of HSP70 in host peripheral blood lymphocytes and xenograft tumours (Mehta et al, 2011). We therefore used the HSF1-dependent induction of HSP70 as an end point to evaluate the ability of 19 sulphoxythiocarbamate analogues of STCA, including 6 new compounds, to inhibit HSP90. To simultaneously test for the involvement of HSF1 in the induction process, we chose as an experimental system MEFs isolated from WT or HSF1-KO mice. Compounds were considered inactive if no induction of HSP70 was observed at concentrations up to 50 μM. Of the 19 sulphoxythiocarbamate compounds tested, 9 were able to induce HSP70 (Table 1). Four of these, that is, S-3, S-4, K-2, and K-3, were more potent than STCA, and they were re-tested at lower concentrations (Figure 6A). Of them, to our knowledge, compounds K-2 and S-4 are new. Induction of HSP70 by S-4 was evident at 2.5 μM, the lowest concentration tested. In all cases, inducer activity is dependent on the presence of HSF1 and is completely abolished in HSF1-deficient cells (Figure 6B). In contrast, upregulation of NQO1, a prototypic NRF2-dependent gene, occurs in both HSF1-WT and HSF1-KO cells, in agreement with our earlier findings for STCA and three other chemically distinct inducers (Zhang et al, 2011). Notably, induction of NQO1 is evident at lower inducer concentrations compared with the concentrations, which cause HSP70 upregulation.
Table 1. Chemical structures, HSP70 inducer (EC), and cell proliferation-inhibitory (IC50) activities of sulphoxythiocarbamates.
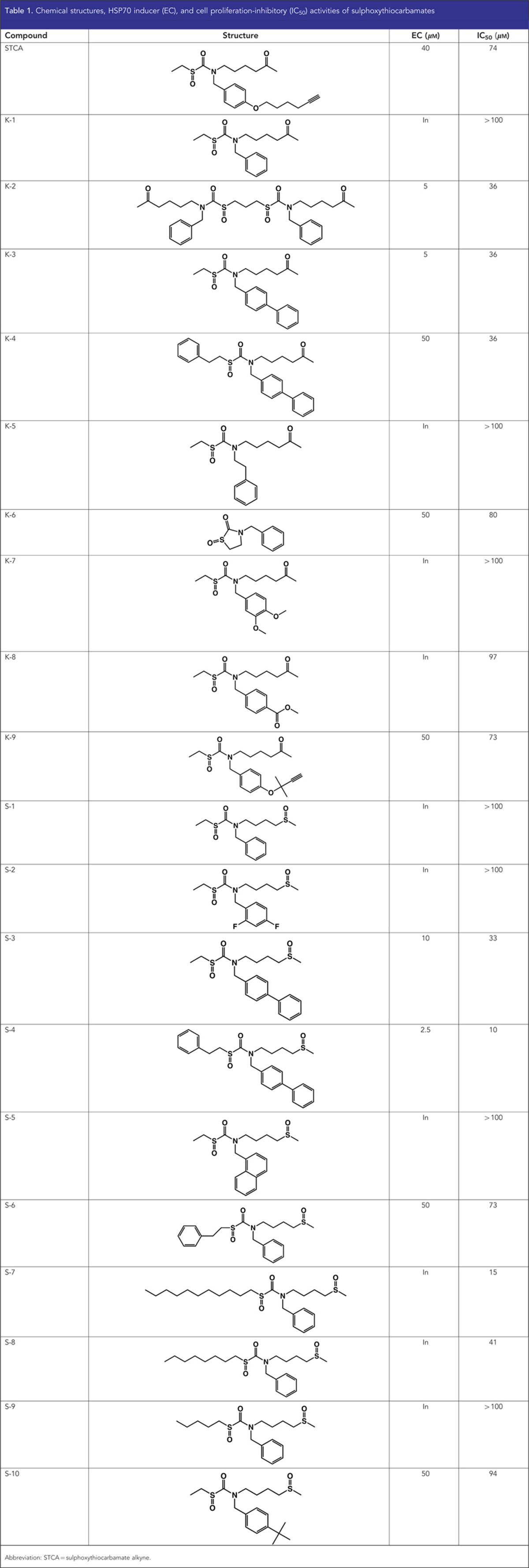
Figure 6.

Induction of HSP70 and NQO1 (A and B) and inhibition of cell proliferation (C and D) by sulphoxythiocarbamates. (A) WT (2.1 × 105 per well) or (B) HSF1-KO (2.5 × 105 per well) MEFs in six-well plates were exposed to vehicle (0.1% acetonitrile) or increasing concentrations of each sulphoxythiocarbamate for 4 h. Cells were lysed in RIPA buffer 20 h later, aliquots from cell lysates were resolved by SDS–PAGE, transferred to immobilon-P, and probed with antibodies against HSP70 and NQO1. GAPDH was used as a loading control. The data are representative from two independent experiments. (C and D) MEFs (104 per well, in 96-well plates) were treated with vehicle (0.1% acetonitrile) or increasing concentrations of sulphoxythiocarbamates of the K-series (C) or the S-series (D) for 24 h. Cell proliferation was assessed using the Alamar Blue fluorometric assay. Data represent means±s.d. from six replicate wells. *P<0.05 relative to vehicle-treated cells.
There were two series of sulphoxythiocarbamate compounds (Table 1): one (named K-series) containing a keto group, and the second (named S-series) containing a sulphoxide group, both groups positioned at an identical distance with respect to the electrophilic functional group. Within the K-series, the presence of an S-phenethyl group in place of an S-ethyl group decreases inducer potency by ∼10-fold (compare K-3 with K-4). However, within the S-series, the same substitution increases the inducer potency (compare S-3 with S-4, and S-1 with S-6), and S-4 is the most potent compound. Addition of a phenyl or a tert-butyl group in tandem to the benzyl substituent at the nitrogen was found to increase the inducer potency for both the sulphoxide and the keto series. Thus, the sulphoxide-containing S-3 and S-10 are active as HSP70 inducers, whereas S-1 is not. This potentiation effect is even more striking when the keto derivatives are compared. Indeed, K-3 induces HSP70 at a concentration of 5 μM, whereas K-1, which only differs from K-3 by the lack of a second aromatic ring at the nitrogen substitution, is inactive. Interestingly, K-2, a dimer of K-1, induces HSP70 at a concentration of 5 μM, indicating that the presence of two reactive centres the same molecule substantially improves the inducer potency. In addition, incorporating the reactive centre within a heterocyclic ring system confers inducer activity (compare K-6 with K-1). Finally, S-7 and S-8, compounds with long aliphatic chains adjacent to the electrophilic functional group are toxic (the protein concentration was ∼50% of the vehicle treated in samples treated with 20 μM of S-7 and 50 μM of S-8, respectively), but completely inactive, demonstrating that toxicity per se is not sufficient for induction. Cell proliferation assays confirmed these conclusions: (i) S-4 was the most potent compound both as an HSP70 inducer and as an inhibitor of cell proliferation, (ii) the more potent inducers of HSP70 were also more potent in inhibiting cell proliferation, and (iii) despite the lack of HSP70 inducer activity, S-7 and S-8 inhibited cell proliferation (Figure 6C and D and Table 1).
Sulphoxythiocarbamates cause transcriptional upregulation of hsp70a1a and hsp70a1b
The ability of STCA to induce transcription of a luciferase reporter under the control of the HSE (Figure 5A) and the requirement for HSF1 for the upregulation of HSP70 by sulphoxythiocarbamates (Figure 6A and B) implied that induction is due to enhanced transcription. To confirm and further quantify inducer potency, we determined the mRNA levels of hsp70a1a and hsp70a1b, the two most commonly inducible genes encoding HSP70. Quantitative RT–PCR analysis showed that the mRNA for both hsp70a1a and hsp70a1b increased in a dose-dependent manner after treatment with STCA, S-3, S-4, K-2, or K-3 (Figure 7A). In full agreement with the results for the HSP70 protein levels (Figure 6A), the most potent and robust inducer was S-4, and the levels of hsp70a1a and hsp70a1b were increased by 24- and 47-fold, respectively by an 8-h exposure to 10 μM S-4. The quantitative nature of this assay allowed us to further rank the compound potencies: S-4>K-2>K-3>S-3>STCA.
Figure 7.

S-4 induces transcription of hsp70a1a and hsp70a1b, causes destabilisation of HSP90 client proteins, and inhibits cell proliferation. (A) WT MEFs (2.1 × 105 per well) in six-well plates were exposed to vehicle (0.1% acetonitrile) or increasing concentrations of each sulphoxythiocarbamate for 8 h. Cells were then lysed and total RNA were extracted. The amount of mRNA for hsp70a1a and hsp70a1b was analysed by quantitative RT–PCR, using β-actin mRNA as an internal control. The mRNA from each sample was measured separately, in triplicate. Data represent means±s.d. and are expressed as ratio of the relative transcription units in treated over control samples. (B–F) Cells seeded in six-well plates at a density of 2.5 × 105 per well (B, D and E) or in 6-cm dishes at a density of 106 per dish (C) were exposed to vehicle (0.1% acetonitrile) or sulphoxythiocarbamates for either 24 h (B and F) or 16 h (C–E). Cells were lysed in RIPA buffer, proteins in aliquots from cell lysates were resolved by electrophoresis, transferred to immobilon-P, and probed with the specified antibodies. The data represent results from two independent experiments. (G) MCF7 cells (5 × 103 per well) in 96-well plates were treated with vehicle (0.1% acetonitrile, ○), 2.5 μM (♦), 5 μM (▴), or 10 μM S-4 (◊) for 24 or 48 h. Cell proliferation was assessed using the Alamar Blue fluorometric assay. *P<0.001 relative to vehicle-treated cells.
S-4 inhibits HSP90 and reduces cell proliferation more potently than STCA
The higher potency of S-4 in inducing HSP70 compared with STCA indicated that the new compound will be more potent in inhibiting HSP90 and reducing cell proliferation. Indeed, we observed destabilisation of RAF1 and HER2 at lower concentrations of S-4 than STCA (compare Figures 1A and B and Figure 7B). In contrast, the levels of RAF1 and HER2 were not affected by K-1 or S-1, which also have no HSP70 inducer activity (Figure 7C). This effect was not limited to the breast cancer cell line MCF7, and was observed in the cervical cancer cell line HeLa, as well as in U2OS osteosarcoma cells (Figure 7D and E). In addition, the levels of the serine/threonine protein kinases CHK1 and CDK1, two other HSP90 client proteins with critical roles in the cell cycle control (Figure 7F), were also reduced by S-4, and at much lower concentrations than those of STCA. Most importantly, S-4 was more potent in inhibiting cell proliferation, with an IC50 value of 10 μM compared with 74 μM for STCA (Figures 2B and 7G).
Discussion
Here we report the identification of sulphhydryl-reactive sulphoxythiocarbamates as a new class of HSP90 inhibitors, which destabilise HSP90's client oncoproteins without interfering with the ATP-binding ability of the chaperone. Using recombinant human HSP90β, we showed that three of its six cysteine residues, that is, Cys412, Cys564, and one of the vicinal Cys589 or Cys590, are direct targets of these mildly electrophilic compounds. Notably, Cys412 and Cys564 are adjacent to Lys411 and Lys565, respectively. Proximity to basic amino acids lowers the pKa value of the cysteine sulphhydryl group, that is, increases its reactivity (Snyder et al, 1981). In rat HSP90β, Cys589 and Cys590 have been shown to be highly reactive (Nardai et al, 2000). In human HSP90α, S-nitrosylation at Cys597 (corresponding to Cys589 in HSP90β) or its substitution with S-nitrosylation-mimicking residues, such as asparagine and aspartic acid, decreases the chaperone activity (Martinez-Ruiz et al, 2005; Retzlaff et al, 2009), and some of the antitumour activity of nitric oxide has been attributed to inhibition of HSP90 (Mollapour and Neckers, 2012). It is therefore highly probable that thiocarbamylation of HSP90 is responsible for the observed inhibition of the chaperone's function by sulphoxythiocarbamates.
In addition to inhibiting HSP90, the sulphoxythiocarbamates are multi-targeting agents and affect other stress response pathways, such as the KEAP1/NRF2 pathway. Activation of the KEAP1/NRF2 pathway leads to induction of cytoprotective genes, including NQO1. The upregulation of NQO1 (Figure 6A and B) may be in part responsible for the observed stabilisation of WT p53 by sulphoxythiocarbamates as NQO1 is a known regulator of p53 stability (Asher et al, 2001). Importantly, although the KEAP1/NRF2 pathway and the heat shock response are two separate cytoprotective pathways, recent evidence suggests that they engage in crosstalk. Thus, HSP90 and KEAP1 interact upon heat shock, leading to activation of NRF2 (Niture and Jaiswal, 2010). It is possible that by direct binding, HSP90 protects the electrophilic stress sensor KEAP1 from potential damage during heat shock, and that the consequent activation of NRF2 is a contributing factor to the protective effect of heat shock against subsequent exposures to other forms of cell stress.
By inhibiting HSP90, sulphoxythiocarbamates activate the HSF1-dependent heat shock response as evidenced by the transcriptional upregulation of endogenous HSP70 and an HSE-dependent luciferase reporter, and by the absolute requirement for HSF1 for induction. Interestingly, in contrast to its mammalian counterpart, yeast HSP90 is devoid of cysteine residues, and specific cysteines of the yeast HSP70 chaperone Ssa1 serve as sensors for electrophiles, leading to HSF1 activation (Wang et al, 2012). Activation of the heat shock response is a common feature of HSP90 inhibitors as HPSP90 is the major negative regulator of HSF1, and presents a major challenge in the development of HSP90 inhibitors and their clinical implementation as anticancer agents. Nonetheless, the destabilisation of mutant p53 and oncogenic kinases, which are overexpressed in tumours with high frequencies, represents an important mechanism of action of HSP90 inhibitors that have been shown to inhibit tumour growth in numerous preclinical cancer models, such as pancreatic, breast, lung, and melanoma, including intracranial metastases (Neckers, 2002; Williams et al, 2007; Moser et al, 2012; Fogliatto et al, 2013; Garon et al, 2013; Haarberg et al, 2013). The increased dependence on HSP90 and higher sensitivity to HSP90 inhibition of cancer cells in comparison with normal cells suggests a paradigm whereby HSP90 inhibitors may selectively target cancer cells while enhancing protection in normal cells via the upregulation of HSF1- and NRF2-dependent genes, collectively termed vitagenes (Calabrese et al, 2011).
The identification of inhibitors of HSP90 as potential anticancer agents is an area of intense scientific research as they offer the attractive possibility of targeting multiple oncogenic pathways simultaneously. The dependence of numerous oncogenic proteins on this chaperone for their stability and function highlights the multitude of effects that inhibition of HSP90 can have on all aspects of tumour development. A number of HSP90 inhibitors have been or currently are in clinical trials, including derivatives of GA and purine analogues, and have shown promising outcomes in breast cancer, non-small-cell lung cancer, and multiple myeloma (for recent reviews, see Jhaveri et al, 2012; Garcia-Carbonero et al, 2013; Zagouri et al, 2013). Our findings demonstrate that targeting specific cysteine residues of HSP90 with mildly electrophilic sulphoxythiocarbamates reduces the stability of its client oncoproteins, promotes apoptosis, and decreases cell proliferation. This type of interfering with the HSP90 function, by targeting its middle domain, the major site of interaction with client proteins, is radically distinct from all of the previously known modes of inhibition of the chaperone, which have been designed to target either its N-terminal or C-terminal domains. As a result of the central role of HSP90 in tumour biology, targeting the chaperone affects not only its own function, but also the ability of large networks of proteins that associate with it to perform their respective roles, further relaying and amplifying the initial ‘cysteine-sensed' signal.
Acknowledgments
We thank Richard I Morimoto (Northwestern University) for his generous gift of the HeLa HSE-luciferase reporter cell line. We acknowledge with gratitude the financial support of Research Councils UK, the Biotechnology and Biological Sciences Research Council (BBSRC, Project Grant BB/J007498/1), Cancer Research UK (C20953/A10270), the NIH, and startup funds from Wayne State University.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Material
References
- Ahn YH, Hwang Y, Liu H, Wang XJ, Zhang Y, Stephenson KK, Boronina TN, Cole RN, Dinkova-Kostova AT, Talalay P, Cole PA. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc Natl Acad Sci USA. 2010;107 (21:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Lotem J, Cohen B, Sachs L, Shaul Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc Natl Acad Sci USA. 2001;98 (3:1188–1193. doi: 10.1073/pnas.021558898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck R, Dejeans N, Glorieux C, Pedrosa RC, Vasquez D, Valderrama JA, Calderon PB, Verrax J. Molecular chaperone Hsp90 as a target for oxidant-based anticancer therapies. Curr Med Chem. 2011;18 (18:2816–2825. doi: 10.2174/092986711796011256. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci USA. 1996;93 (16:8379–8383. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Cornelius C, Cuzzocrea S, Iavicoli I, Rizzarelli E, Calabrese EJ. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol Aspects Med. 2011;32 (4-6:279–304. doi: 10.1016/j.mam.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8 (2:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315 (1:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- Chen L, Shi Y, Jiang CY, Wei LX, Wang YL, Dai GH. Expression and prognostic role of pan-Ras, Raf-1, pMEK1 and pERK1/2 in patients with hepatocellular carcinoma. Eur J Surg Oncol. 2011;37 (6:513–520. doi: 10.1016/j.ejso.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Fogliatto G, Gianellini L, Brasca MG, Casale E, Ballinari D, Ciomei M, Degrassi A, De Ponti A, Germani M, Guanci M, Paolucci M, Polucci P, Russo M, Sola F, Valsasina B, Visco C, Zuccotto F, Donati D, Felder E, Pesenti E, Galvani A, Mantegani S, Isacchi A. NMS-E973, a novel synthetic inhibitor of Hsp90 with activity against multiple models of drug resistance to targeted agents, including intracranial metastases. Clin Cancer Res. 2013;19 (13:3520–3532. doi: 10.1158/1078-0432.CCR-12-3512. [DOI] [PubMed] [Google Scholar]
- Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14 (9:e358–e369. doi: 10.1016/S1470-2045(13)70169-4. [DOI] [PubMed] [Google Scholar]
- Garon EB, Finn RS, Hamidi H, Dering J, Pitts S, Kamranpour N, Desai AJ, Hosmer W, Ide S, Avsar E, Jensen MR, Quadt C, Liu M, Dubinett SM, Slamon DJ. The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther. 2013;12 (6:890–900. doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarberg HE, Paraiso KH, Wood E, Rebecca VW, Sondak VK, Koomen JM, Smalley KS. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol Cancer Ther. 2013;12 (6:901–912. doi: 10.1158/1535-7163.MCT-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144 (5:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hwang Y, Thompson PR, Wang L, Jiang L, Kelleher NL, Cole PA. A selective chemical probe for coenzyme A-requiring enzymes. Angew Chem Int Ed Engl. 2007;46 (40:7621–7624. doi: 10.1002/anie.200702485. [DOI] [PubMed] [Google Scholar]
- Itoh H, Ogura M, Komatsuda A, Wakui H, Miura AB, Tashima Y. A novel chaperone-activity-reducing mechanism of the 90-kDa molecular chaperone HSP90. Biochem J. 1999;343 (Pt 3:697–703. [PMC free article] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. Heat shock factor 1 attenuates 4-Hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J Biol Chem. 2007;282 (46:33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. Systems analysis of protein modification and cellular responses induced by electrophile stress. Accounts Chem Res. 2010;43 (5:673–683. doi: 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823 (3:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VA, Singh EK, Nazarova LA, Alexander LD, McAlpine SR. Macrocyclic inhibitors of hsp90. Curr Top Med Chem. 2010;10 (14:1380–1402. doi: 10.2174/156802610792232088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Rockliffe N, Johnson GG, Sherrington PD, Pettitt AR. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene. 2008;27 (17:2445–2455. doi: 10.1038/sj.onc.1210893. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92 (3:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA. 2005;102 (24:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta PP, Whalen P, Baxi SM, Kung PP, Yamazaki S, Yin MJ. Effective targeting of triple-negative breast cancer cells by PF-4942847, a novel oral inhibitor of Hsp 90. Clin Cancer Res. 2011;17 (16:5432–5442. doi: 10.1158/1078-0432.CCR-11-0592. [DOI] [PubMed] [Google Scholar]
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823 (3:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G, Verkhivker G, Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput Biol. 2009;5 (3:e1000323. doi: 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser C, Lang SA, Hackl C, Wagner C, Scheiffert E, Schlitt HJ, Geissler EK, Stoeltzing O. Targeting HSP90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 2012;32 (7:2551–2561. [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, Vu LP, Zhao X, Zatorska D, Taldone T, Smith-Jones P, Alpaugh M, Gross SS, Pillarsetty N, Ku T, Lewis JS, Larson SM, Levine R, Erdjument-Bromage H, Guzman ML, Nimer SD, Melnick A, Neckers L, Chiosis G. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7 (11:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardai G, Sass B, Eber J, Orosz G, Csermely P. Reactive cysteines of the 90-kDa heat shock protein, Hsp90. Arch Biochem Biophys. 2000;384 (1:59–67. doi: 10.1006/abbi.2000.2075. [DOI] [PubMed] [Google Scholar]
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8 (4 Suppl:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- Niture SK, Jaiswal AK. Hsp90 interaction with INrf2(Keap1) mediates stress-induced Nrf2 activation. J Biol Chem. 2010;285 (47:36865–36875. doi: 10.1074/jbc.M110.175802. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nociari MM, Shalev A, Benias P, Russo C. A novel one-step, highly sensitive fluorometric assay to evaluate cell-mediated cytotoxicity. J Immunological Methods. 1998;213 (2:157–167. doi: 10.1016/s0022-1759(98)00028-3. [DOI] [PubMed] [Google Scholar]
- Prodromou C. The ‘active life' of Hsp90 complexes. Biochim Biophys Acta. 2012;1823 (3:614–623. doi: 10.1016/j.bbamcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009;10 (10:1147–1153. doi: 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhagen MC, Soti C, Schmidt U, Wochnik GM, Hartl FU, Holsboer F, Young JC, Rein T. The heat shock protein 90-targeting drug cisplatin selectively inhibits steroid receptor activation. Mol Endocrinol. 2003;17 (10:1991–2001. doi: 10.1210/me.2003-0141. [DOI] [PubMed] [Google Scholar]
- Santagata S, Xu YM, Wijeratne EM, Kontnik R, Rooney C, Perley CC, Kwon H, Clardy J, Kesari S, Whitesell L, Lindquist S, Gunatilaka AA. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem Biol. 2012;7 (2:340–349. doi: 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Kimura Y, Mukai A, Mori H, Ito S, Asaka Y, Oe S, Tanaka H, Takahashi T, Uchida K. Transthiocarbamoylation of proteins by thiolated isothiocyanates. J Biol Chem. 2011;286 (49:42150–42161. doi: 10.1074/jbc.M111.308049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers RL, Galligan JJ, Stewart BJ, Petersen DR. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem Biol Interact. 2011;192 (1-2:107–112. doi: 10.1016/j.cbi.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder GH, Cennerazzo MJ, Karalis AJ, Field D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry. 1981;20 (23:6509–6519. doi: 10.1021/bi00526a001. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11 (7:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10 (8:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosten-Hawle P, Porter RS, Morimoto RI. Regulation of organismal proteostasis by transcellular chaperone signaling. Cell. 2013;153 (6:1366–1378. doi: 10.1016/j.cell.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21 (2:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gibney PA, West JD, Morano KA. The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol Biol Cell. 2012;23 (17:3290–3298. doi: 10.1091/mbc.E12-06-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5 (10:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Williams CR, Tabios R, Linehan WM, Neckers L. Intratumor injection of the Hsp90 inhibitor 17AAG decreases tumor growth and induces apoptosis in a prostate cancer xenograft model. J Urol. 2007;178 (4 Pt 1:1528–1532. doi: 10.1016/j.juro.2007.05.120. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999;18 (21:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Koga F, Tatokoro M, Kawakami S, Fujii Y, Kumagai J, Neckers L, Kihara K. Low-dose Hsp90 inhibitors tumor-selectively sensitize bladder cancer cells to chemoradiotherapy. Cell Cycle. 2011;10 (24:4291–4299. doi: 10.4161/cc.10.24.18616. [DOI] [PubMed] [Google Scholar]
- Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: A systematic review. Breast. 2013;22 (5:569–578. doi: 10.1016/j.breast.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ahn YH, Benjamin IJ, Honda T, Hicks RJ, Calabrese V, Cole PA, Dinkova-Kostova AT. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem Biol. 2011;18 (11:1355–1361. doi: 10.1016/j.chembiol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Blagg BS. Novobiocin analogues with second-generation noviose surrogates. Bioorg Med Chem Lett. 2013;23 (2:552–557. doi: 10.1016/j.bmcl.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.