

Abstract
Patients with aneurysmal subarachnoid hemorrhage (SAH) frequently have deficits in learning and memory that may or may not be associated with detectable brain lesions. We examined mediators of long-term potentiation after SAH in rats to determine what processes might be involved. There was a reduction in synapses in the dendritic layer of the CA1 region on transmission electron microscopy as well as reduced colocalization of microtubule-associated protein 2 (MAP2) and synaptophysin. Immunohistochemistry showed reduced staining for GluR1 and calmodulin kinase 2 and increased staining for GluR2. Myelin basic protein staining was decreased as well. There was no detectable neuronal injury by Fluoro-Jade B, TUNEL, or activated caspase-3 staining. Vasospasm of the large arteries of the circle of Willis was mild to moderate in severity. Nitric oxide was increased and superoxide anion radical was decreased in hippocampal tissue. Cerebral blood flow, measured by magnetic resonance imaging, and cerebral glucose metabolism, measured by positron emission tomography, were no different in SAH compared with control groups. The results suggest that the etiology of loss of LTP after SAH is not cerebral ischemia but may be mediated by effects of subarachnoid blood such as oxidative stress and inflammation.
Keywords: cognitive dysfunction, LTP, rats, subarachnoid hemorrhage, synaptic plasticity
Introduction
Survivors of aneurysmal subarachnoid hemorrhage (SAH) often suffer memory and cognitive deficits that affect their quality of life.1 Brain magnetic resonance imaging (MRI) of patients after SAH shows that there may be atrophy in the medial temporal lobes and that this may correlate with neurocognitive deficits in the absence of any focal structural lesions.2, 3 The hippocampal formation in the medial temporal lobe has been postulated to be involved in cognitive functions such as learning and memory. Long-term potentiation (LTP), a form of synaptic plasticity, is believed to be a cellular substrate of memory and learning.4 We previously demonstrated cognitive dysfunction5 and loss of LTP in the hippocampal synapses after SAH in rats.6 We found SAH caused significant vasospasm of the middle cerebral artery and decreased postsynaptic responses and loss of LTP at CA3–CA1 synapses. However, the cause of the loss of LTP was not worked out.
Here we use a prechiasmatic rat model of SAH5, 6 to test the hypothesis that alterations of proteins that participate in the subcellular maintenance of LTP contribute to loss of LTP after experimental SAH. Positron emission tomography (PET), MRI, and immunohistochemistry were used to investigate neuronal cell death, cerebral blood flow (CBF), and brain metabolism to determine if ischemia contributed to the loss of LTP. There are several different models of SAH in rats, including the endovascular perforation model7 and the cisterna magna injection models.8, 9, 10 The prechiasmatic SAH model was used, as this deposits blood around the anterior circle of Willis where most aneurysmal SAH occurs in humans. The model also has less variability than the endovascular perforation model.11
Materials and Methods
Animals
All procedures performed on animals were approved by the institutional Animal Care Committee and complied with regulations of Canadian Council on Animal Care. Male Sprague–Dawley rats (350 to 450 g, n=80) were randomly allocated to one of four groups: naïve control, sham-operated control, saline (0.9% NaCl)-injected control, and blood-injected SAH. Animals were housed in temperature and humidity controlled environments with a 12-hour light–dark cycle.
Subarachnoid Hemorrhage
The SAH model was created as previously reported.5 We injected 300 μL non-heparinized autologous blood into the prechiasmatic cistern of rats. Rats were anesthetized with isofluorane by inhalation (1% to 2%) and were positioned in a stereotactic frame (David Kopf Instruments, Tujunga, CA, USA). The scalp was prepared sterilely with povidone iodine. Subarachnoid hemorrhage was produced by injection of 300 μL blood withdrawn from the tail artery, through a 27-gauge spinal needle with a rounded tip and side hole, which was inserted into the prechiasmatic cistern. The injections were made over 20 seconds using a syringe pump (Harvard Apparatus, Holliston, MA, USA). The prechiasmatic injection model of SAH was modified from the model initially described by Prunell et al9, 11 The injection speed was similar to what was used in their original studies, in which 200 μL of blood was injected manually over 12 seconds. The injection speed was initially set aiming to maintain the intracranial pressure at the same level as the mean arterial blood pressure during the injection.9, 11 A syringe pump was used to keep the injection time consistent. The needle was angled 40° caudally in the sagittal plane, and placed through a burr hole placed just off the midline, 7.5 mm anterior to the bregma. Correct location of the needle in the subarachnoid space was obtained by advancing the needle until it touched the skull base and then withdrawing it 0.5 mm. CBF was monitored with a laser Doppler flow probe (Transonics model BLF21, type N flow probe, Ithaca, NY, USA) that was placed over an area of the left side of the skull just posterior to the bregma line. Care was taken to avoid large vessels. The tail artery was catheterized with PE10 tubing that was sutured in place and connected to a pressure transducer (World Precision Instruments, Sarasota, FL, USA) with rigid tubing filled with 0.9% NaCl for blood pressure measurement. Body temperature was maintained and monitored with a heating pad (Homeothermic systems, Harvard Apparatus, Holliston, MA, USA) and rectal temperature probe at 37°C. Baseline CBF, blood pressure, and body temperature were recorded for 15 minutes. Saline-control animals underwent injection of 300 μL of 0.9% NaCl. After completing these procedures, 5 mL of 0.9% NaCl was injected subcutaneously to prevent dehydration and the rats were returned to an incubator maintained at 26±1°C for 48 hours with access to soft food. Buprenorphine (0.05 mg/kg) was given twice daily for 48 hours after surgery. There was no mortality. Saline-injected animals were injected with 300 μL saline. Sham-operated controls were subjected to all procedures without injecting anything. Naive controls did not undergo any surgical procedures. Rats were killed under general anesthesia induced by administration of ketamine/xylazine (120/10 mg/kg) on day 0, 2, or 6 after surgery.
For biochemical assays, the brains were isolated immediately after killing. For histology, animals were perfused through the left cardiac ventricle with 0.9% NaCl for 2 minutes, followed by 250 mL of 4% paraformaldehyde in phosphate-buffered saline (PBS). The brains were removed and fixed in 4% paraformaldehyde for another 48 hours. Sections were obtained from coronal cuts of the brain centered at 5 mm interaural (∼−4 mm bregma). They were processed and embedded in paraffin. Sections 7 μm thick were cut using a microtome (Leica, Wetzlar, Germany). The coronal hippocampal slides were from the vicinity of bregma, −4 mm area on a 2-mm block of the brain.
Transmission Electron Microscopy
Quantification of synapses was done using transmission electron microscopy (TEM) as described previously.12 Six days after surgery, SAH, or saline-injected control rats (n=4 per group) were perfused with saline for 2 minutes followed by 2% paraformaldehyde/2.5% glutaraldehyde. The brains were removed and sectioned in the coronal plane (1 mm) on a vibratome (VT1200, Leica Microsystems, Richmond Hill, Canada). Samples were taken from the region of the CA1 layer in the hippocampus on the 1 mm brain slices using a biopsy punch (1.5 mm diameter, Acuderm, Fort Lauderdale, FL, USA). The tissue blocks (1.5 mm circular block of 1 mm thickness) were then processed.12 Tissue blocks were fixed with 2% osmium tetroxide/1.5% potassium ferricyanide. They were embedded in epon and 1-μm sections were cut from a block of embedded tissue from each rat using an ultramicrotome. The sections were collected on slotted, copper grids and stained with 2% uranyl acetate followed by lead citrate. The slides were examined by TEM (JEM 1011, JEOL, Munchen, Germany).
Double Immunofluorescence Staining
To verify the data from TEM, we carried out double immunofluorescent staining using synaptophysin (a presynaptic marker) and MAP2 (neuronal and postsynaptic marker). Active caspase-3/NeuN double staining was used for detection of neuronal cell death. Immunofluorescence staining was performed as previously described.12 Sections were deparaffinized and rehydrated, followed by antigen retrieval in a 96°C water bath for 25 minutes using Vector antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA). Sections were permeabilized with 0.3% Triton X-100 for 15 minutes and blocked by incubating with 10% normal goat serum in 1% BSA for 60 minutes. Primary antibodies were mouse monoclonal anti-NeuN (1:200 clone A60, Millipore, Billerica, MA, USA), rabbit polyclonal anti-human active caspase-3 (1:400, BD Pharmingen, San Diego, CA, USA), mouse monoclonal anti-rat synaptophysin (1:10, Millipore), and rabbit polyclonal anti-MAP2 (1:200, Abcam, Cambridge, MA, USA). After incubation overnight at 4°C, the slides were washed four times in PBS. They were then incubated at room temperature for 1 hour with Alexa Fluor 488-conjugated goat anti-mouse IgG (1:1000, Life Technologies, Burlington, ON, Canada) to visualize synaptophysin or NeuN and Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:1000, Life Technologies) to visualize MAP2 or caspase-3. Sections were then rinsed three times in PBS. Slides were rinsed two times in PBS and cover-slipped with anti-fading mounting medium. The stained sections were examined with a confocal microscope (Olympus BX50 confocal microscope, Olympus, Richmond Hill, ON, Canada).
Immunohistochemistry for GluR1/2, CaMK-II, and Myelin Basic Protein
For GluR2 staining, slides were deparaffinized and rehydrated through xylene and ethanol solutions and antigen was retrieved in a 96°C water bath for 25 minutes using Vector antigen unmasking solution (Vector Laboratories). Endogenous peroxidase activity was quenched using 0.3% hydrogen peroxide in water for 45 minutes. Sections were blocked with 10% horse serum in 1% bovine serum albumin in PBS for 40 minutes, incubated with mouse polyclonal anti-rat GluR2 (1:400 in 1% bovine serum albumin (BSA)/PBS, Millipore) for 90 minutes. They were then washed with PBS and incubated with biotinylated secondary antibody (horse anti-mouse 1:200 in 1% BSA/PBS, Vector) for 30 minutes. Staining was visualized with VIP using the VECTASTAIN ABC Kit (Vector) and counterstained with 0.5% methyl green. For immunohistochemical staining of GluR1, CaMK-II, and MBP, the above protocol was repeated where samples were blocked with 10% goat serum in 1% bovine serum albumin, then incubated with the primary antibodies rabbit polyclonal anti-rat GluR1 (1:200 in 1% BSA/PBS, Millipore), rabbit polyclonal anti-CaMK-II (1:200 in 1% BSA/PBS, Santa Cruz, Dallas, TX, USA), and rabbit polyclonal anti-rat MBP (1:500, Boster Biological Technology, Wuhan, China, in 1% BSA/PBS). For the secondary antibody goat anti-rabbit biotinylated antibody was used (1:200 in 1% BSA/PBS, Vector).
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling and Fluoro-Jade Staining
Deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was done as reported.12, 13 Rats injected with kainic acid (10 mg/kg) were used as positive controls. Apoptosis was assessed using TUNEL in accordance with the manufacturer's protocol (DeadEnd Flurometric kit, Promega, Fitchburg, WI, USA). Slides were counterstained with 4′,6-diamidino-2-phenylindole, washed, and cover-slipped with a water-based mounting medium, and sealed with nail polish.
Fluoro-Jade B (Histo-Chem, Jefferson, AR, USA) staining was used as a marker for neuronal injury. After deparaffinization, rehydration, and subsequent incubation with deionized water, the slides were incubated in 0.06% potassium permanganate (VWR International, Strasbourg, France) for 15 minutes with gentle shaking. Slides were then rinsed in deionized water to remove excess permanganate and immersed in 0.001% Fluoro-Jade B working solution (0.1% acetic acid) for 30 minutes. They were washed and dried in an incubator (60°C) for 15 minutes. Sections were cleared in xylene and cover-slipped with a non-aqueous, low fluorescence, styrene based mounting medium (DPX, Sigma-Aldrich, St Louis, MO, USA). Slides were examined with a fluorescence microscope (Leica). Images were taken with the same parameters for all sections.
Nitric Oxide and Superoxide Anion Radical Detection
The biochemical assay was performed as described.13 After killing, the brain was removed and placed on dry ice. A coronal section of the frontal cortex was cut and placed in an Eppendorf tube and frozen at −81°C. Bilateral hippocampal sections were dissected and transferred to −81°C as well for homogenization at a later date.
Superoxide anion radical (O2-) and nitric oxide (NO) were detected in homogenized fresh or deep frozen brain tissue using spectrophotometric methods.13 To detect NO, cell-permeable DAF-2DA (fluorophore 4,5-diaminofluorescein-2-diacetate, Alexis Biochemicals, Gruenberg, Germany) was used and a chemiluminescence probe, 2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidazo[1,2-]-pyrazin-3-one (MCLA) was used for O2− detection. Ten microliters μL of homogenized cortical or hippocampal tissues were incubated with either 10 mmol/L DAF-2DA for 30 minutes or 4 mmol/L MCLA for 10 minutes in transparent 96-well plates (Fisher, Ottawa, ON, Canada) at room temperature in the dark. DAF-2DA was excited at 495 nm and emission read at 515 nm in a spectrofluorometer (SpectraMAX-Gemini, Molecular Devices, Sunnyvale, CA, USA). The luminescence signal of MCLA was read directly at 495 nm. Experiments were repeated three times for each sample. MCLA crosses cell membranes and therefore the O2− detected is from both intracellular and extracellular sources. Nitric oxide values are expressed as relative fluorescence units. Superoxide anion radical values are expressed as relative luminescence unit.
Imaging
Rats underwent MRI and PET imaging before the killing time. The basis for co-registered MRI/PET was the multi-modal Minerve beds (Bioscan, Washington, DC, USA) so that rats could be transported between modalities. While on the Minerve bed, rats were oriented in the prone position with anesthesia delivered by inhalation (1.5% to 2% isoflurane). A pneumatic pillow was placed under the animal to enable respiratory monitoring while within the MRI suite (SA Instruments, Stony Brook, NY, USA).
Position Emission Tomography
On the day of euthanasia, rats were fasted (water access allowed) for at least 6 hours. Rats were anesthetized with 2% isoflurane and put in a supine position on the surgical table. The tail artery was exposed and cannulated with a 0.75-inch 24-gauge catheter (AngioCath, BD Biosciences, San Jose, CA, USA) to allow arterial blood sampling during the dynamic scan. A lateral tail vein was cannulated with polyethylene (PE10) tubing filled with heparinized saline for radiotracer injection. The rats were then secured on the bite bar of a warmed rat Minerve bed.
Rats were then advanced into a Focus 220 microPET system (Siemens Medical Systems, Erlangen, Germany), which had been cross-calibrated to the dose calibrator by imaging a known source of 18F and determining a quantification calibration factor. Transmission brain scans were acquired for 8 minutes using a 57Co point source to allow attenuation correction to be performed after the dynamic scan. Rats were then injected with 0.25 mL containing 75 MBq of 18F-FDG over 3 to 6 seconds and the 60-minute dynamic scan started at the same time as 18F-FDG injection. Multiple arterial blood samples were obtained to enable determination of a whole blood time activity curve, which then allowed calculation of cerebral glucose metabolic rates (CMRglc). After injection, the syringes and tubing line with residual radiotracer were measured in the dose calibrator to determine the net injected amount of radiotracer at injection time.
Magnetic Resonance Imaging
After completion of the PET acquisition, rats were transported to a 7 Tesla Biospec MRI system (Bruker Corporation, Ettlingen, Germany), fitted with the B-GA12 gradient coil insert, 7.2 cm inner diameter linearly polarized volume resonator for radiofrequency transmission, and dedicated flat surface coil for radiofrequency reception. The flat coil was fixed superior to the Minerve bed without compression of the rat head.
The rat brain was first visualized as a stack of transverse slices using a T2-weighted rapid acceleration relaxation enhancement technique (echo time=72 milliseconds; repetition time=4,800 milliseconds, 150 × 150 × 1,000 μm voxels; 30 × 30 mm field-of-view; 16 slices; 2-minute 40-second scan time). These images were used for PET registration. Three transverse slices separated by 3 mm at the level of the corpus callosum were then selected for CBF measurements based on a flow-alternating-inversion-recovery technique (two-dimensional fat-suppressed single-shot echo planar imaging; echo time=13.4 milliseconds; repetition time=17 seconds; 400 × 400 × 1,000 μm voxels; 30 × 30 mm field-of-view; 18 inversion times ranging from 25 milliseconds to 6,825 milliseconds separated by 400 milliseconds; 227 kHz effective readout bandwidth, 10-minute 12- second scan time). The flow-alternating-inversion-recovery acquisition quantifies CBF as the difference in T1 relaxation rate between measurements with global and slice-selective inversion pulses, as per equation 1.
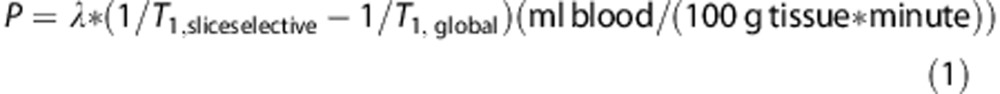 |
where P is perfusion and λ is the blood–brain partition coefficient for water (60 mL/(100 g*minute)).
Data Analysis
All data were from animals killed 6 days after surgery, except PET and MRI, which was from day 0, 2, or 6 as labeled in the corresponding figures and tables. All data was expressed as means±s.d.. Comparison of the groups was analyzed by Student's t-test or one-way analysis of variance with post hoc Holm–Sidak test using SigmaPlot 12.0. P⩽0.05 was considered significant.
For TEM, four squares of the copper grids in the CA1 dendritic area were chosen for taking images. Five images were acquired under × 15,000 magnification. Synapses were counted by an investigator who was masked to the animal group (SB and JD). Unbiased stereological methods for counting were used, and the final number used was the mean of 20 images.
Double stained sections (MAP2/synaptophysin) were imaged using a laser confocal microscope (Axiovert 200, Carl Zeiss, Hawthorn, PA, USA). Data were analyzed with imaging software (Fiji: an open-source platform for biologic-image analysis14) that determines the coefficients of the colocalization of the double staining, and which represents functional synapses.15 The quantification of colocalization was expressed as Manders' coefficient, which is the portion of signals above background threshold from both channels.
For GluR1, GluR2, CamK-II, and MBP quantification, masks of positive stained signals were generated using ImageJ (NIH, Bethesda, MD, USA), and the total area of positive staining was quantified and averaged against total neurons in the field. Raw SAH data were further normalized to that from saline controls. Six images of right and left CA1 subfields were taken under consistent light and exposure conditions at × 400 magnification by an operator masked to the animal group (HW). Images were color-matched against a randomly selected image to ensure a consistent background, and converted into an 8-bit image. The region of interest (ROI) and threshold for positive signal was predefined, and the percent of positive signal in each ROI was quantified using ImageJ. For CAMK2, GluR1, and GluR2, the amount of positive signal was normalized to the number of methyl green-positive cells in the ROI.
Magnetic resonance imaging/positron emission tomography co-registration used Inveon Research Workplace (Siemens Medical Systems), and PET ROI placement was guided by the rat brain atlas of Paxinos, et al16 Image analysis of PET activity used three-dimensional-ROIs drawn manually over five bilateral regions (frontal cortex, parietal cortex, caudate-putamen, thalamus, and hippocampus) by a single observer who was masked to the animal group (GK).
The Patlak graphical analysis method was used to determine CMRglc,17 based on equation 2:
In this equation, Cp is the arterial plasma glucose concentration, LC is the lumped constant in the method of Sokoloff, et al,18 and K is the constant slope.19 The LC value used was 0.625 for the normoglycemic range: 6.0 to 19.7 mmol/L.19 The interval of 30 to 60 minutes was used to estimate K.
Magnetic resonance imaging cerebral blood flow analysis used a custom mMtlab script for flow-alternating-inversion-recovery map generation, and MIPAV (NIH) for manual segmentation and histogram analysis of ROIs within cortex, sub-cortex, and hippocampus.
Results
Subarachnoid hemorrhage Reduces the Hippocampal Synapses
The quantification with TEM showed that the number of synapses in the dendritic layer of CA1 was significantly decreased in SAH animals as compared to saline controls (55±4.7 versus 74±3.1, P<0.01, Figure 1B). Similar to the data from TEM, the portion of MAP2 and synaptophysin colocalization in the immunochemical double staining was significantly decreased in the dendritic area of CA1 pyramidal layer of SAH rats as compared with that of saline and naive controls (Figure 2B). The Manders' coefficients were 0.24±0.05 for SAH, 0.38±0.07 for saline-injected and 0.37±0.1 for naïve controls (P⩽0.05, Figure 2B).
Figure 1.

Quantification of synapses using transmission electron microscopy (TEM). (A) Representative images of TEM showing synapses in CA1 area of the hippocampus (red arrow heads). (B) Bar graph showing significantly decreased number of synapses in subarachnoid hemorrhage (SAH) group as compared with saline controls (P⩽0.01, t-test). Data are mean±s.d. All animals were killed 6 days after surgery.
Figure 2.

Manders' coefficient analysis of active synapses in the hippocampus. (A) Representative images showing the colocalization of presynaptic marker synaptophysin and postsynaptic marker microtubule-associated protein 2 (MAP2) (white areas as indicated by white arrows in c, f, and i). Decreased colocalization (white area in images) is apparent in subarachnoid hemorrhage (SAH) group (i). (B) Subarachnoid hemorrhage induced a significant decrease of Manders' coefficient on the colocalization of synaptophysin/MAP2 (P⩽0.05, analysis of variance). Data are mean±s.d. All animals were killed 6 days after surgery.
Subarachnoid Hemorrhage Decreased Postsynaptic GluR1, CaMK-II, and Presynaptic Myelin Basic Protein, but Increased Postsynaptic GluR2
The loss of synapses prompted us to investigate whether there was a change in the expression of key proteins that mediate maintenance of LTP. Immunohistological staining for GluR1, GluR2, and CaMK-II showed that there was a significant decrease in the immunoreactivity of CaMK-II and a near significant decrease of GluR1 in the dendritic layer of CA1 pyramidal cells in comparison with those of saline-control and naïve rats (Figure 3). The normalized CamK-II positive signal in SAH group was significantly decreased (0.53±0.13 as compared with saline 1±0.13, P⩽0.001, Figure 3B). Similarly, the normalized GluR1 signal in the SAH group was 0.74±0.23, as compared with saline control (1±0.12, P=0.077, Figure 3B). In contrast, the normalized GluR2 positive staining in the SAH group was 1.2±0.07, which was significantly increased as compared with saline (1±0.17, P⩽0.05, Figure 3B).
Figure 3.

Immunohistological staining of GluR1, GluR2, and CaMK-II. Representative images are shown in panel A, decreased signal of GluR1 and CaMK-II staining, but increased signal of GluR2 staining was seen in subarachnoid hemorrhage (SAH) group as compared with that in naive or saline controls. Quantified data are shown in panel B. Data are mean±s.d. All animals were killed 6 days after surgery. *P<0.05, #P<0.001.
To determine the involvement of a presynaptic component (axons) in the loss of synapses, we stained for myelin basic protein (MBP). We found that there was a decrease in immunoreactivity to MBP in the CA1 area of hippocampus of the SAH group in comparison with saline-control rats (Figure 4). The normalized total area of MBP positive area in SAH rats was significantly lower than in saline controls (1±0.09, n=4 for saline, 0.73±0.06, n=6 for SAH, P⩽0.05).
Figure 4.

Immunohistological staining of myelin basic protein (MBP). (A) Images of MBP-stained slices from saline and subarachnoid hemorrhage (SAH) group. A mask of the positive signals was generated for each image, and the total area of the signal was quantified in panel B. There was significantly decreased MBP signal in the SAH group (P⩽0.05, t-test). Data are mean±s.d. All animals were killed 6 days after surgery.
Subarachnoid Hemorrhage did not Cause Significant Neuron Death
To determine whether the decrease in number of synapses and related proteins in CA1 area of the hippocampus was associated with neuron death, we assessed the brains of naïve, saline-injected, and SAH rats using three different stains. We did not find positive cells in TUNEL, caspase-3/NeuN, or Fluoro-Jade B staining in the CA3–CA1 area of the hippocampus of rats from any of the groups (Figure 5). Qualitative examination of other brain regions like entorhinal cortex also did not show neuron death.
Figure 5.

Neuronl death stained by deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL), Fluoro-Jade B (B), and caspase-3/NeuN (C). No apparent neuronal cell death in subarachnoid hemorrhage (SAH) animals was detected by any method. All animals were killed 6 days after surgery. Kainate-treated animals were used as positive controls in TUNEL staining (A, a to c, white arrows).
Subarachnoid Hemorrhage Induces Large Artery Vasospasm
Vasospasm was evaluated in the anterior cerebral artery using a ratio of lumen diameter/wall thickness. There was no significant difference among the seven groups (analysis of variance). There was a trend for vasospasm in the SAH group as compared with naive controls on day 6 (Figure 6, lumen/wall ratio was 18±5.5 for naive control, 12.5±7.3 for SAH day 6, P=0.065).
Figure 6.

Vasospasm of anterior cerebral artery (ACA). (A) Representative images showing ACA from different groups of animals. (B) Quantification of lumen diameter/wall thickness ratio in all animal groups. There is no statistical significant difference among the groups (P⩾0.05 analysis of variance). However, there was a trend toward vasospasm in subarachnoid hemorrhage (SAH) day 6 animals (P=0.065 as compared with naive controls). Data are mean±s.d. All animals were killed 6 days after surgery.
Nitric Oxide and Superoxide Anion Radical Detection
We measured NO and O2− in the hippocampus and cerebral cortex to assess the involvement of oxidative stress. There were no significant changes in NO or O2− in the cerebral cortex (Figures 7B and 7D). However, there was a significant increase of NO and decrease of O2− in the hippocampus after SAH as compared with saline controls. Nitric oxide is 1252±79 relative fluorescence units for SAH compared with 518±118 relative fluorescence units for saline controls (Figure 7A, P⩽0.001, t-test). O2− was 377±22 relative luminescence unit for SAH and 590±49 RLU for saline control (Figure 7C, P⩽0.001, t-test).
Figure 7.

Nitric oxide and superoxide anion radical (O2−) analysis. Significantly increased NO was found in the hippocampus (A), P⩽0.001, Student's t-test) but not cerebral cortex (B) in subarachnoid hemorrhage (SAH) group as compared with saline controls. In contrast, significantly decreased O2− was found in the hippocampus (C), P⩽0.001, Student's t-test) but not cerebral cortex (D) in SAH group as compared with saline controls. Data are mean±s.d. All animals were killed 6 days after surgery.
Cerebral Blood Flow and Metabolism
Using laser Doppler flow measurements, we found a dramatic decrease in CBF ranging from 5% to 30% of baseline immediately after injection of blood or saline (Supplementary Figure 1, P⩽0.001 as compared with sham controls). Cerebral blood flow recovered and stabilized at around 80% of the baseline level by the time of transfer to the Minerve bed. There was no significant difference among the groups in acute CBF recovery after the injection (Supplementary Figure 1). Similarly, we did not find any significant difference between any of the groups (SAH and naïve, sham-operated, and saline-injected controls) at any time points (day 0, 2, and 6 after SAH) in MRI measurements of CBF and PET measurements of glucose metabolism (Supplementary Figure 2, P⩾0.05, analysis of variance; Supplementary Tables 1 and 2).
Discussion
New Findings
The new findings of this study are that SAH in rats was associated with a significant decrease of synapses in neurons in the CA1 area. This was accompanied by a significant reduction in several proteins involved in LTP, includingCamK II and MBP, and a trend towards reduction of GluR1. There was an increase in GluR2. These changes may be responsible for the loss of LTP after SAH. Importantly, histologic studies with TUNEL, active caspase-3/NeuN and Fluoro-Jade B staining did not show significant neuron death, suggesting the loss of LTP in the hippocampus, which we observed in a prior study in the same model,6 could occur in the absence of cell death. Although there was a significant decrease of CBF at the time point of saline or blood injection, there was no significant CBF or metabolism change at various time points after SAH (1 hour to 6 days), suggesting that the effects of SAH are not mediated by ischemia. Oxidative stress may have a role in mediating the dysfunction of the hippocampal neurons.
Cognitive Impairment after Subarachnoid Hemorrhage
Cognitive impairment, measured on the Montreal Cognitive Assessment, was detected in 40% of patients followed up after SAH.20 Few animal studies have examined cognitive function using neurobehavioral testing or electrophysiology.5, 6, 21, 22 Takata, et al22 created SAH in rats by two injections into the cisterna magna. Subarachnoid hemorrhage was associated with significantly increased escape latency and swimming distance compared with saline-injected or sham-operated animals in the Morris water maze. Similarly, we found that the escape latency and swimming distance were significantly increased in rats with SAH as compared with controls. Subarachnoid hemorrhage rats also tended to do poorly on accuracy in spatial and working memory tests.5 Similar findings were reported by two other groups using an endovascular perforation model of SAH.21, 23
The discrepancy between our studies may be due to differences in methods. In the current study, rats were housed in an intensive care incubator (at 26°C, with easily accessible soft food and water for 48 hours) before their return to a room temperature cage. In comparison, Jeon, et al5 returned injured rats directly to their normal room temperature cage after recovery from anesthesia. Mortality decreased from 30% to less than 0% between these studies, despite the same blood injection volume (300 μL).
It must be noted that the current results reflect early neuronal changes after experimental SAH. Future studies should examine neuronal changes weeks after SAH, given the fact that cognitive/memory deficits in patients often persist for long period of time.1 In other studies, rats with SAH did have cognitive/memory dysfunction up to 5 weeks after ictus.21, 22, 23
Mechanism of Long-Term Potential Loss in Subarachnoid and Other Neurologic Diseases
Our previous study showed that the induction phase of LTP did not change after SAH but that the maintenance phase of LTP was diminished.6 This suggests that the molecular machinery for induction of LTP such as N-methyl-D-aspartate (NMDA) receptors is intact, but the molecular components for maintenance of LTP such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic (AMPA) receptors and their associated trafficking, CamK-II activation, and other downstream processes, are impaired.24 The current results are consistent with this hypothesis. It seems that a decrease in key proteins affects the function of these receptors, resulting in loss of plasticity of the synapses, and that neuron loss in the hippocampus is not necessary. This is consistent with studies in humans with SAH that show that cognitive dysfunction may be associated with temporal lobe volume loss in the absence of infarctions.3, 20 Animal studies in models of traumatic brain injury also found memory impairment in the absence of hippocampal cell death.25
Role of Key Proteins in Long-Term Potentiation
Enhanced expression of phosphorylated GluR1 by CamK-II is a key molecular event for induction and maintenance of LTP.26 In the current study, the decreased expression of CamK-II (and a trend towards decreased expression of GluR1) in CA1 area of the hippocampus could explain the loss of LTP after SAH.6 Impaired cognitive function and loss of LTP associated with decreased expression or dysfunction of GluR1 and CamK-II were found in other diseases such as Fragile X syndrome and Alzheimer disease27, 28 and in GluR1 and CamK-II knockout mice29, 30 but not GluR2 or 3 knockout mice.31 Interestingly, IL-1β, a proinflammatory cytokine was found to impair both memory and AMPA receptor expression.32 This is of interest because IL-1β was significantly increased in cerebrospinal fluid and blood from SAH patients and animals.33
Etiology of Hippocampal Changes after Subarachnoid Hemorrhage
Subarachnoid hemorrhage can cause ischemia due to increased intracranial pressure or in the form of delayed cerebral ischemia. We measured CBF with MRI and glucose metabolism with PET at different time points after SAH to determine if ischemia might mediate the hippocampal changes we observed. Experimental evidence showed that persistent synaptic failure may result from mild or moderate cerebral ischemia involving both pre- and postsynaptic components of the hippocampus.34 Although there was an acute decrease in CBF at the time of blood or saline injection, we did not find differences between CBF or glucose metabolism in SAH groups as compared with saline-injected controls at any time (1 to 2 hours, 2 days, and 6 days after injection). Thus, ischemia may not be contributing to the decreased expression of LTP-related proteins in this SAH model. One question is whether microcirculatory constriction or thrombosis could cause the changes in the absence of detectable CBF changes. There are reports that microthrombi mediating focal ischemia are likely formed in situ rather than from the breakdown of distal larger emboli, and that these microthrombi were accompanied by arteriolar vasoconstriction.12
If the hippocampal changes are not due to ischemia, then they must be mediated by the subarachnoid blood. In the current model, the blood was injected in the prechiasmatic cistern. If the blood is directly responsible for the hippocampal changes, one possibility is that the blood flows retrogradely into the ventricles. We did see blood in the ventricles from SAH rats in histology (data not shown) and iron deposition based on Perl staining. In the T2-weighted data sets, some rats display abnormal patterns of intraventricular T2 contrast, and some also present with bright intraventricular signal, which is characteristic of blood. However, the specificity of the T2 contrast is not good enough to distinguish blood retention from cerebrospinal fluid in the ventricles.
Subarachnoid blood has been suggested to cause oxidative stress.35 In this study, we found increased NO and decreased O2−, which was opposite to what we observed in our previous studies in a mouse model of SAH.13 This may be due to species differences or to the different times at which these parameters were assessed (two days after SAH in mice, 6 days after SAH in rats). Yatsushige, et al,36 found NO levels increased above basal levels at 24 hours after SAH created by endovascular perforation in rats. This was believed to have detrimental effects on the brain because increased NO could act as a free radical and form peroxynitrite that can damage the cell membrane of endothelial and smooth muscle cells.37 At this point, the etiology of the changes in the hippocampus remains uncertain. Potential etiologies may include cortical spreading depolarization, oxidative stress, and inflammation.35, 38 Interestingly, cortical spreading depolarization has been reported to enhance LTP,39 suggesting depolarization waves invading the hippocampus could be a mechanism of endogenous protection antagonizing the mechanisms that cause the reduction of LTP after SAH.38, 40
Conclusions
The anterior circulation model of SAH in rat causes loss of LTP, which is independent of global ischemia and neuron loss but which is associated with a decreased number of synapses, and a diminished density of AMPA receptors and CamK-II proteins at the Schaffer collateral—CA1 synapses in the hippocampus. The loss of these key proteins and possibly their functions in mediating LTP maintenance may account for, or at least contribute to, the cognitive and neurologic impairments of patients with SAH. These key proteins may be used as potential target to enhance or restore cognitive function in SAH patients.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This study was supported by an operating grant to Dr Jinglu Ai as the North Shore University Hospital, Brain Aneurysm Center Chair of Research from the Brain Aneurysm Foundation with Dr RLoch Macdonald as the mentor. RL Macdonald receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Stroke Network and the Heart and Stroke Foundation of Ontario. RL Macdonald is a consultant for Actelion Pharmaceuticals and Chief Scientific Officer of Edge Therapeutics, Inc.
Supplementary Material
References
- Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke. 2010;41:e519–e536. doi: 10.1161/STROKEAHA.110.581975. [DOI] [PubMed] [Google Scholar]
- Bendel P, Koivisto T, Kononen M, Hanninen T, Hurskainen H, Saari T, et al. MR imaging of the brain 1 year after aneurysmal subarachnoid hemorrhage: randomized study comparing surgical with endovascular treatment. Radiology. 2008;246:543–552. doi: 10.1148/radiol.2461061915. [DOI] [PubMed] [Google Scholar]
- Tam AK, Kapadia A, Ilodigwe D, Li Z, Schweizer TA, Macdonald RL. Impact of global cerebral atrophy on clinical outcome after subarachnoid hemorrhage. J Neurosurg. 2013;119:198–206. doi: 10.3171/2013.3.JNS121950. [DOI] [PubMed] [Google Scholar]
- Shapiro ML, Eichenbaum H. Hippocampus as a memory map: synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Jeon H, Ai J, Sabri M, Tariq A, Macdonald RL. Learning deficits after experimental subarachnoid hemorrhage in rats. Neuroscience. 2010;169:1805–1814. doi: 10.1016/j.neuroscience.2010.06.039. [DOI] [PubMed] [Google Scholar]
- Tariq A, Ai J, Chen G, Sabri M, Jeon H, Shang X, et al. Loss of long-term potentiation in the hippocampus after experimental subarachnoid hemorrhage in rats. Neuroscience. 2010;165:418–426. doi: 10.1016/j.neuroscience.2009.10.040. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086–1091. doi: 10.1161/01.str.26.6.1086. [DOI] [PubMed] [Google Scholar]
- Piepgras A, Thome C, Schmiedek P. Characterization of an anterior circulation rat subarachnoid hemorrhage model. Stroke. 1995;26:2347–2352. doi: 10.1161/01.str.26.12.2347. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Svendgaard NA. A new experimental model in rats for study of the pathophysiology of subarachnoid hemorrhage. Neuroreport. 2002;13:2553–2556. doi: 10.1097/00001756-200212200-00034. [DOI] [PubMed] [Google Scholar]
- Solomon RA, Antunes JL, Chen RY, Bland L, Chien S. Decrease in cerebral blood flow in rats after experimental subarachnoid hemorrhage: a new animal model. Stroke. 1985;16:58–64. doi: 10.1161/01.str.16.1.58. [DOI] [PubMed] [Google Scholar]
- Prunell GF, Mathiesen T, Diemer NH, Svendgaard NA. Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery. 2003;52:165–175. doi: 10.1097/00006123-200301000-00022. [DOI] [PubMed] [Google Scholar]
- Sabri M, Ai J, Lakovic K, D'abbondanza J, Ilodigwe D, Macdonald RL. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience. 2012;224:26–37. doi: 10.1016/j.neuroscience.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Sabri M, Ai J, Knight B, Tariq A, Jeon H, Shang X, et al. Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2011;31:190–199. doi: 10.1038/jcbfm.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40:101–111. doi: 10.1267/ahc.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Rat Brain in Stereotaxic Coordinates. Academic Press: San Diego; 2001. [Google Scholar]
- Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J Cereb Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Toyama H, Ichise M, Liow JS, Modell KJ, Vines DC, Esaki T, et al. Absolute quantification of regional cerebral glucose utilization in mice by 18F-FDG small animal PET scanning and 2-14C-DG autoradiography. J Nucl Med. 2004;45:1398–1405. [PubMed] [Google Scholar]
- Schweizer TA, Al-Khindi T, Macdonald RL. Mini-Mental State Examination versus Montreal Cognitive Assessment: rapid assessment tools for cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. J Neurol Sci. 2012;316:137–140. doi: 10.1016/j.jns.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Sherchan P, Lekic T, Suzuki H, Hasegawa Y, Rolland W, Duris K, et al. Minocycline improves functional outcomes, memory deficits, and histopathology after endovascular perforation-induced subarachnoid hemorrhage in rats. J Neurotrauma. 2011;28:2503–2512. doi: 10.1089/neu.2011.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Sheng H, Borel CO, Laskowitz DT, Warner DS, Lombard FW. Long-term cognitive dysfunction following experimental subarachnoid hemorrhage: new perspectives. Exp Neurol. 2008;213:336–344. doi: 10.1016/j.expneurol.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Silasi G, Colbourne F. Long-term assessment of motor and cognitive behaviours in the intraluminal perforation model of subarachnoid hemorrhage in rats. Behav Brain Res. 2009;198:380–387. doi: 10.1016/j.bbr.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- Lyeth BG, Jenkins LW, Hamm RJ, Dixon CE, Phillips LL, Clifton GL, et al. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Hsiao YH, Kuo JR, Chen SH, Gean PW. Amelioration of social isolation-triggered onset of early Alzheimer's disease-related cognitive deficit by N-acetylcysteine in a transgenic mouse model. Neurobiol Dis. 2012;45:1111–1120. doi: 10.1016/j.nbd.2011.12.031. [DOI] [PubMed] [Google Scholar]
- Li J, Pelletier MR, Perez Velazquez JL, Carlen PL. Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol Cell Neurosci. 2002;19:138–151. doi: 10.1006/mcne.2001.1085. [DOI] [PubMed] [Google Scholar]
- Andrasfalvy BK, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR1-deficient mice. J Physiol. 2003;552:35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds HL, Tonegawa S, Malinow R. CA1 long-term potentiation is diminished but present in hippocampal slices from alpha-CaMKII mutant mice. Learn Mem. 1998;5:344–354. [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39:163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter-Levin G, et al. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus. 2003;13:826–834. doi: 10.1002/hipo.10135. [DOI] [PubMed] [Google Scholar]
- Hendryk S, Jarzab B, Josko J. Increase of the IL-1 beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuro Endocrinol Lett. 2004;25:141–147. [PubMed] [Google Scholar]
- Hofmeijer J, van Putten MJ. Ischemic cerebral damage: an appraisal of synaptic failure. Stroke. 2012;43:607–615. doi: 10.1161/STROKEAHA.111.632943. [DOI] [PubMed] [Google Scholar]
- Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31:151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsushige H, Calvert JW, Cahill J, Zhang JH. Limited role of inducible nitric oxide synthase in blood-brain barrier function after experimental subarachnoid hemorrhage. J Neurotrauma. 2006;23:1874–1882. doi: 10.1089/neu.2006.23.1874. [DOI] [PubMed] [Google Scholar]
- Ayer RE, Zhang JH. Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir Suppl. 2008;104:33–41. doi: 10.1007/978-3-211-75718-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Wernsmann B, Pape HC, Speckmann EJ, Gorji A. Effect of cortical spreading depression on synaptic transmission of rat hippocampal tissues. Eur J Neurosci. 2006;23:1103–1110. doi: 10.1111/j.1460-9568.2006.04643.x. [DOI] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.