

Abstract
The disappointing clinical outcomes of neuroprotectants challenge the relevance of preclinical stroke models and data in defining early cerebrovascular events as potential therapeutic targets. The kinetics of blood–brain barrier (BBB) leakage after reperfusion and the link with parenchymal lesion remain debated. By using in vivo and in vitro approaches, we conducted a kinetic analysis of BBB dysfunction during early reperfusion. After 60 minutes of middle cerebral artery occlusion followed by reperfusion times up to 24 hours in mice, a non-invasive magnetic resonance imaging method, through an original sequence of diffusion-weighted imaging, determined brain water mobility in microvascular compartments (D*) apart from parenchymal compartments (apparent diffusion coefficient). An increase in D* found at 4 hours post reperfusion concurred with the onset of both Evans blue/Dextran extravasations and in vitro BBB opening under oxygen-glucose deprivation and reoxygenation (R). The BBB leakage coincided with an emerging cell death in brain tissue as well as in activated glial cells in vitro. The co-culture of BBB endothelial and glial cells evidenced a recovery of endothelium tightness when glial cells were absent or non-injured during R. Preserving the ischemic brain parenchymal cells within 4 hours of reperfusion may improve therapeutic strategies for cerebrovascular protection against stroke.
Keywords: Blood–brain barrier, brain ischemia, cell culture, magnetic resonance imaging (MRI), reperfusion
Introduction
Despite numerous studies of supposedly neuroprotective agents in the field of stroke, the disappointing clinical outcomes reflect the multiplicity of the cellular and molecular mechanisms involved in this disease,1, 2 challenging the relevance of preclinical stroke data and models.3, 4 However, recent efforts have sought to enhance thrombolytic therapy and thrombectomy or provide new protection strategies by using hypothermia or agents with pleiotropic activity in relation to the neurovascular component of stroke.5 To achieve a suitable clinical outcome, it is essential to intervene as early as possible after stroke and act on the pathophysiological cascade. This implies a good understanding of when and how the blood–brain barrier (BBB) may become damaged, in relation to brain tissue injury, as the efficacy of therapeutic interventions is assumed to rely on the consideration of the kinetics of mechanisms.6 Indeed, brain tissue reperfusion is considered to have harmful effects, such as the leakage and then complete disruption of the BBB. This disruption enhances vasogenic brain edema and is thought to promote hemorrhagic transformation. Although post-stroke disruption of the BBB as a cause of brain edema has been well documented, the time course of early-phase BBB leakage, biphasic or progressive, remain subject to debate.7, 8 Examination of the impact of reperfusion is thus of prime importance in research that seeks to limit the risk of intracerebral hemorrhage in the acute phase,9 in line with a recently suggested need for more integrated approaches combining both in vivo and in vitro assessments.3
The BBB is a highly complex, dynamic interface between blood and the central nervous system, widely recognized as responsible for the maintenance of brain homeostasis. The BBB is formed by brain capillary endothelial cells endowed with barrier properties. These properties are induced and maintained by astroglial cells, and influenced by other perivascular cells in heath and disease states.10, 11 In vivo experimental access to this interface remains a technical challenge, hampering investigations of BBB function and intercellular crosstalk. Although magnetic resonance imaging (MRI) is a powerful tool for assessing focal brain lesions, the early dynamics of BBB function cannot be visualized by conventional MRI sequences.
Here, on a middle cerebral artery occlusion (MCAO) model, we applied a sequence of diffusion-weighted imaging (DWI, a non-invasive clinical MR sequence) assessing the random motion of water molecules in both the microvascular (D*) and the parenchymal (ADC: apparent diffusion coefficient) compartments of the ischemic brain area. The aim was to examine early disturbances in brain water status possibly accounting for BBB leakage and brain infarct constitution, the latter being observed through T2-weighted imaging considered as the result of increase of water content in brain tissue that is concurrent with the development of brain edema. However, in a view of understanding better the link between the permeable BBB endothelium and injury of brain parenchyma, we aimed at observing early appearance of necrosis, microvascular alterations, and astroglial activation in the brain slices. In parallel, BBB transendothelial permeability measurements and cell–cell communication studies were carried out at the same time points as in vivo, using an in vitro model that reproduces BBB characteristics.
Thus, in the present study, by using in vivo and in vitro approaches based on the same mouse strain, we evidence that post-stroke BBB endothelial leakage is initiated from 4 hours after reperfusion, and is driven by brain parenchymal cell death.
Materials and Methods
Animals
All experiments were performed in accordance with the European Community legislation (86/609/CEE November 24 1986). The local Ethics Committee approved the experiments ('Comité d'Ethique en Expérimentation Animale'). All mice in this study were males of C57Bl/6N genetic background (Janvier SAS, Le Genest-St-Isle, France) weighting 28 to 32 g according to MCAO model. Animals were housed under controlled laboratory conditions, with a 12-hour dark–light cycle, a temperature of 21±2°C, and a humidity of 60% to 70%. The animals had ad libitum access to standard chow and tap water. For the in vivo study, 90 mice were randomly distributed in two groups: 54 mice for the assessment of brain water status along the reperfusion kinetic (2 hours, 4 hours, 8 hours, 16 hours, 24 hours), and 36 mice for histologic studies. To limit the neuroprotective effect of repetitive anesthesia, subgroups of 10 mice were dedicated to the different reperfusion periods. Cerebral infarcts were produced by a 60-minute MCAO and subsequent reperfusion, as described previously.12 Surgery was performed on the daytime at 9.00 a.m. under a surgical microscope with isoflurane anesthesia (induction by 2% and maintained at 1% to 1.5%). After MCAO surgery, surviving animals were checked for success of MCAO through MRI. Details of the experimental design are described in Supplementary Figure S1.
Magnetic Resonance Imaging
All MRI experiments were performed on a Biospec 7.0T/20 cm machine with a horizontal magnet (Bruker, Ettlingen, Germany). We combined two MRI sequences including T2 relaxometry (T2r) with DWI. Animals were first anesthetized with isoflurane and then placed in a dual-coil small animal restrainer that contained a volume coil for transmitting and a surface quadratic coil for receiving. Coil-to-coil electromagnetic interaction was actively decoupled. The respiration rates and waveforms were continuously monitored via a force transducer. The rectal temperature was monitored and maintained at 37±1°C via a feedback-regulated circulating water pad.
Anatomic T2-weighted images were acquired using the fast spin-echo rapid acquisition relaxation-enhanced (TurboRARE) pulse sequence with the following parameters: repetition time: 2500 milliseconds; echo time: 33 milliseconds; matrix size: 256 × 256 pixels; field of view: 20 × 20 mm; acquisition time: less than 2 minutes. For coronal images, 12 non-overlapping 0.5 mm-thick slices were acquired. The DWI was performed on a unique axial slice positioned on the cortical and subcortical regions (defined on coronal T2-weighted images). The DWI sequence was performed in three directions [1, 0, 0], [0, 1, 0], [0, 0, 1] for which 11 b values were used to characterize signal decay. The b values were defined as follows: 1, 10, 20, 30, 40, 60, 80, 100, 400, 600, 800, and 1,000 seconds/mm2. The sequence parameters were as follows: field of view: 20 mm × 20 mm; matrix size: 128 × 128; slice thickness: 1 mm; repetition time: 3,000 milliseconds; echo time: 33 milliseconds; acquisition time: 13 minutes. To assess T1 and T2 values (relaxometry), a RARE sequence was realized with different repetition time (5,000, 3,000, 1,500, 800, 400, and 200 milliseconds) and echo time (11, 33, 55, 77, and 99 milliseconds) values. Geometric parameters were imported from the diffusion sequence to allow perfect comparison. The acquisition time for this sequence was 17 minutes.
Working Hypothesis and Magnetic Resonance Imaging Data Analysis
Diffusion-weighted imaging relies on the definition of diffusion gradients. The b value is related to tissue ultrastructural constraints of tissues and reflects water mobility. For example, diffusion gradients defined by b values ranging from 100 to 1,000 seconds/mm2 (including b=0) enable the assessment of water mobility in brain tissues (according to the ADC). Calculation of the diffusion coefficient remains complex, particularly for non-linear regression of the MRI signal decay with mono-, bi- or multi-exponential mathematical models.13 In a general view, mathematical models can be applied to the signal decay as a function of the ultrastructural constraint: 0<b value<100 seconds/mm2 for microvessels, 100<b value<1,000 seconds/mm2 for tissues (as in our study), and b value >1,500 seconds/mm2 for intra- and extracellular components (as in cell swelling, cell size variations, and the interaction between extra- and intracellular water molecules). For low b values (<100 seconds/mm2)—in which an intense signal level and fast decay are observed—the water diffusion response is related to the water mobility of the microvascular component referred to as the pseudo-diffusive component (D*) described by Le Bihan et al.14
In this study, we assumed that variations of D* signal refer to variations of the water molecules displacement in the cerebral microvascular bed, reflecting different states of the brain microvascular integrity. From our experimental diffusion data sets, we applied the bi-exponential fit model to characterize diffusion coefficients of both microvascular and tissue components. Hence, we have modified our diffusion sequence parameters by expanding the number of b values (with 6 values for 0<b values<100 seconds/mm2 and 5 values for 100<b values<1,000 seconds/mm2) and by optimizing the calculation of D* with the following equation according to Le Bihan et al14:
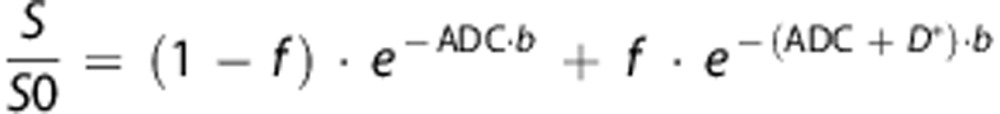 |
where S is the MRI signal, S0 is the signal intensity at b=0, b is the diffusion gradient intensity (b value) and f is the blood volume fraction (f<0.05). Considering brain edema formation during stroke, relaxation times of brain tissue (T2) may have significant differences. We thus included a correction in the data analysis for low b values, the latter being sensitive to T2r. The equation was corrected as follows:
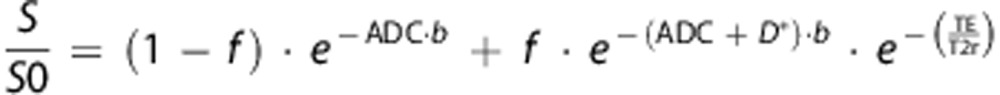 |
where TE is the echo time (33 ms), and T2r the relaxation of brain tissue without diffusion gradients (b=0). Figure 1 shows an example of the fit obtained for diffusion data sets. Non-linear regressions of diffusion data sets were performed with GraphPad software (Prism, GraphPad Software, La Jolla, CA, USA) according to the Levenberg–Marquardt method.
Figure 1.

Diffusion data set analysis. The diffusion sequence parameters were modified by expanding the number of b values (6 values ranging from 0 to 100 seconds/mm2 and 5 values ranging from 100 to 1000 seconds/mm2) to optimize the calculation, within regions of interest (1 to 4, open circles), of water mobility for both microvessels (D*) and brain tissue (apparent diffusion coefficient). From diffusion data sets, the bi-exponential fit model was applied to the signal decay as a function of the ultrastructural constraint related to diffusion gradients (b values): 0<b value<100 seconds/mm2 for the microvascular diffusion component and 100<b value<1000 seconds/mm2 for the brain tissue diffusion component. ADC, apparent diffusion coefficient.
In Vivo Blood–Brain Barrier Integrity Assessments
Evans blue perfusion
At the time of reperfusion, 2% Evans blue in physiologic saline solution (6 mL/kg bodyweight) was injected into the penis vein and allowed to circulate for 1 hour. Mice were deeply anesthetized with isoflurane and transcardially perfused with saline solution until colorless fluid was obtained from the right atrium. Brains were removed and placed in ice-cold saline solution for 10 minutes before cutting 300-μm-thick slices with a brain slicer (Vibroslice, World Precision Instruments, Sarasota, FL, USA). Evans blue extravasations were observed and considered from a purely qualitative standpoint.
FITC-Dextran perfusion
Two minutes before killing, at each time of reperfusion, 5% fluorescein isothiocyanate (FITC)-Dextran (Sigma-Aldrich, Saint-Quentin Fallavier, France) in physiologic saline solution was injected into the penis vein for 2 minutes to allow fluorescence-based visualization of BBB leakage through FITC-Dextran extravasation. After injection, brains were extracted and snap-frozen in isopentane on dry ice. The brains were cut coronally in 20-μm frozen sections. To visualize the diffusion of FITC-Dextran out of the capillaries, the samples were post fixed in −20°C methanol, stained with Hoechst, and mounted in Vectashield medium (Vector Laboratories, Burlingame, CA, USA).
In Vitro Blood–Brain Barrier Model
Mouse brain capillary endothelial cells (MBCECs) were isolated and cultured as previously described.16, 17 The resulting capillary suspension was seeded at 51,000 digested capillaries/cm2 onto Matrigel-coated (BD Biosciences, Le Pont de Claix, France) Transwell polyester membrane inserts (pore size: 0.4 μm) obtained from Costar (Corning, New York, NY, USA). The growing medium was Dulbecco's modified Eagle's medium supplemented with 15% calf serum (Hyclone Laboratories, Logan, UT, USA), 50 × amino acids, 100 × vitamins (Sigma-Aldrich), 50 μg/mL gentamicin, 2 mM glutamine, and 1 ng/mL basic fibroblast growth factor. As this model consists of a co-culture, the 24-hour seeded MBCEC monolayer inserts were set in 12-well culture plates containing a stabilized primary culture of glial cells (GCs) (confluence achieved after 3 weeks) extracted according to Booher and Sensenbrenner's method.18 This GC culture contained not only astrocytes (84%), but also microglia (6%) and oligodendrocytes (10%). Mouse brain capillary endothelial cells were also characterized for endothelial tightness (transendothelial electrical resistance=777.67±14.8 Ω/cm2) and other BBB features as described by Coisne et al.17 The medium was renewed daily until the day of experiment. Under these conditions, MBCECs migrated from digested capillaries and reached confluence ∼4 days after plating. Experiments were carried out 4 days after the co-culture setting.
Oxygen-Glucose Deprivation–Reoxygenation Studies
To avoid undue depletion of glucose in the culture medium, experiments were performed 24 hours after the last medium renewal. To simulate ischemia-like conditions in vitro, co-cultures were subjected to oxygen-glucose deprivation (OGD) as described by Mysiorek et al.16 Briefly, co-cultures were exposed to OGD for 4 hours, by replacing the culture medium with a glucose- and serum-free medium that had been equilibrated in an anaerobic atmosphere (at <0.1% O2, 5% CO2, and 95% N2) inside a temperature-controlled hypoxic workstation (Hypoxystation H35, Don Whitley Scientific, Shipley, West Yorkshire, UK). For normoxic controls, co-cultures were incubated for 4 hours in glucose-containing (1 g/L), serum-free medium equilibrated with air. Throughout the experiments (whether normoxic or with OGD), the pH of the medium remained constant. As reoxygenation (R) consisted of a return to the initial culture conditions after OGD, co-cultures were withdrawn from the hypoxic workstation (Hypoxystation H35, Don Whitley Scientific) and the OGD medium was replaced with glucose- and serum-containing culture medium. In parallel, normoxic controls were submitted to the same R procedure, to check the baseline BBB permeability after medium renewal.
Transendothelial Permeability Studies
To assess transendothelial permeability, we used the well-known fluorescent marker of BBB integrity Lucifer Yellow CH dilithium (LY, Sigma-Aldrich) at a concentration of 50 μmol/L. Endothelial cell monolayer inserts were transferred to 12-well plates containing Ringer-HEPES solution (150 mmol/L NaCl, 5.2 mmol/L KCl, 2.2 mmol/L CaCl2, 0.2 MgCl2, 6 mmol/L NaHCO3, 2.8 mmol/L glucose, 5 mmol/L HEPES). The medium on the upper side of the insert was removed and replaced by Ringer-HEPES containing 50 μmol/L LY. At 30 and 60 minutes, inserts were transferred to other wells containing Ringer-HEPES to prevent a flux of LY from the abluminal compartment to the luminal compartment. For each experimental condition, three inserts were used. Using a fluorimeter (Fluoroskan Ascent FL, Thermo Lab Systems, Issy-Les-Moulineaux, France), the fluorescence of LY was measured at each time point in aliquots of 200 μL drawn from the abluminal compartment and aliquots of 20 μL drawn from the luminal compartment and was compared with the value for the initial solution. To obtain a concentration-independent transport parameter, the clearance principle was used. The slopes of the clearance curves for the cultured insert and the control filters were referred to as PSt and PSf, respectively, where PS=permeability × surface area product. The permeability of endothelial cell monolayers (PSe) was calculated according to the following equation: 1/PSe=1/PSt−1/PSf. The permeability coefficient of endothelial cells (Pe) was obtained by dividing PSe by the insert surface area (1 cm2 for a 12-well Transwell filter). For the OGD experiments, the LY permeability assay was performed in a hypoxic workstation and with glucose-free Ringer-HEPES buffer.
Immunofluorescent Stainings
Immunofluorescent staining was performed in vivo on non-perfused, snap-frozen tissues, 20-μm frozen sections fixed in −20°C methanol, or in vitro on filters containing fixed MBCECs according to Mysiorek et al.16 To stain tight junctions (TJs), specific rabbit anti-human Claudin-5 primary antibody was used in vivo and in vitro (Zymed Laboratories, San Francisco, CA, USA). For GCs, specific rabbit primary antibody against cow glial fibrillary acidic protein (GFAP Dakocytomation S.A., Trappes, France) was used in vitro according to Coisne et al.,17 and mouse monoclonal anti-GFAP antibody conjugated with Cy3 was used in vivo (Sigma-Aldrich).
Ultrastructural Analysis
For ultrastructural analysis of MBCEC TJs, wheat germ agglutinin-conjugated horseradish peroxidase (Sigma-Aldrich) was used as an electron-dense tracer for BBB leakage according to Hamm et al19 with slight modifications. Ultrathin sections (75 nm) were obtained from the central part of the insert filter using an Ultracut UCT ultramicrotome (Leica Microsystems, Wetzlar, Germany), contrasted with uranyl acetate in H20, and examined under a Jeol 1011 transmission electronic microscope at an accelerating voltage of 100 kV.
Statistics
All results were reported as the mean±s.e.m.. Statistical analyses were performed using GraphPad Prism version 5.00 (Prism, GraphPad Software, La Jolla, CA, USA, www.graphpad.com). Inter-group differences included in vivo areas (ipsilateral versus contralateral brain infarct location) or in vitro conditions (normoxic versus OGD) and time-dependent comparisons for both. The mean scores were tested in a two-way ANOVA with Bonferroni post hoc tests. The threshold for statistical significance was set to P<0.05.
Results
Characterization of Brain Water Status during Reperfusion
On the basis of anatomic T2-weighted images, we investigated several quantitative MRI parameters over the course of reperfusion (2, 4, 8, 16, and 24 hours post occlusion; see Figure 2) including T2r parameters (the tissue water content, Figure 2A) and diffusion (water mobility, Figure 2B). We focused on the post reperfusion modifications of brain water status to better understand the dynamics of the BBB permeability in vivo in relation to brain tissue lesion. Quantitative T2-weighted relaxometry is based on changes in tissue relaxation (expressed in milliseconds) and evaluates the tissue water content in relation to the time of reperfusion. Figure 2A shows the progressive increase of the tissue water content from 55.0±2.8 milliseconds at 2 hours to 74.5±5.8 milliseconds at 24 hours in the ipsilateral cortex and from 58.2±2.2 milliseconds at 2 hours of reperfusion to 88.2±4.3 ms at 24 hours in the ipsilateral subcortex. A significant increase in T2r was observed at 4 hours after reperfusion (2 hours: 58.2±2.2 versus 4 hours: 64.0±1.0; P<0.001; n=7), possibly corresponding to a water entry into the brain tissue (beginning of brain edema), before higher values were reached at 48 hours to 72 hours (Supplementary Figure S2), which accounted for maturation of the brain lesion.
Figure 2.

Postischemic modifications of brain water status. (A) Time course of brain tissue water content. Assessment of magnetic resonance imaging quantitative parameters during reperfusion (2, 4, 8, 16, and 24 hours post occlusion) including T2-weighted (tissue water content) imaging. The tissue water content (T2r) is expressed as the mean±s.e.m. tissue relaxation time (ms) calculated for contralateral (open symbols) and ipsilateral (closed symbols) regions of interest (ROIs) in the cortex (circles) and in the subcortex (triangles). The mean scores were significantly different according to a two-way analysis of variance (ANOVA) and subsequent Bonferroni post hoc tests: ipsilateral versus contralateral cortex for each reperfusion time §P<0.05, §§P<0.01, and §§§P<0.001; ipsilateral versus contralateral subcortex for each reperfusion time ***P<0.001 (2 hours: n=7; 4 hours: n=7; 8 hours: n=8; 16 hours: n=6; 24 hours: n=9). (B) Time course of tissue water mobility. Diffusion-weighted imaging assessed random motion of water molecules in the brain. Both apparent diffusion coefficients (ADC) and D* were extracted from bi-exponential fits and are expressed as the mean±s.e.m. (.10−3 mm2/second) for contralateral (open symbols) and ipsilateral (closed symbols) ROIs of the cortex (circles) and the subcortex (triangles). Apparent diffusion coefficient values were significantly different according to a two-way ANOVA and subsequent Bonferroni post hoc tests: ipsilateral versus contralateral cortex for each reperfusion time §P<0.05, §§P<0.01, and §§§P<0.001; ipsilateral versus contralateral subcortex for each reperfusion time **P<0.01 and ***P<0.001 (2 hours: n=7; 4 hours: n=7; 8 hours: n=8; 16 hours: n=6; 24 hours: n=9). Reperfusion time points of cortical D* coefficients (left panel) were significantly different according to a two-way ANOVA and subsequent Bonferroni post hoc tests: ipsilateral cortex D* at 4 hours versus each other time point (§P<0.05 and §§P<0.01). The multiple comparison between each D* coefficient in subcortex area showed no difference (middle panel; NS: non-significant). No difference was found in the contralateral cortical and subcortical areas (right panel). (C) Time course of blood–brain barrier (BBB) leakage. Representative brain slices from Evans blue-infused animals (correspondence with ADC maps shown in panel B) at each reperfusion time point. Extravasation of Evans blue in the ipsilateral brain area was found clearly visible at 4 hours of reperfusion, compared with the next reperfusion times. The right panel displays a schematic model of the possible link between the microvascular component of the dynamic water distribution (D*) and the BBB leakage. When the BBB starts to leak after ischemia/reperfusion (‘leaky'), water molecules still circulate within the vessels (jagged arrows) but the transcapillary filtration rate is also much enhanced (dotted arrows), leading to an increase in D* and then T2r because water enters the brain parenchyma.
Apparent diffusion coefficients (ADC) for cortical and subcortical areas were calculated from the bi-exponential fit model. Contralateral ADC values were reproducible and not significantly different for the whole reperfusion period (cortex: 0.65±0.04; subcortex: 0.64±0.05; Figure 2B). After 2 hours of reperfusion, brain tissue water mobility respectively decreased by 30% and 31% in the ipsilateral cortex (contralateral: 0.73±0.05 versus ipsilateral: 0.51±0.08; P<0.05 n=7) and the ipsilateral subcortex (contralateral: 0.71±0.05 versus ipsilateral: 0.49±0.05; P<0.05 n=7). This decrease in ADC revealed brain injury for the earliest reperfusion times and validated the occurrence of brain ischemia induced by the MCAO. At the end of the 24-hour reperfusion period, ADC values were still in the same range of (0.48±0.01) for cortical area and (0.45±0.02) for subcortical area (n=9).
Increase in Microvascular Water Mobility Coincides with Blood–Brain Barrier Leakage In Vivo
In terms of microvascular water mobility, the D* coefficient (expressed as the mean±s.e.m. 10−3 mm2/second) clearly showed a climax at 4 hours after reperfusion in the ipsilateral cortex (34.8±12.4 at 2 hours and 111.4±26.2 at 4 hours; P<0.01; n=7, Figure 2B). This increase was not significant in the ipsilateral subcortex (24.9±9.1 at 2 hours and 71.4±15.9 at 4 hours; NS; n=7, Figure 2B). The values then progressively returned to baseline for both cortical and subcortical areas (with the exception of the subcortical D*, which tended to a second increase at 24 hours (61.4±20.0; n=9; NS). Contralateral D* values were reproducible and not significantly different throughout the whole reperfusion period (cortex: 31.1±8.0; subcortex: 40.4±13.7).
To test whether the dynamic water distribution was potentially related to BBB leakage, we used a standard, in vivo administration of Evans blue for 1 hour over a similar reperfusion period. Our analysis of Evans blue extravasation remained qualitative, as the objective of such a short time infusion was to determine the starting time of the loss of BBB integrity. We drew ADC maps to assess the brain ischemic site before Evans blue experiments. Extravasation of Evans blue in the ipsilateral brain region was not found in sham animals (Supplementary Figure S3), but was clearly visible at 4 hours of reperfusion, as indicated by the bluish-colored tissue, compared with the next reperfusion time points (Figure 2C). Figure 2C right panel illustrates our hypothesis linking D* and BBB function.
As the ischemic cortex was heterogeneously stained in contrast to the subcortical region (Figure 2C), we have further scaled down the ROI to microscopic areas (‘micro-ROIs') where BBB leakage could be visualized under the microscope, to determine the D* values corresponding to ‘tight' or ‘leaky' BBB microvessels. FITC-Dextran (77 kDa) was used as a fluorescent tracer for assessing BBB TJ function. The tracer was kept in the lumen of tight capillaries, revealing the vascular segments, but diffused out of the leaky capillaries, which were then surrounded by a blurred signal alongside the vascular segments (see inner windows in Figure 3A). Leaky capillaries were mostly present in ipsilateral cortices at 4 hours and 24 hours after reperfusion (Figure 3A), where heterogeneous D* values ranged from 0.3 to 299 × 10−3 mm2/second at 4 hours, and from 1.8 to 140.9 × 10−3 mm2/second at 24 hours (Figure 3B). A more intense fluorescence signal was globally noticed at 4 hours and 24 hours after reperfusion in microvessels of both ipsilateral and contralateral hemispheres, compared with the microvessels at 2 hours after reperfusion and the ones at 24 hours after sham operation. Some leaky capillaries were sparsely found in contralateral cortices at 4 hours and 24 hours, although the fluorescent signal was clearly located alongside the capillary segments at 24 hours only (see inner windows in Figure 3A). Moreover, no leaky capillary was observed in both hemispheres of the sham-operated animals at 24 hours, accounting for the absence of leakage with time after sham operation. Tight capillaries were observed in both hemispheres of sham-operated animals, mainly in the contralateral cortices of ischemic brains and in the ipsilateral cortices of ischemic brains at 2 hours, but also sometimes at 4 hours and 24 hours. D* values were less variable at 2 hours since ranging from 0.5 to 36 × 10−3 mm2/second.
Figure 3.

Heterogeneity in microvascular water mobility (D*) and blood–brain barrier (BBB) leakage. (A) Visualization of BBB function. Apparent diffusion coefficient maps were drawn to localize brain infarction for each reperfusion time: 2 hours (n=4), 4 hours (n=4), and 24 hours (n=3). Brain slices from fluorescein isothiocyanate (FITC)-Dextran-infused animals illustrate the different states of BBB function in contralateral and ipsilateral cortices observed (scale bar, 50 μm) from indicated fields of view (square windows). Extravasation of fluorescein isothiocyanate (FITC)-Dextran was clearly visible through a blurred fluorescence signal alongside the capillary segments at 4 hours and 24 hours of reperfusion. No extravasation was observed in hemispheres of 24-hour sham-operated animals. Magnified images (inner windows) illustrate typical morphology of capillaries in each field of view, as revealed by intravascular FITC-Dextran (for tight capillaries) or extravasated FITC-Dextran alongside the leaky capillaries (scale bar, 10 μm). (B) D* calculations from diffusion-weighted imaging (DWI). As a case study, within each microscope field of view (rectangles), 72 micro-regions of interest (ROIs) (circles) were defined in six zones along the cortex (contralateral: 3; ipsilateral: 3). In each zone, 12 micro-ROIs were drawn. Results in vertical scatter plots give the heterogeneous repartition of D* values at each reperfusion time (2, 4, and 24 hours).
Early Blood–Brain Barrier Leakage under Ischemia/Reperfusion is Linked to Brain Tissue Injury, but not to Astroglial Activation
To better understand the link between the BBB permeability changes and the brain lesional status, Nissl staining was first performed after 2, 4, and 24 hours of reperfusion, and showed cell rarefaction in the brain tissue at 24 hours of reperfusion, that was slightly observable at 4 hours but not at 2 hours (Figure 4A). Then, we performed propidium iodide injections to highlight dead cells, i.e. those that have undergone necrosis. Hoechst is a hydrophobic molecule able to stain all cell nuclei, whereas propidium iodide is a hydrophilic molecule that only stains the nucleus of dead cells (i.e. those with a permeable cell membrane). Propidium iodide-positive cells were visible in the ipsilateral area both at 4 hours and 24 hours of reperfusion, but not at 2 hours (Figure 4B).
Figure 4.

Lesional status of ischemic brain tissue and blood–brain barrier cells. (A) Structural integrity of brain tissue. After the determination of brain lesions through the drawing of apparent diffusion coefficient (ADC) maps, tissue damage was visualized by Cresyl violet staining in sham or ischemic animals after 2 hours, 4 hours, or 24 hours of reperfusion. (B) Necrotic cell death in brain tissue. Brain cell necrosis was assessed in sham or ischemic animals after 2 hours (n=4), 4 hours (n=3), or 24 hours (n=3) of reperfusion, by double nuclear staining: Hoechst 33258 (blue) stained all cells, and propidium iodide (PI, red) stained dead cells. Scale bar, 50 μm. In an inner window, a magnified image of Hoechst/PI-stained nuclei (scale bar, 5 μm). (C) Structure of blood–brain barrier endothelial tight junction network. Immunostaining of Claudin-5 tight junction protein was performed in sham or ischemic animals after 2 hours (n=2), 4 hours (n=2) or 24 hours (n=2) of reperfusion. Nuclei were stained by Hoechst 33258 (blue). Scale bar, 100 μm. In inner windows, a magnified typical image shows continuous Claudin-5 network (scale bar, 5 μm). (D) Astroglial activation in brain tissue. Immunostaining of glial fibrillary acidic protein (GFAP) was performed in sham or ischemic animals after 2 hours (n=2), 4 hours (n=2), 24 hours (n=2) or 72 hours (n=3) of reperfusion. Nuclei were stained by Hoechst 33258 (blue). Scale bar, 100 μm. In the inner windows, a magnified image shows the rare astroglial cells activated in the cortex (scale bar, 5 μm). The corpus callosum is indicated by an asterisk. H: Hoechst. In panels B, C, D, merged images of ipsilateral cortex are represented.
The endothelial Claudin-5 TJ protein was immunostained in a view to observe the impact of stroke on the structure of BBB capillaries. A continuous network was observed in the capillary segments of both ipsilateral and contralateral hemispheres at all three time points (Figure 4C).
As the BBB endothelium is almost entirely connected to the brain tissue through the end-feet of astrocytes, a glial cell type considered as a major part of the cell population in the rodent brain,20 the impact of the deprived conditions on astroglial cells was first assessed in vivo. Immunostaining of GFAP was undergone at the same time points as previously in an attempt to observe early astroglial activation in response to ischemia/reperfusion. The results presented in Figure 4D show a very few isolated cells stained in the ipsilateral but not in the contralateral cortices at 2 hours, 4 hours, and 24 hours. At 72 hours, massive staining was found throughout the ipsilateral cortex, and thus shown as a positive control for astroglial activation. The edge of corpus callosum displayed a steady signal of astroglial activation in all hemispheres and conditions, thus considered as not relevant to ischemia. Sham-operated animals did not display activated astrocytes (see Supplementary Figure S4).
As the GFAP staining revealed a sparse pattern of activated astrocytes, the input of these BBB-influencing cells on the endothelium early leakage was then investigated in vitro, i.e. in a simplified stroke paradigm. A co-culture cell model of the BBB was used in experimental conditions simulating some of the immediate consequences of ischemia/reperfusion, named as ‘OGD-reoxygenation'. This model was based on cells from the same mouse strain as used in our MRI studies. After incubating the co-culture under OGD conditions for 4 hours, we evidenced a ninefold increase in BBB MBCEC permeability to LY (versus control) (Figure 5A), a tracer molecule widely considered to enable assessment of variations in BBB permeability.21 This initial increase was followed by several permeability variations over the course of R. After 2 hours of R, a decrease in MBCEC permeability was observed, leading to a baseline permeability value of 0.61 × 10−3 cm/minute (i.e. below our model tightness threshold of 1 × 10−3 cm/minute, according to Culot et al.21). The reduction in permeability after 2 hours of R was followed by a second increase after 4 hours of R (9.3-fold versus control). The permeability tended to be lower after 8 hours and 16 hours of R but was still above the baseline (i.e. >1 × 10−3 cm/minute). We did not observe any significant increase in permeability during R in normoxic controls.
Figure 5.

Blood–brain barrier response to oxygen-glucose deprivation (OGD)–reoxygenation in vitro. The permeability of a mouse brain capillary endothelial cells (MBCEC) monolayer to Lucifer Yellow (LY) after incubation of the co-culture for 4 hours under normoxic conditions (open bars) or OGD conditions (filled bars), followed by 2 hours, 4 hours, 8 hours, 16 hours, or 24 hours of reoxygenation (A). In other experiments, glial cells (GCs) were removed during reoxygenation (B) or replaced by GCs derived from a co-culture that had not been submitted to OGD (C). The results are presented as the mean fold-increase versus control±s.e.m. The control permeability corresponds to a Pe value of 0.34 × 10−3 cm/minute (A), 0.24 × 10−3 cm/minute (B), and 0.34 × 10−3 cm/minute (C) for LY after 4 hours of normoxia. Culture medium renewal during reoxygenation did not alter the baseline permeability under control conditions. The mean scores were significantly different, according to a two-way analysis of variance and subsequent Bonferroni post hoc tests. *P<0.05, **P<0.01, ***P<0.001 (n=9 endothelial cell (EC) monolayers per treatment (A–C) and n=6 EC monolayers per treatment (B)). NS, non-significant, C, control, R, reoxygenation.
Preserved Glial Cells Allow Recovery of the Blood–Brain Barrier Endothelium Tightness during Reoxygenation In Vitro
To study the influence of GCs on MBCECs permeability during R, GC cultures were first removed from the in vitro system after OGD. After 4 hours of OGD, the MBCEC-containing filter insert was transferred to an empty well plate containing culture medium. After the initial sevenfold permeability increase induced by OGD, the removal of GCs during R prompted the MBCEC permeability to return to baseline values (0.82 × 10−3 cm/minute) between 8 hours and 24 hours of R. Only the permeability value after 2 hours and 4 hours of R (2.52 × 10−3 cm/minute and 1.47 × 10−3 cm/minute, respectively) remained high (i.e. above the tightness threshold of >1 × 10−3 cm/minute) (Figure 5B).
In order to clarify whether the GC injury in vitro is the status that promotes the BBB reopening during R, a non-ischemic culture of GCs was used during 2 hours to 24 hours of R. After incubation of the co-culture under OGD conditions, the MBCEC-containing filter insert was transferred to a new well plate containing GCs that had not been exposed to OGD. A the first (5.6-fold) increase in MBCEC permeability induced by OGD, we observed a threefold reduction of MBCEC permeability at 2 hours of R. The return to a baseline permeability value (of 0.85 × 10−3 cm/minute) was maintained below the tightness threshold of 1 × 10−3 cm/minute through to 24 hours of R (0.48 × 10−3 cm/minute, Figure 5C).
Whereas no specific pattern of activated astroglia was seen around the ischemic areas up to 24 hours of reperfusion in vivo, the BBB in vitro model enabled the assessment of astroglial activation and GC viability, as the system consists of a co-culture of GCs and MBCECs in separate compartments (well and insert). A GFAP staining revealed activated astrocytes in normoxic as well as in OGD-reoxygenation conditions (Figure 6). Glial cell viability was studied with both Hoechst/propidium iodide and LDH release assays. Propiod iodide stained GC nuclei both in OGD and R conditions. The control value of 1% LDH rose by 70% after OGD, evidencing a high rate of cell death (Figure 6). The remaining GCs continued to die up until 24 hours of R.
Figure 6.

In vitro post oxygen-glucose deprivation(OGD)/reoxygenation (R) glial cell (GC) lesional status. Glial cell necrosis was assessed by double nuclear staining: Hoechst 33258 (blue, left-handed lane) stained all cells, and propidium iodide (PI) (red, middle lane) stained dead cells, and merged images on the next lane to the right. Astrocyte morphology was observed by glial fibrillary acidic protein (GFAP) staining (right-handed lane). This experiment was completed by a measurement of activity of the lactate dehydrogenase (LDH) released into the culture medium (n=9). The death rate under normoxic or OGD conditions is expressed as a percentage of a totally lysed GC well (full kill) and indicated in the top, right-hand corner of images. Cell death was observed with either method. Images are representative of cell populations under each set of experimental conditions. Scale bar, 25 μm.
As the BBB leakage in vivo was observed without interruption of the Claudin-5 TJ network, a study of both structure and ultrastructure of TJs was performed from our MBCECs to validate the recovery of BBB tightness observed in vitro when non-ischemic GCs were present during R. Claudin-5 immunostaining revealed a continuous TJ network both when the BBB was tight in normoxia or opened in OGD (Figure 7A, left-handed lane). By contrast, the following 24 hours of R caused an interruption in the staining of Claudin-5 protein, evidencing an alteration of the TJ network (Figure 7A, middle lane). In the presence of non-ischemic GCs during R, the Claudin-5 network was continuous again (Figure 7A, right-handed lane). Furthermore, an ultrastructural analysis of the TJs (using wheat germ agglutinin-conjugated horseradish peroxidase tracer) enabled us to have a magnified view of endothelial TJ states in our experimental conditions. Transmission electron microscopy (TEM) analysis revealed subtle, early-onset permeabilization of the TJs in the MBCEC monolayers. Surprisingly, 60% of TJs were permeable to wheat germ agglutinin-conjugated horseradish peroxidase and 40% were still impermeable following OGD (Figure 7B). By contrast, after 24 hours of R, the opened BBB had disorganized TJs since 50% were completely disrupted and 50% were permeable. The use of non-ischemic GCs allowed the observation of 97% of impermeable TJs and 3% of permeable TJs, illustrating the recovery of BBB tightness previously evidenced through LY transport experiments.
Figure 7.

Analysis of tight junction (TJ) integrity in blood–brain barrier endothelial cells in vitro. (A) Structure of endothelial tight junction network in vitro. Immunostaining of Claudin-5 (Green) after 4 hours of normoxia or oxygen-glucose deprivation (OGD) (left-handed lane), followed by 24 hours of reoxygenation with ischemic glial cells (GCs) (reoxygenation (R), middle lane) or non-ischemic GCs (right-handed lane). Interruptions in the staining are indicated by white arrows and were observed only after 24 hours of R with ischemic GCs. Nuclei are stained with Hoechst (blue). Scale bar, 50 μm. (B) Ultrastructure of endothelial tight junctions in vitro. Transmission electron microscopy observations were performed after wheat germ agglutinin-conjugated horseradish peroxidase (WGA-HRP) incubation to account for TJ function. The morphology of the endothelial cell (EC) monolayer as grown on filter inserts (filter) in normoxic control conditions is presented in the top left panel. Two endothelial cells, (EC1 and EC2) are shown, with a magnified view of the TJ in the top right corner of the picture. The different TJ states observed from EC monolayers in normoxic or OGD-R conditions are presented in the lower panel, and the ones for both early onset OGD and late (after 24 hours of R) blood–brain barrier (BBB) openings are rated in the histograms in the top right panel, compared with tight BBB conditions when in normoxia or after 24 hours of R in the presence of non-ischemic GCs, respectively. When the TJ was functional, WGA-HRP (black stain) could not diffuse between the cells (arrowhead) and the junction was considered as impermeable. The picture was obtained from a monolayer after 4 hours of normoxia, and is representative of impermeable junctions observed in endothelial monolayers under normoxia, or under 4 hours of OGD. In contrast, non-functional (i.e. permeable) TJs allowed the WGA-HRP to penetrate through the inter-endothelial cleft and to reach the EC abluminal membrane (black arrow). The picture was obtained from a monolayer after 4 hours of OGD, and is representative of permeable junctions observed in endothelial monolayers under 4 hours of OGD followed or not by 24 hours of R. In the worst case, after 24 hours of R, physical separation of the ECs corresponded to a disrupted junction. The picture was obtained from a monolayer after 4 hours of OGD followed by 24 hours of R, and is representative of disrupted junctions observed only in this condition. When the ECs were incubated with non-ischemic GCs during R, the recovered ECs displayed impermeable TJs. The picture was obtained from a monolayer after 4 hours of OGD followed by 24 hours of R with non-ischemic GCs, and is representative of impermeable TJs observed in this condition. The EC luminal side is indicated by an asterisk.
Discussion
Studying the dynamics of post-stroke BBB permeability in vivo or in vitro is still a major challenge. The various experimental stroke models (animal species, occlusion methods, stress duration) combined with the BBB integrity measurement techniques, may give different BBB integrity kinetics, leading to either linear22 or biphasic23 opening schemes, with no correlation with in vitro BBB permeability so far. In currently used in vivo stroke models assessed by MRI, gadolinium-based contrast agents barely cross the BBB in the first 24 hours of reperfusion unless the BBB is physically disrupted (by electrocoagulation, for example).24, 25 Hence, we hypothesized that use of DWI to assess water molecule movements in the microvascular compartment would be a rapid, non-invasive technique. Our study is the first to report post ischemic variations in the microvascular component of diffusion (D*) in the brain. For that purpose, we have ranged our b values from 0 to 1,000 second/mm2 so as to refer to the movements of water molecules in both brain tissue and microvasculature without including high b values (from 1,500 s/mm2), which would refer to the movements between extra- and intracellular compartments (as in cell swelling). A link between the increase in D* and the start of BBB leakage at 4 hours of reperfusion may be proposed, in view of the observed peak in D* and Evans blue extravasation in the brain ischemic area. These two parameters matched in the subcortical region of the ipsilateral hemisphere, but not in the cortex where sparse spots of Evans blue were seen within the ROI. Such a heterogeneous pattern of BBB leakage is in line with the complex cerebrovascular dysfunction suggested to occur within the penumbra, which evolves heterogeneously toward infarction.26, 27 This idea was recently supported by clinical evidences of a highly variable rate of progression of the brain tissue to irreversible injury between human individuals.28 At the next time of reperfusion in our study, the diffusion of the dye into the parenchyma may be slowed down the increased tissue hydrostatic pressure after enhanced water entry, possibly limiting visual detection of the dye after 1 hour of infusion.29 This early-onset, non-linear BBB leakage suggests that the microvascular mechanisms contributing to the initiation of edema have more a complex time course than the progressive development of edema found here from the T2r signals detected up to 72 hours in vivo (Supplementary Figure S2), in accordance with the literature.30 Diffusion-weighted imaging is commonly used in clinical practice to determine (according to diffusion–perfusion mismatch) eligibility of patients for thrombolysis. Our observation of the D* signal suggested that early-onset BBB leakage can occur while the ADC signal just slightly changes in the short reperfusion times of our study. The use of fluorescent Dextran injection resulted in the observation of leaky capillaries with increased D* values, although highly heterogeneous at 4 hours, in ischemic cortex, confirming the potential of D* to account for early BBB leakage. By using the same diffusion sequence, D* could be a tool for revealing the BBB integrity before 4 hours of reperfusion, which coincides with the therapeutic window for rtPA (recombinant tissue-type plasminogen activator).31 However, much research is needed to figure out whether D* could be a diagnostic tool for thrombolysis.
A link between BBB leakage and brain infarction is suggested here through both the increase in T2r signal (Figure 2B), and the parenchymal necrosis (Figures 4A and 4B) slightly detected from 4 hours (but not seen at 2 hours) after reperfusion. These data may not be in accordance with a recent study on rats from Jin et al.32 who showed major BBB leakage in non-infarcted areas after 2 or 3 hours of MCAO followed by 10 minutes of reperfusion. As the experimental stroke model was quite different from ours, the results of that study suggest a different stroke severity and potentially different mechanisms than the ones occurring here to open the BBB from 4 hours after reperfusion. The cellular events underlying the early-onset dysfunction of the BBB remain unclear, especially the intercommunications between the BBB endothelial cells and the damaged parenchyma in this time frame. Microvascular Claudin-5 network seemed to be preserved with time of reperfusion up to 24 hours, whereas astroglial activation was almost absent (Figures 4C and 4D). This last finding is in accordance with a recent work of Barreto et al.,33 evidencing a sparse pattern of activated astrocytes in early times after stroke, an event considered by the author as significant only from 48 hours after stroke. The BBB, although known to be induced and maintained by astroglial cells, belongs to an even more complex functional unit comprising a range of different cell types, termed the neurovascular unit or neurogliovascular unit. All these brain cell types may finally die from ischemia through a yet undefined time profile, except for neurons, which first undergo excitotoxic cell death. To reduce the complexity of this cellular unit as well as of ischemic stroke, we tried to use an in vitro model conjoining BBB ECs with GCs, and to study, in this environment, the contribution of GCs to the early leakage of BBB endothelium. Still, the relevance of modeling the consequences of stroke in vitro on the BBB cells is a major issue.34 The in vitro BBB model used in the present study displayed the main characteristics of a functional BBB.17 This syngeneic mouse model is a primary culture and has been used to examine drug-induced protection of the BBB under OGD conditions.16 Although the possible loss of some BBB characteristics in vitro cannot be ruled out, our model has proved to be valuable in permeability as well as cell–cell communication studies.16, 17 Our present objective was to apply some of the effects of reperfusion on these co-cultures by re-introducing, after OGD, glucose- and serum-containing, air-equilibrated medium to cells from mice with the same genetic background as in our MRI study. Our present in vitro study confirmed the occurrence of a BBB opening event at 4 hours of R, in agreement with our in vivo DWI D* data and the Evans blue extravasation time profile. This early leakage may be a critical event that precedes an intensified BBB breakdown as suggested by the greatly enhanced water content in the brain in vivo at 48 hours and 72 hours (Supplementary Figure S2). In vivo, although the dysfunction of capillary TJs in ischemic territories (Figure 3) seemed to expand with time, their structure (Claudin-5 network) did not show any difference between sham and ischemic time points. In vitro, the enhanced disruption of TJs observed at 24 hours after R under photon microscope, compared with OGD (Figure 7A), might evoke a higher degree of BBB damage. But as the permeability values at 24 hours were not higher than the ones under OGD, where no disruption of the TJ network was seen, one cannot simply regard the TJs as the only and real-time quantitative markers of BBB breakdown in our early conditions, as recently suggested.35 This idea is further supported by the observation under TEM of impermeable TJs and permeable TJs in the same EC monolayers subjected to OGD, and of permeable TJs and disrupted TJs in the same EC monolayers after 24 hours of R. This suggests a non-synchronous response of ECs to stress, suggesting that the extent of brain edema is more related to the duration of BBB leakage rather than a mere and uniform increase in BBB endothelium permeability. However, in our early ischemic paradigm, our in vitro experiments confirm the transient nature of the BBB early leakage observed through a recovery at 2 hours of R, possibly underlying reversible mechanisms that could be interesting therapeutic targets.
Given that the 4 hours BBB opening was observed in our in vitro model in which GC viability is compromised, and that parenchymal necrosis detection started at the same time point after reperfusion in vivo, we propose that the damaged brain tissue triggers this early BBB leakage. In this process, the input of GCs was hypothesized, as these cells represent a major proportion of the cell population in the rodent brain20 and the prime maintainers of BBB integrity.36 Unexpectedly, the sparse pattern of activated astrocytes observed in vivo is not in accordance with the substantial death of our consistently activated astrocytes in vitro. Indeed in our in vitro system, astrocytes are constitutively activated, as GFAP staining is present both in normoxic and stroke-like conditions (Figure 6). As activated astrocytes were present in normoxic co-cultures displaying low BBB baseline permeability values, one may theorize that astroglial activation may not be invariably associated with a BBB-increasing event. Still, the great GC death observed here in vitro is surprising, as these cells are generally regarded as resistant to injury compared with neurons. Our severe anoxic (<0.1% O2) condition applied on the cells may accelerate GC death, and would then mimic a grave injury, which may be rather occurring heterogeneously within the ischemic area in vivo. Besides, as neurons are absent from our in vitro system, the contribution of neuronal damage on the BBB leakage must not be excluded in vivo, despite the more considered impact of GCs, either deleterious or protective.37, 38 Hence, it is tempting to speculate that the constitutive activation of astrocytes in our in vitro system could increase their susceptibility to OGD, enabling them to die in the early hours of stroke as neurons may do in vivo, and causing BBB leakage with the same time profile. Furthermore, this put forward the idea that a general signal from necrotic brain parenchymal cells may act as a BBB-permeabilizing stimulus, regardless of the brain parenchymal cell type. However, our in vitro model enabled us to observe that MBCECs were capable to recover their baseline permeability transiently (at 2 hours of R) when the GCs were injured or permanently (up to 24 hours of R) when the GCs were removed. Such a recovery is often argued with complex mechanisms involving dynamic TJ phosphorylations.7 These observations support the hypothesis that our ECs may not die under our in vitro stroke-like conditions, in contrast to our GCs. Nevertheless, possible EC death cannot be ruled out, owing to the complexity of the neurogliovascular unit pathophysiology,39 still cautioning that the different cells supporting neurons may be also impacted by stroke, generating specific signaling cascades.40 In addition, as the use of non-ischemic GCs during R promoted earlier MBCEC recovery, we suggest that in a non-lesional context, the BBB endothelium may be able to recover its baseline permeability. But one cannot rule out that in vivo, other features of reperfusion such as hemodynamics (i.e hyperemia), that are not present in our in vitro system, may enhance the BBB leakage in lesioned areas, or promote it in non-lesioned areas, although hyperemia is known to act mainly within minutes after reperfusion.7 As the astroglial cells of our in vitro model are not in contact with the BBB endothelium, we may also propose that our in vitro BBB endothelium leakage has occurred independently of any astrocyte end-feet detachment, though widely recognized as a typical mechanism of BBB breakdown,7, 41 or sometimes of recovery.42 As cells can communicate only via soluble factors in our in vitro system, we speculate that necrotic secretions of the injured parenchyma may contribute to the BBB leakage.
Finally, our study emphasizes the great therapeutic potential of preserving the brain parenchymal cells from ischemia/reperfusion injury in the 2–4 hours time frame post reperfusion. The interest in considering such an early BBB permeability increase has been recently suggested through a pharmacological intervention using neurotrophic factors in the 3–7 hours time frame, which improved neurovascular outcome after 24 hours of transient stroke, but without unveiling the targeted tissue.43 A recent preconditioning study using a different in vitro BBB model suggested increasing the GC resistance to ischemia preventively.44 As the necrotic secretions of the ischemic brain parenchyma may comprise multiple molecules, broad-based approaches such as secretomics or magnetic resonance spectroscopy may be of value so as to efficiently find the key molecule(s) that permeabilize(s) the BBB in this 2–4 hours time frame, which could be ultimately targeted for protecting the neurogliovascular unit.
In conclusion, this work is the first to evidence an in vivo/in vitro BBB dysfunction time profile throughout early reperfusion after ischemic stroke in mice. At 4 hours after reperfusion, the BBB may be leaky upon parenchymal damage, emphasizing the key role of brain tissue in BBB endothelium opening within the thrombolytic time window. Furthermore, the possibility of a collective BBB-breaking response to ischemia, shared by the different parenchymal cell types when necrosis is involved, may open new perspectives in brain protection, as encouraged in the literature.36 However, additional investigations are needed to identify the key molecule(s) of this response, and to validate them as potential target(s) for protecting the brain efficiently in the early phases of stroke.
Acknowledgments
The authors would like to thank Dr Elodie Vandenhaute, Professor Marie-Pierre Dehouck, and Professor Brigitte Arbeille for the interesting discussions related to electron microscopy pictures obtained from the RIO Electron Microscopy Facility of Francois Rabelais University. In addition, we would like to thank Mr Florent Auger and Mr Nicolas Durieux for their technical contribution to the MRI study, Ms Camille Pottey and Mrs Meryem Tardivel for their great technical assistance in fluorescent Dextran experiments and confocal microscopy (BICel-IFR114 imaging platform). To end, we would like to thank Dr Grégory Lestiennes for his helpful assistance in mathematics related to the bi-exponential fit model applied to MRI data.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreements no. 201024 and no. 202213 (European Stroke Network). Mélanie Kuntz and Caroline Mysiorek have received a doctoral fellowship from the 'Conseil régional du Nord-Pas-de-Calais'.
Supplementary Material
References
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Mehra M, Henninger N, Hirsch JA, Chueh J, Wakhloo AK, Gounis MJ. Preclinical acute ischemic stroke modeling. J Neurointerv Surg. 2012;4:307–313. doi: 10.1136/neurintsurg-2011-010101. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Tymianski M. Nonhuman primate models of stroke for translational neuroprotection research. Neurotherapeutics. 2012;9:371–379. doi: 10.1007/s13311-012-0115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Chen M, Prabhakaran S. New devices for treating acute ischemic stroke. Recent Pat CNS Drug Discov. 2010;5:118–134. doi: 10.2174/157488910791213176. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37:3–23. doi: 10.1111/j.1365-2990.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke. 2004;35:2220–2225. doi: 10.1161/01.STR.0000138023.60272.9e. [DOI] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonskiy DA, Sukstanskii AL. Theoretical models of the diffusion weighted MR signal. NMR Biomed. 2010;23:661–681. doi: 10.1002/nbm.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan D. The 'wet mind': water and functional neuroimaging. Phys Med Biol. 2007;52:57–90. doi: 10.1088/0031-9155/52/7/R02. [DOI] [PubMed] [Google Scholar]
- Le Bihan D, Breton E, Lallemand D, Aubin ML, Vignaud J, Laval-Jeantet M. Separation of diffusion and perfusion in intravoxel incoherent motion MR imaging. Radiology. 1988;168:497–505. doi: 10.1148/radiology.168.2.3393671. [DOI] [PubMed] [Google Scholar]
- Mysiorek C, Culot M, Dehouck L, Derudas B, Staels B, Bordet R, et al. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr Neurovasc Res. 2009;6:181–193. doi: 10.2174/156720209788970081. [DOI] [PubMed] [Google Scholar]
- Coisne C, Dehouck L, Faveeuw C, Delplace Y, Miller F, Landry C, et al. Mouse syngenic in vitro blood-brain barrier model: a new tool to examine inflammatory events in cerebral endothelium. Lab Invest. 2005;85:734–746. doi: 10.1038/labinvest.3700281. [DOI] [PubMed] [Google Scholar]
- Booher J, Sensenbrenner M. Growth and cultivation of dissociated neurons and glial cells from embryonic chick, rat and human brain in flask cultures. Neurobiology. 1972;2:97–105. [PubMed] [Google Scholar]
- Hamm S, Dehouck B, Kraus J, Wolburg-Buchholz K, Wolburg H, Risau W, et al. Astrocyte mediated modulation of blood-brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell Tissue Res. 2004;315:157–166. doi: 10.1007/s00441-003-0825-y. [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S, Mota B, Lent R. Cellular scaling rules for rodent brains. Proc Natl Acad Sci USA. 2006;103:12138–12143. doi: 10.1073/pnas.0604911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culot M, Lundquist S, Vanuxeem D, Nion S, Landry C, Delplace Y, et al. An in vitro blood-brain barrier model for high throughput (HTS) toxicological screening. Toxicol In Vitro. 2008;22:799–811. doi: 10.1016/j.tiv.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, Pedrono E, et al. The blood-brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153:175–181. doi: 10.1016/j.neuroscience.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Witt KA, Mark KS, Sandoval KE, Davis TP. Reoxygenation stress on blood-brain barrier paracellular permeability and edema in the rat. Microvasc Res. 2008;75:91–96. doi: 10.1016/j.mvr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, et al. Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 2000;31:1965–1972. doi: 10.1161/01.str.31.8.1965. [DOI] [PubMed] [Google Scholar]
- Stoll G, Kleinschnitz C, Meuth SG, Braeuninger S, Ip CW, Wessig C, et al. Transient widespread blood-brain barrier alterations after cerebral photothrombosis as revealed by gadofluorine M-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2009;29:331–341. doi: 10.1038/jcbfm.2008.129. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Sharp FR, Heiss WD, Albers GW. Heterogeneity in the penumbra. J Cereb Blood Flow Metab. 2011;31:1836–1851. doi: 10.1038/jcbfm.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss WD. The ischemic penumbra: how does tissue injury evolve. Ann N Y Acad Sci. 2012;1268:26–34. doi: 10.1111/j.1749-6632.2012.06668.x. [DOI] [PubMed] [Google Scholar]
- Fisher M, Albers GW. Advanced imaging to extend the therapeutic time window of acute ischemic stroke. Ann Neurol. 2013;73:4–9. doi: 10.1002/ana.23744. [DOI] [PubMed] [Google Scholar]
- Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–268. doi: 10.1016/S1474-4422(07)70055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virley D, Beech JS, Smart SC, Williams SC, Hodges H, Hunter AJ. A temporal MRI assessment of neuropathology after transient middle cerebral artery occlusion in the rat: correlations with behavior. J Cereb Blood Flow Metab. 2000;20:563–582. doi: 10.1097/00004647-200003000-00015. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Jin X, Liu J, Yang Y, Liu KJ, Liu W. Spatiotemporal evolution of blood brain barrier damage and tissue infarction within the first 3 h after ischemia onset. Neurobiol Dis. 2012;48:309–316. doi: 10.1016/j.nbd.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Barreto GE, White RE, Xu L, Palm CJ, Giffard RG. Effects of heat shock protein 72 (Hsp72) on evolution of astrocyte activation following stroke in the mouse. Exp Neurol. 2012;238:284–296. doi: 10.1016/j.expneurol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25:59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger M, Hartig W, Reichenbach A, Bechmann I, Michalski D. Blood-brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS One. 2013;8:e56419. doi: 10.1371/journal.pone.0056419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. 2012;18:3624–3644. doi: 10.2174/138161212802002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50:307–320. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodriguez JJ, Parpura V. Astroglia in neurological diseases. Future Neurol. 2013;8:149–158. doi: 10.2217/fnl.12.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Ahmad A, Taboada CB, Gassmann M, Ogunshola OO. Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J Cereb Blood Flow Metab. 2011;31:693–705. doi: 10.1038/jcbfm.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci. 2012;1268:21–25. doi: 10.1111/j.1749-6632.2012.06691.x. [DOI] [PubMed] [Google Scholar]
- Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. 2011;71:1018–1039. doi: 10.1002/dneu.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis CL, Nolan CC, Reith SN, Lister T, Prior MJ, Guerin CJ, et al. Focal astrocyte loss is followed by microvascular damage, with subsequent repair of the blood-brain barrier in the apparent absence of direct astrocytic contact. Glia. 2004;45:325–337. doi: 10.1002/glia.10333. [DOI] [PubMed] [Google Scholar]
- Pillai DR, Shanbhag NC, Dittmar MS, Bogdahn U, Schlachetzki F. Neurovascular protection by targeting early blood-brain barrier disruption with neurotrophic factors after ischemia-reperfusion in rats(*) J Cereb Blood Flow Metab. 2013;33:557–566. doi: 10.1038/jcbfm.2012.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesuete R, Orsini F, Zanier ER, Albani D, Deli MA, Bazzoni G, et al. Glial cells drive preconditioning-induced blood-brain barrier protection. Stroke. 2011;42:1445–1453. doi: 10.1161/STROKEAHA.110.603266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.