

Abstract
Although heavy alcohol consumption has deleterious effects on heart health, moderate drinking is thought to have cardioprotective effects, reducing the risk of coronary artery disease and improving prognosis after a myocardial infarction. It still is unclear, however, if this effect can be achieved with all types of alcoholic beverages and results from the alcohol itself, from other compounds found in alcoholic beverages, or both. For example, the polyphenolic compound resveratrol, which is found particularly in red wine, can reduce the risk of atherosclerosis; however, it is not clear if the resveratrol levels present in wine are sufficient to achieve this result. Alcohol itself contributes to cardioprotection through several mechanisms. For example, it can improve the cholesterol profile, increasing the levels of “good” cholesterol and reducing the levels of “bad” cholesterol. Alcohol also may contribute to blood clot dissolution and may induce a phenomenon called pre-conditioning, whereby exposure to moderate alcohol levels (like short bouts of blood supply disruption [i.e., ischemia]), and result in reduced damage to the heart tissue after subsequent prolonged ischemia. Finally, the enzyme aldehyde dehydrogenase (ALDH) 2, which is involved in alcohol metabolism, also may contribute to alcohol-related cardioprotection by metabolizing other harmful aldehydes that could damage the heart muscle.
Keywords: Alcohol consumption, light drinking, moderate drinking, effects and consequences of alcohol and other drug use, beneficial moderate alcohol consumption, risk and protective factors, risk-benefit, cardiovascular system, cardioprotection, wine, red wine, French Paradox, cholesterol, resveratrol, aldehyde dehydrogenase 2
Although the deleterious effects of chronic heavy alcohol consumption on the cardiovascular system—including hypertension, cardiomyopathy,1 and arrhythmias—have been documented as early as the late 19th century (Bollinger 1884), interest in possible beneficial effects of moderate drinking only arose about two decades ago. In November 1991, CBS correspondent Morley Safer presented a segment during the news show “60 Minutes” that discussed what became known as the “French Paradox”—the fact that the French, despite their life style of eating diets high in saturated fats and having a very high rate of smoking, only suffer about one-quarter the rate of coronary heart disease (CHD) compared with the U.S. population. The news segment attributed this to the French people’s consumption of wine, especially red wine. As a result of the broadcast, wine sales in the United States skyrocketed, and interest in research on the cardiovascular effects of alcohol increased. Thus, the National Institute on Alcohol Abuse and Alcoholism (NIAAA) issued several requests for research in this area, initiating serious investigation of this issue.
Epidemiological studies performed in various countries over the last decades have described the relationship between alcohol intake and mortality as a J-shaped curve—that is, light-to-moderate drinking is associated with decreased mortality, whereas heavy alcohol intake has a detrimental effect. Moderate drinking in the United States is defined as no more than one drink per day for women and no more than two drinks per day for men. (For more information, see the textbox.) Thus, light-to-moderate drinking consistently has been associated with a reduced risk of coronary artery disease and death compared with abstinence (e.g., Fuchs et al. 1995; Lindeman et al. 1999; Mukamal et al. 2010; Renaud et al. 1999). This observation has led to several questions, including the following:
What is the effect of variations in drinking patterns (i.e., quantity and frequency of drinking, binge drinking)?
Does the observed beneficial effect result from the alcohol itself or from other compounds in alcoholic beverages?
What are the mechanisms underlying such effects?
Alcohol Consumption Levels and Their Relationship With Heart Health.
Animal studies have found that blood alcohol levels of 10 to 17 mM ethanol induce cardioprotection against damage caused by insufficient blood supply (i.e., ischemic damage) (Miyamae et al. 1998a, b). Similar alcohol levels are found in humans after consumption of 1 to 2 glasses of wine, cans of beer, or shots of liquor, and they therefore can be considered physiological. In the United States, consumption of no more than one drink per day for women and no more than two drinks per day for men is considered moderate drinking. Consumption at these levels appears beneficial to human health. Conversely, substantially higher levels of consumption are considered excessive and are associated with many health risks, including cardiac damage and heart failure. However, the definition of moderate and excessive drinking varies between countries, and the definition of what constitutes moderate versus excessive alcohol consumption therefore must be clarified, particularly when comparing different studies.
This review briefly addresses the first two questions, before expanding more extensively on the third question regarding the mechanism of cardioprotection, of which much has been learned in the past 10 years. The article will focus on recent data identifying a phenomenon called ethanol-induced preconditioning as well as on an unexpected player in alcohol-induced cardioprotection, the alcohol-metabolizing mitochondrial enzyme aldehyde dehydrogenase 2 (ALDH2).
Alcohol—Friend or Foe?
The World Health Organization (WHO) has estimated that every American over 15 years of age consumes more than 6.1 liters of pure alcohol (i.e., ethanol) per year (WHO 2007). Whereas excessive ethanol consumption has a negative impact on health, acute and chronic moderate consumption appears to have beneficial effects, especially on the heart (Gaziano et al. 2000; Renaud et al. 1998; Rimm et al. 1991; Stampfer et al. 1988; Thun et al. 1997; also see the textbox). Specifically, patients who consume moderate amounts of alcohol before and after an acute heart attack (i.e., myocardial infarction) have an improved prognosis (Chen et al. 1999b; Krenz et al. 2001a; Mukamal et al. 2001; Muntwyler et al. 1998). In addition to causing beneficial changes in lipid levels in the blood (see Renaud and de Lorgeril 1992; Rimm et al. 1999), alcohol produces protective effects through a phenomenon called preconditioning (Chen et al. 1999b; Guiraud et al. 2004; Krenz et al. 2001a; Miyamae et al. 1997). Both of these mechanisms will be discussed in more detail in the section “Mechanisms of Alcohol-Induced Cardioprotection.”
Influence of Quantity and Frequency of Drinking
To date, most epidemiological studies analyzing associations between beneficial or detrimental effects and drinking levels have used average volume of alcohol consumption as a measure. However, little research has investigated the effects of specific drinking pattern (i.e., the quantity and frequency of drinking), such as binge drinking. Yet this distinction is crucial in dissecting the effects of moderate drinking, because the health effects of an average consumption of seven drinks per week (or one drink per day) can differ vastly depending on whether the drinker really has one drink on every day of the week or consumes all seven drinks in one sitting on a Saturday night (i.e., is a binge drinker). Thus, studies found out that binge drinking seems to be more hazardous to cardiovascular health (Pletcher et al. 2005). Furthermore, Knupfer and colleagues (1987) reported that most light drinkers do not drink daily, and most daily drinkers are not moderate drinkers, further emphasizing the need to differentiate between drinking patterns. For more information on the relationship between various dimensions of alcohol consumption and outcome, the reader is referred to Rehm and colleagues (2010); in addition, van de Wiel and Lange (2008) have discussed the importance of measuring quantity and frequency of alcohol consumption.
Does Type of Beverage Play a Role?
Although a beneficial effect of moderate drinking repeatedly has been shown, questions remain whether the effect differs between types of beverages and whether it is the alcohol itself or other compounds found in the beverages that are responsible for these effects. The majority of epidemiological studies have pooled data for all types of alcoholic beverages when reporting beneficial effects of moderate consumption; however, some studies also have provided beverage-specific risk estimates for CHD. For example, Grønbaek and colleagues (2000, 2004) reported that wine intake may have a more beneficial effect on all-cause mortality than other alcoholic beverages, with a relative risk of death of 0.66 for light wine drinkers compared with 0.90 for light drinkers who avoided wine. A meta-analysis of the effects of wine and beer consumption (Di Castelnuovo et al. 2002) indicated that consumption of red wine lead to a greater reduction in risk of CHD (32 percent) than did beer consumption (22 percent). In contrast, an advisory by the American Heart Association (Goldberg et al. 2001) indicated no distinguishable effect of wine from other alcoholic beverages. Finally, some studies have attributed the apparent beneficial effects of wine consumption to the fact that wine drinking was significantly associated with a higher socioeconomic status and lifestyle (Mortensen et al. 2001) and a healthier diet (Tronneland et al. 1999), which might have conferred cardioprotection.
Similarly, disagreement still exists regarding the compounds responsible for the beneficial effects. Some investigators have attributed the added “bonus” effect of wine to the presence of antioxidants and polyphenolic compounds, such as resveratrol. For example, Opie and Lecour (2007) claimed that red wine potentially has beneficial effects beyond alcohol. Conversely, Belleville (2002) maintained that “alcohol per se rather than compounds specific to certain beverages reduces mortality risk.”(p. 173) The potential role of resveratrol is explored in the following section.
Alcohol, Resveratrol, and Sirtuins
Resveratrol is a polyphenolic compound (see figure 1) with antifungal properties that primarily is present in the skin of grapes. It is renowned for its beneficial effects on atherosclerosis and cancer (for a review, see Vidavalur et al. 2006). Moreover, resveratrol and other polyphenols in wine have been claimed to have a positive synergistic effect with alcohol on some risk factors for atherosclerosis (for a review, see Cooper et al. 2004). Based on in vitro studies, it has been advocated that polyphenols in wine can reduce risk of atherosclerosis through several mechanisms:
They prevent the blood-clot–promoting (i.e., prothrombotic) effects of a cholesterol-rich diet (De Curtis et al. 2005);
They reduce the susceptibility of low-density lipoprotein (LDL; “bad” cholesterol”) to chemical modification (i.e., oxidation) that initiates the formation of atherosclerotic plaque;
They enhance endothelium-dependent relaxation of the blood vessels, which reduces the risk of blood vessel obstruction (Carrero et al. 1998; Deckert et al. 2002; Wallerath et al. 2003); and
They inhibit platelet aggregation (de Lange et al. 2007).
Figure 1.
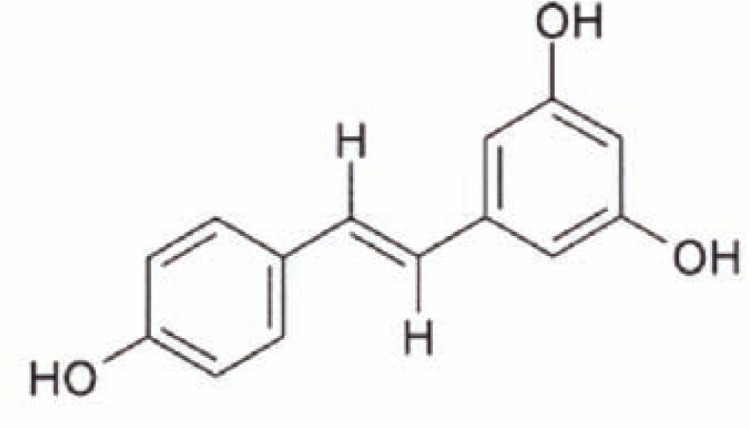
Molecular structure of resveratrol. Resveratrol is a polyphenol—that is, it contains several ring-like molecular building blocks known as phenols.
In all of these studies, however, the concentrations of polyphenols used were several magnitudes higher than those encountered after human alcohol consumption, limiting their generalizability.
In addition to resveratrol, white wine also is rich in two other phenolic compounds, called tyrosol and hydroxytyrosol. These agents also may confer cardioprotection (Dudley et al. 2008).
Mechanism of Action of Resveratrol
The key molecular targets for resveratrol are the sirtuins, a family of enzymes (SIRT1 through SIRT7) that remove an acetyl chemical group from (i.e., deacylate) the acetylated forms of the amino acid lysine found in some proteins. Sirtuins serve as sensors for cellular energy availability. Recent studies by Zhang and colleagues (2008) demonstrated that SIRT1 produced by endothelial cells has antiatherosclerotic effects because in a certain strain of mice it attenuates the formation of atherosclerotic plaque in the aorta induced by a high-fat diet. In addition, SIRT1 suppresses the development of atherosclerosis by enhancing the activity of an enzyme called endothelial nitric oxide synthase (eNOS)2 and by suppressing inflammation and reactive oxygen species (Brandes 2008; Borradaile et al. 2009). Consequently, SIRT1 activators, such as resveratrol, can produce cardioprotection.
Resveratrol likely activates SIRT1 by altering the enzyme’s structure (i.e., conformation) so that a molecule required for deacetylation can bind more easily to SIRT1. However, there still is some debate if the resveratrol can directly activate SIRT1 or if additional compounds are required (Borra et al. 2005; Kaeberlein et al. 2005). The crucial role of SIRT1 in heart health was demonstrated in studies showing that hearts from diabetic mice produced less SIRT1 protein, impairing the heart muscle’s ability to contract and leading to heart failure (Dong and Ren 2007).
Other studies have demonstrated that resveratrol can inhibit platelet aggregation and has antioxidant effects on cholesterol metabolism (for a review, see Markus and Morris 2008). Thus, resveratrol has been advocated for heart health. However, it still is unclear whether the amount present in wine is enough to confer cardioprotection, and this issue merits proper clinical investigation.
Mechanisms of Alcohol-Induced Cardioprotection
The apparent cardioprotective effect of moderate alcohol consumption observed in epidemiological studies has spurred biochemical studies to determine whether the alcohol (i.e., ethanol) itself confers these effects and to understand the molecular mechanisms underlying it. Several mechanisms have been proposed, including the following (for more information, see Lakshman et al. 2009):
Increase in high-density lipoprotein (HDL) cholesterol (i.e., “good” cholesterol);
Increased fibrinolysis; and
Activation of protein kinase C epsilon (ɛPKC)-mediated preconditioning of the myocardium as a protective mechanism from injury induced by myocardial infarction.
Effects on HDL and LDL Cholesterol
Cholesterol is transported in the body by compounds known as lipoproteins. These can be classified according to their density, with LDL and HDL representing two of the groups. These groups differ in their functions in the body (i.e., in the types of lipids they transport). Thus, LDLs transport cholesterol from the liver to the cells of the body, whereas HDL acts as a cholesterol scavenger, transporting it back to the liver where it is converted to bile acids and/or eliminated. Excessively high levels of cholesterol in the blood, reflected by lower-than-normal HDL levels and higher-than-normal LDL levels, are a risk factor for cardiovascular disease and atherosclerosis. Several epidemiological studies have shown that moderate drinking is associated with higher concentrations of HDL cholesterol compared with abstinence (e.g., Bazzano et al. 2008; Rimm et al. 1999). In fact, about 50 percent of the cardioprotective effect associated with moderate drinking was attributed to this increase in HDL cholesterol (Gaziano et al. 1993, Grønbaek et al. 2002) and a concurrent decrease in LDL cholesterol (Savolainen et al. 1995).
The mechanisms by which HDL carries cholesterol to the liver is called reverse cholesterol transport (RCT). It involves three main steps:
HDL interacts with a receptor called ABCA1 on various cells, causing the release (i.e., efflux) of unesterified cholesterol from the cells.
The cholesterol is esterified by an enzyme called lecithin-cholesterol acyltransferase (LCAT), leading to the maturation of the newly produced HDL particles.
Mature HDL particles are removed from the circulation by binding to a receptor called SR-B1 that is found on the main type of liver cells (i.e., the hepatocytes). The HDL then is modified by the exchange of the esterified cholesterol for another lipid—a reaction that is mediated by an enzyme called cholesteryl ester transfer protein (CETP).
The relationship between CETP function and atherosclerosis risk is complex; however, it is clear that reduction in CETP activity results in an increase in HDL cholesterol levels. The alcohol-induced increase in HDL cholesterol also may be mediated at least in part by CETP because studies found that moderate drinking reduces CETP activity (Hagiage et al. 1992). Moreover, Hannuksela and colleagues (1994) and Fumeron and colleagues (1995) reported that heavy drinkers have lower CETP activity. Finally, Valimaki and colleagues (1993) found that CETP activity was low in alcoholic women but increased after cessation of drinking.
More recently, other functions of HDL have been reported, including anti-inflammatory and antioxidant properties, raising the possibility that some of the antiatherosclerotic effect of HDL may be attributed to functions beyond RCT. Some research in this area has focused on an HDL-associated enzyme called paraoxonase-1 (PON1) that protects LDL from oxidative modification and is central to the anti-inflammatory and antioxidative properties of HDL. Oxidation of LDL currently is believed to be central to the initiation and progression of atherosclerosis (Mackness et al. 2000). Consequently, an increase in PON1 activity would be expected to decrease inflammation and atherosclerosis. Studies in rats found that moderate but not high doses of alcohol increased PON1 activity in the blood and liver by 20 to 25 percent as well as increased the levels of PON1 mRNA in the liver by 59 percent (Rao et al. 2003). In humans, daily moderate consumption of beer, wine, or spirits also resulted in higher PON1 activity compared with consumption of water, and this increase correlated with concomitant increases in HDL cholesterol and one of its major components, apolipoprotein A1 (Sierksma et al. 2002; van der Gaag 1999; van Tol and Hendriks 2001). Thus, the antiatherosclerotic effects of moderate alcohol consumption also may be mediated by alcohol’s effects on PON1.
Effects on Fibrinolysis
Blood clots, which can lead to obstruction of blood vessels and ischemia, are formed by the clumping together of certain blood cells (i.e., platelets) and a protein called fibrin, which is produced from a precursor called fibrinogen. The dissolution of these clots normally is achieved through a process called fibrinolysis. This process is mediated by an enzyme called plasmin that cuts the blood clot in various places. However, plasmin is found in the body only as an inactive precursor molecule, plasminogen. The conversion of plasminogen to plasmin is stimulated by various compounds, including tissue plasminogen activator (t-PA) and urokinase-type PA (u-PA), and is inhibited by a compound known as PA inhibitor type 1 (PAI1).
Moderate alcohol consumption can mediate additional cardioprotection by promoting the dissolution of blood clots through its actions on one or more of the components of the fibrinolytic system (i.e., t-PA, u-PA, PAI1, and fibrinogen). For example, Ridker and colleagues (1994) demonstrated an association between alcohol consumption and t-PA levels in healthy volunteers; moreover, t-PA levels were significantly higher in drinkers than in nondrinkers. In addition, studies on cultured cells obtained from the human umbilical vein showed that small doses of ethanol (less than 0.1 percent volume/volume) increased the cells’ fibrinolytic activity and that this effect was closely associated with the altered expression of t-PA, u-PA, and PAI1 (Aikens et al. 1997; Grenett et al. 1998).
Preconditioning
Preconditioning is a phenomenon first observed in the setting of disrupted blood supply (i.e., ischemia). Researchers found that if they exposed tissue or an organ to short bouts of ischemia, the tissue or organ subsequently could survive even prolonged ischemia that normally would have had a damaging effect (Murry et al. 1986). Several studies (Chen et al. 1999b; Guiraud et al. 2004; Krenz et al. 2001a; Miyamae et al. 1997) have demonstrated that alcohol exerts its cardioprotective effects in part by inducing preconditioning-like mechanisms. Thus, treatment of isolated heart muscle cells (i.e., cardiomyocytes) or isolated hearts with 50 mM ethanol diminished injury associated with subsequent prolonged ischemia and improved cardiac function (Chen et al. 1999b; Hale and Kloner 2001; Krenz et al. 2001a). Other studies (Bellows et al. 1996; Hale and Kloner 2001; Itoya et al. 1998), however, did not show cardioprotective effects following acute alcohol treatment. These conflicting reports likely result from differences in the timing and dose of alcohol treatment, which appear to influence alcohol-induced protection. For example, alcohol administration before prolonged ischemia is more protective if there is a delay between alcohol exposure and the ischemic injury, as shown in both experimental models (Krenz et al. 2001b, 2002a, b) and in clinical studies (Mukamal and Mittleman 2001; Niccoli et al. 2008).
More than 10 years ago, several studies demonstrated that the protective effects of alcohol administration require activation of the enzyme ɛPKC (Miyamae et al. 1998b). Protein kinases are enzymes that add a phosphate group to other proteins. This phosphorylation can change the protein’s activity, location in the cell, or association with other proteins. Accordingly, protein phosphorylation by kinases (and also dephosphorylation by other enzymes) plays important roles in many regulatory and signaling processes. Numerous protein kinases have been identified that can be classified according to the specific amino acids in their target proteins to which they add the phosphate group. For example, the PKC family of protein kinases, which consists of more than 10 members, adds the phosphate to the amino acids serine and threonine.
The role of ɛPKC in alcohol-induced preconditioning was demonstrated in guinea pigs that were exposed to ethanol (i.e., 25 to 50 mM) in their drinking water for several weeks, resulting in blood alcohol levels of about 1 percent (10 mg/ml). When hearts isolated from these animals were subjected to global ischemia, cardiac damage was reduced by about 50 percent and cardiac functions were improved correspondingly compared with hearts from control animals (Miyamae et al. 1998b). Moreover, sustained activation of ɛPKC was found in the hearts of the alcohol-exposed animals (Miyamae et al. 1998b). The delayed protective effects of acute alcohol exposure also involve actions of another member of the PKC family called PKC delta (δPKC), which stimulates the release of adenosine, resulting in ɛPKC activation (Chen and Mochly-Rosen 2001; Inagaki and Mochly-Rosen 2005). The observed role of the different PKCs depends on the experimental system used. Thus, treatment of rats with alcohol levels of more than 1.5 g/kg body weight induced the activation of the damage-associated δPKC in the animals (Chen and Mochly-Rosen 2001; Inagaki and Mochly-Rosen 2005). Conversely, treatment of isolated cardiomyocytes and isolated hearts with low amounts of alcohol (0.5 g/kg, or 10 mM) induced activation of ɛPKC and cardiac protection (Chen et al. 1999b; Miyamae et al. 1998b; Zhou et al. 2002). However, the specific protein(s) that are phosphorylated by ɛPKC following acute alcohol treatment have not yet been identified.
Role of Mitochondrial Aldehyde Dehydrogenase 2
Following ischemic preconditioning as described above, researchers have observed that several proteins in the mitochondria, the cell’s “power plants,” also are phosphorylated by ɛPKC. These proteins include the mitochondrial ATP-sensitive K+ channel (Costa et al. 2005; Jaburek et al. 2006), cytochrome-c oxidase (complex IV; COIV) (Guo et al. 2007), and the permeability transition pore (MPTP) (Juhaszova et al. 2004). However, it is not clear whether these proteins have any role in alcohol-mediated preconditioning (Krenz et al. 2001a, 2002a).
Chen and colleagues (2008) used an unbiased approach to identify all proteins in heart cells that were phosphorylated (and thereby activated) under conditions inducing cardiac cytoprotection (i.e., after exposure to alcohol or direct activation of ɛPKC). To their surprise, one of the proteins they identified was mitochondrial ALDH2, which, as described below, plays a pivotal role in alcohol metabolism. In addition, both ALDH2 phosphorylation and cytoprotection induced by alcohol or direct ɛPKC activation could be inhibited by first treating the hearts with an ɛPKC-selective inhibitor. Finally, ALDH2 activity was inversely correlated with infarct size. These data together suggested that ALDH2 activation is required for cardioprotection, leading to the question, What of mechanism(s) underlie this relationship?
The ALDH2 enzyme, which consists of four identical subunits and is located in the interior of the mitochondria (Goedde and Agarwal 1990), mediates the rate-limiting step in ethanol metabolism—the conversion of toxic acetaldehyde (which is produced from ethanol by the enzyme alcohol dehydrogenase) into nontoxic acetate (Zakhari 2006). Several variants of this enzyme exist and one of them, called ALDH2*2, is inactive—that is, it cannot metabolize acetaldehyde. In this variant, a mutation in the gene encoding the protein causes the exchange of a single amino acid from a glutamine to lysine in the region where the four subunits interact with each other (Larson et al. 2005). The prevalence of this inactive variant differs significantly among ethnic groups. For example, approximately 40 percent of east Asians carry at least one copy of the gene encoding the defective ALDH2*2 variant. In people who carry two copies of the defective gene (i.e., who are homozygous for ALDH2*2/*2), the activity of the ALDH enzyme is reduced by more than 95 percent compared with people who are homozygous for the normal, active form of the enzyme (i.e., ALDH2*1). Even in people who carry only one copy of the mutant gene and one copy of the normal gene (i.e., who are heterozygous ALDH2*1/*2) the activity of the resulting enzyme only is about 40 percent of the normal ALDH2.
The ALDH2 protein a person produces determines to some extent how much alcohol he or she will drink. Presence of even one mutant gene leads to reduced acetaldehyde metabolism and acetaldehyde accumulation, which results in highly unpleasant effects, such as facial flushing, increased heart rate, palpitation, and dizziness (Luu et al. 1995; Takeshita and Morimoto 1999). As a result, people carrying one or two copies of the defective gene tend to drink less alcohol, and ALDH2*2/*2 homozygotes are less likely to be alcoholics (Higuchi et al. 1994). Similarly, the presence of another less active variant, ALDH2*A, which is found in Jewish people of Ashkenazi descent, also is associated with reduced alcohol consumption (Fischer et al. 2007). In heterozygous people with only one defective ALDH2*2 gene, however, the potential deterring effect on excessive ethanol consumption appears not to be strong enough to fully prevent drinking or alcoholism. In fact, social pressure and changes in cultural acceptance in recent years are thought to contribute to the dramatic rise in alcoholism among ALDH2*2/*1 heterozygotes in Asia (Chen et al. 1999a; Higuchi et al. 1994; Yokoyama et al. 2002), even though these people still experience substantial and dose-dependent acetaldehyde accumulation and the resulting effects.
The assumption that ALDH2 may play a role in cardio-protection from myocardial infarctions has been supported by studies comparing the effect of 0.5 g/kg ethanol on ALDH2*1/*1 homozygotes and ALDH2*1/*2 heterozygotes. The investigators found that presence of the defective variant was associated with a higher incidence of ischemia/reperfusion injury (Jo et al. 2007). Therefore, it is important to further explore the connection between ALDH2 activity and cardiac protection from ischemia and to determine how people with the mutant ALDH2*2 variant can be protected.
However, it is important to note that the reported tight correlation between cardiac protection and ALDH2 activity does not prove that ALDH2 is indeed critical to the process. To demonstrate that ALDH2 activation is sufficient to induce cytoprotection, researchers needed a direct and specific agonist of this enzyme—that is, a molecule that could activate ALDH2 and induce the same cardioprotective effects without the presence of alcohol. A high-throughput screening of a large number of small molecules identified a class of structurally similar compounds called Aldas (for aldehyde dehydrogenase activator), including one called Alda-1.3 This molecule activates the defective ALDH2 enzyme by acting as a structural chaperone. The study demonstrated that ALDH2 activation by Alda-1 reduced infarct size by 60 percent in vivo (Chen et al. 2008).
As mentioned earlier, ALDH2 had been identified as a candidate involved in cardioprotection by screening for proteins that were phosphorylated by ɛPKC or in response to alcohol treatment, suggesting that ALDH2 activation was the result of alcohol-induced ɛPKC activation. If this was the case, then alcohol’s cardioprotective effect should be absent in genetically modified mice that lacked ɛPKC (i.e., ɛPKC-null mice). Consistent with this hypothesis, studies exposing hearts from ɛPKC-null mice to ischemia and reperfusion found that these hearts were not protected by alcohol but were protected by pretreatment with Alda-1 (Churchill et al. 2009). These data suggest that ALDH2 activation results from ethanol-induced ɛPKC activation and that direct activation of ALDH2 by Alda-1 was sufficient to confer cardioprotection in the absence of ɛPKC. Furthermore, the ability of Alda-1 to protect ɛPKC-null mice (which cannot be preconditioned by either mild ischemia or ethanol [Gray et al. 2004]) made it unlikely that Aldas work simply by mimicking the process of preconditioning (i.e., by delivering a mild toxic insult to the heart prior to ischemia onset).
How does ALDH2 Contribute to Cardioprotection?
According to a recent commentary (Karliner 2009), toxic aldehydes and their modulation by ALDH2 play a central role in cardiac injury following acute myocardial infarction. For example, increased expression of ALDH2 was found to attenuate acetaldehyde-induced cardiomyocyte injury (Li et al. 2006). But why does a decrease in ALDH2 activity contribute to ischemic damage, and how does ALDH2 activation by moderate alcohol consumption result in cardiac protection? ALDH2 metabolizes not only acetaldehyde, but also other short- and long-chain aliphatic and aromatic aldehydes (Reichard et al. 2000; Vasiliou and Nebert 2005; Vasiliou et al. 2004; also see figure 2). These aldehydes often are produced by the interactions of reactive oxygen species (ROS) with the lipids found in the membranes surrounding the cell. One common aldehyde formed this way is 4-hydroxy-2-nonenal (4HNE), which is metabolized by ALDH2; it has been implicated in tissue damage after cardiac ischemia (Lucas and Szweda 1998). 4HNE (as well as other aldehydes that accumulate during ischemia) are very reactive and form reaction products (i.e., adducts) with other macromolecules in the cell (e.g., proteins or DNA); these adducts then can interfere with normal cell function (see figure 2). For example, 4HNE adducts inhibit cell components called proteasomes that are responsible for the elimination of proteins that are damaged or no longer needed (Farout et al. 2006; Ferrington and Kapphahn 2004). In addition, 4HNE adducts interfere with the mitochondria’s ability to produce the energy-storing compound adenosine triphosphate (ATP) (Yan and Sohal 1998) and with mitochondrial integrity (Echtay and Brand 2007; Kristal et al. 1996). Therefore, rapid metabolism of these reactive aldehydes into nonreactive acids likely is critical for protecting the heart muscle (i.e., myocardium) from the oxidative stress associated with cardiac ischemia. Indeed, Alda treatment (which activates ALDH2) prior to cardiac ischemia reduced formation of 4HNE adducts in the ischemic hearts (Chen et al. 2008). Further, greater-than-normal production of aldehyde dehydrogenase-2 in mice reduced heart failure–like symptoms following chronic alcohol consumption.
Figure 2.

Schematic demonstrating some of the steps leading to cellular damage as a result of reactive oxygen species (ROS). (1) ROS interact with lipids in the cell membrane, (2) resulting in the formation of the aldehyde 4-hydroxynonenal (4HNE) (brown zigzag line). (3) Many proteins are inactivated as a result of 4HNE-induced adduct formation, and their removal by the proteasome (black and gray structure) is critical. However, 4HNE directly inactivates the proteasome (Farout et al. 2006). (4) Therefore, protein aggregates accumulate, further increasing oxidative stress in the cell. (5) Several mitochondrial proteins also are inactivated by 4HNE (Chen et al. 1995; Echtay and Brand 2007; Kristal et al. 1996) including those involved in electron transfer chain (ETC), (6) the citric acid cycle α-ketoglutarate dehydrogenase (KGDH), and (8) mitochondrial integrity. (9) This leads to increased mitochondria-induced ROS production, decreased ATP generation during reperfusion (Inagaki 2003), and reduced repair of the cells from the oxidative damage. This, in turn, leads to further cardiac function loss. This schematic demonstrates a critical role for enzymes that remove the toxic aldehydes, such as the aldehyde dehydrogenase (ALDH)-1 enzyme, which is found in the fluid filling the cells (i.e., the cytosol), and the ALDH2 enzyme, which is found in the mitochondria. (7) However, these ALDHs are inactivated themselves by the toxic 4HNE (Doorn et al. 2006; Luckey et al. 1999). A compound that could protect ALDH from inactivation and decrease the amounts of aldehydes in the cell should protect from damage induced by ischemia and reperfusion and other oxidative stress-induced injuries.
Alda-1 and ALDH2*2
An important feature of Alda-1 is its ability to increase the activity of the inactive ALDH2 variant, ALDH2*2. Thus, studies found that Alda-1 increased the enzyme’s activity in ALDH2*2 homozygous animals by 900 percent, resulting in about 40 percent of the activity of the normal enzyme (Chen et al. 2008). The ability of Alda-1 to partially complement or restore the activity of ALDH2*2 is striking because small molecules rarely can specifically reverse (i.e., rescue) a mutation in humans. Recently, Perez-Miller and colleagues (2010) examined the crystal structures of the complexes between Alda-1 and normal ALDH2 and between Alda-1 and mutant ALDH2*2. This structural comparison revealed that Alda-1 activates the normal enzyme and restores the activity of ALDH2*2 by acting as a structural chaperone. Thus, Alda-1 binds to the “tunnel” formed by the enzyme’s four subunits, in which the acetaldehyde is converted into acetic acid. Binding of Alda-1 does not, as one might assume, block the access of acetaldehyde to the active site. Instead, it may protect critical cysteine amino acids within the tunnel from interacting with the acetaldehyde, which would lead to adduct formation and inactivation of ALDH2 (see figure 2, step 7) (Perez-Miller et al. 2010). These findings explain the data from a previous study, in which Alda-1 prevented 4HNE-induced ALDH2 inactivation, and explain how a brief treatment with Alda-1 can reduce infarct size by about 60 percent in vivo (Chen et al. 2008).
Finally, the structural analysis showed that in the case of the defective ALDH2*2, Alda-1 binding does not just simply increase the effective concentration of acetaldehyde within the active site; instead, it directly promotes the structural and functional rescue of ALDH2*2 (Perez-Miller et al. 2010). This rescue happens without Alda-1 directly interacting with the altered amino acid in ALDH2*2. Thus, it represents a new pharmacological agonist because it increases the interaction between the enzyme and the molecule it acts on (i.e., the aldehyde) and at the same time it protects the enzyme from aldehyde-induced inactivation.
What Did We Learn From the French Paradox?
Although the antioxidant activities of some ingredients in red wine (e.g., resveratrol) may have a role in the cardioprotective effect of moderate red wine consumption, ethanol itself also clearly initiates specific molecular events that lead to better handling of oxidative stress following myocardial infarction. The discussion here has focused on ethanol’s benefits in reducing arteriosclerotic plaques, reducing blood clotting, and protecting cardiac myocytes, thereby reducing the risk for CHD. Whether other components in alcoholic beverages also contribute to the cardioprotection remains to be determined. However, it is clear that moderate alcohol consumption reduces pathological processes associated with cardiac diseases. Nevertheless, the risk of excessive alcohol consumption and alcohol addiction make the medical use of this commonly used agent a challenge.
Acknowledgments
This study was supported in part by National Institutes of Health grant NIAAA–11147 to Dr. Mochly-Rosen.
Footnotes
Financial Disclosure
The authors declare that they have no competing financial interests. Dr. Mochly-Rosen is the founder of KAI Pharmaceuticals, Inc. However, none of the work at her laboratory is supported by the company and the company had no access to information about unpublished research.
For a definition of this and other technical terms, see the glossary, pp. 161–164.
eNOS is an enzyme that produces nitric oxide in the blood vessels; appropriate amounts of nitric oxide help protect organs from damage caused by insufficient blood supply.
The chemical name for Alda-1 is N-(1,3-benzodioxol-5-ylmethyl)-2,6-dichlorobenzamide; other ALDH2 activators have a similar structure except that the chloride atoms on the benzamide ring are substituted by bromide or fluoride.
Refrerences
- Aikens ML, Benza RL, Grenett HE, et al. Ethanol increases surface-localized fibrinolytic activity in cultured endothelial cells. Alcoholism: Clinical and Experimental Research. 1997;1997;21:1471–1478. [PubMed] [Google Scholar]
- Bazzano LA, Gu D, Reynolds K, et al. Alcohol consumption and risk of coronary heart disease among Chinese men. International Journal of Cardiology. 2009;135:78–85. doi: 10.1016/j.ijcard.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belleville J. The French paradox: Possible involvement of ethanol in the protective effect against cardiovascular diseases. Nutrition. 2002;18:173–177. doi: 10.1016/s0899-9007(01)00721-3. [DOI] [PubMed] [Google Scholar]
- Bellows SD, Hale SL, Kloner RA. Acute ethanol does not protect against ischemic/reperfusion injury in rabbit myocardium. Journal of Thrombosis and Thrombolysis. 1996;3(3):181–184. doi: 10.1007/BF00181659. [DOI] [PubMed] [Google Scholar]
- Bollinger O. Ueber die Haufigkeit und Ursachen der idiopathischen Herzhypertrophie in Muenchen. Deutsche Medizinische Wochenschrift. 1884;10:180–181. [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. Journal of Biological Chemistry. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Borradaile NM, Pickering JG. NAD(+), sirtuins, and cardiovascular disease. Current Pharmaceutical Design. 2009;15(1):110–117. doi: 10.2174/138161209787185742. [DOI] [PubMed] [Google Scholar]
- Brandes RP. Activating SIRT1: A new strategy to prevent atherosclerosis? Cardiovascular Research. 2008;80(2):163–164. doi: 10.1093/cvr/cvn245. [DOI] [PubMed] [Google Scholar]
- Britton A, Marmot MG, Shipley MJ. How does variability in alcohol consumption over time affect the relationship with mortality and coronary heart disease? Addiction. 2010;105(4):639–645. doi: 10.1111/j.1360-0443.2009.02832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Mochly-Rosen D. Opposing effects of delta and xi PKC in ethanol-induced cardioprotection. Journal of Molecular and Cellular Cardiology. 2001;33(3):581–585. doi: 10.1006/jmcc.2000.1330. [DOI] [PubMed] [Google Scholar]
- Chen CC, Lu RB, Chen YC, et al. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. American Journal of Human Genetics. 1999a;65(3):795–807. doi: 10.1086/302540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321(5895):1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Gray MO, Mochly-Rosen D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: Role of epsilon protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1999b;96(22):12784–12789. doi: 10.1073/pnas.96.22.12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Bertrand H, Yu BP. Inhibition of adenine nucleotide translocator by lipid peroxidation products. Free Radical Biology & Medicine. 1995;19(5):583–590. doi: 10.1016/0891-5849(95)00066-7. [DOI] [PubMed] [Google Scholar]
- Chen YC, Peng GS, Tsao TP, et al. Pharmacokinetic and pharmacodynamic basis for overcoming acetaldehyde-induced adverse reaction in Asian alcoholics, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenetics and Genomics. 2009;19(8):588–599. doi: 10.1097/FPC.0b013e32832ecf2e. [DOI] [PubMed] [Google Scholar]
- Churchill EN, Disatnik MH, Mochly-Rosen D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilonPKC and activation of aldehyde dehydrogenase 2. Journal of Molecular and Cellular Cardiology. 2009;46(2):278–284. doi: 10.1016/j.yjmcc.2008.09.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KA, Chopra M, Thurnham DI. Wine polyphenols and promotion of cardiac health. Nutrition Research Reviews. 2004;17(1):111–130. doi: 10.1079/NRR200482. [DOI] [PubMed] [Google Scholar]
- Costa AD, Garlid KD, West IC, et al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circulation Research. 2005;97(4):329–336. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- de Curtis A, Murzilli S, di Castelnuovo A, et al. Alcohol-free red wine prevents arterial thrombosis in dietary-induced hypercholesterolemic rats: Experimental support for the ‘French paradox’. Journal of Thrombosis and Haemostasis. 2005;3(2):346–350. doi: 10.1111/j.1538-7836.2005.01126.x. [DOI] [PubMed] [Google Scholar]
- de Lange DW, Verhoef S, Gorter G, et al. Polyphenolic grape extract inhibits platelet activation through PECAM-1: An explanation for the French paradox. Alcoholism: Clinical and Experimental Research. 2007;31(8):1308–1314. doi: 10.1111/j.1530-0277.2007.00439.x. [DOI] [PubMed] [Google Scholar]
- Deckert V, Desrumaux C, Athias A, et al. Prevention of LDL alpha-tocopherol consumption, cholesterol oxidation, and vascular endothelium dysfunction by polyphenolic compounds from red wine. Atherosclerosis. 2002;165(1):41–50. doi: 10.1016/s0021-9150(02)00189-2. [DOI] [PubMed] [Google Scholar]
- Di Castelnuovo A, Rotondo S, Iacoviello L, et al. Meta-analysis of wine and beer consumption in relation to vascular risk. Circulation. 2002;105(24):2836–44. doi: 10.1161/01.cir.0000018653.19696.01. [DOI] [PubMed] [Google Scholar]
- Dong F, Ren J. Fidarestat improves cardiomyocyte contractile function in db/db diabetic obese mice through a histone deacetylase Sir2-dependent mechanism. Journal of Hypertension. 2007;25(10):2138–2147. doi: 10.1097/HJH.0b013e32828626d1. [DOI] [PubMed] [Google Scholar]
- Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chemical Research in Toxicology. 2006;19(1):102–110. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- Dudley JI, Lekli I, Mukherjee S, et al. Does white wine qualify for French paradox? Comparison of the cardioprotective effects of red and white wines and their constituents: Resveratrol, tyrosol, and hydroxytyrosol. Journal of Agricultural Food Chemistry. 2008;56(20):9362–9373. doi: 10.1021/jf801791d. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Brand MD. 4-hydroxy-2-nonenal and uncoupling proteins: An approach for regulation of mitochondrial ROS production. Redox Report. 2007;12(1):26–29. doi: 10.1179/135100007X162158. [DOI] [PubMed] [Google Scholar]
- Endo J, Sano M, Katayama T, et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circulation Research. 2009;105(11):1118–1127. doi: 10.1161/CIRCRESAHA.109.206607. [DOI] [PubMed] [Google Scholar]
- Farout L, Mary J, Vinh J, et al. Inactivation of the proteasome by 4-hydroxy-2-nonenal is site specific and dependant on 20S proteasome subtypes. Archives of Biochemistry and Biophysics. 2006;453(1):135–142. doi: 10.1016/j.abb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Kapphahn RJ. Catalytic site-specific inhibition of the 20S proteasome by 4-hydroxynonenal. FEBS Letters. 2004;578(3):217–223. doi: 10.1016/j.febslet.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Fischer M, Wetherill LF, Carr LG, et al. Association of the aldehyde dehydrogenase 2 promoter polymorphism with alcohol consumption and reactions in an American Jewish population. Alcoholism: Clinical and Experimental Research. 2007;31(10):1654–1659. doi: 10.1111/j.1530-0277.2007.00471.x. [DOI] [PubMed] [Google Scholar]
- Fuchs CS, Stampfer MJ, Colditz GA, et al. Alcohol consumption and mortality among women. New England Journal of Medicine. 1995;332(19):1245–1250. doi: 10.1056/NEJM199505113321901. Erratum in: New England Journal of Medicine 1997;336(7):523. [DOI] [PubMed] [Google Scholar]
- Gaziano JM, Buring JE, Breslow JL, et al. Moderate alcohol intake, increased levels of high-density lipoprotein and its subfractions, and decreased risk of myocardial infarction. New England Journal of Medicine. 1993;329(25):1829–1834. doi: 10.1056/NEJM199312163292501. [DOI] [PubMed] [Google Scholar]
- Gaziano JM, Gaziano TA, Glynn RJ, et al. Light-to-moderate alcohol consumption and mortality in the Physicians’ Health Study enrollment cohort. Journal of the American College of Cardiology. 2000;35(1):96–105. doi: 10.1016/s0735-1097(99)00531-8. [DOI] [PubMed] [Google Scholar]
- Grenett HE, Aikens ML, Torres JA, et al. Cardiprotective benefit of low ethanol: Down-regulation of PAI-1 gene expression and increased fibrinolytic activity in cultured human endothelial cells (ECs) (Abstract) Alcoholism: Clinical and Experimental Research. 1998;22:66a. [Google Scholar]
- Goedde HW, Agarwal DP. Pharmacogenetics of aldehyde dehydrogenase (ALDH) Pharmacology & Therapeutics. 1990;45(3):345–371. doi: 10.1016/0163-7258(90)90071-9. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ, Mosca L, Piano MR, et al. AHA Science Advisory: Wine and your heart: A science advisory for healthcare professionals from the Nutrition Committee, Council on Epidemiology and Prevention, and Council on Cardiovascular Nursing of the American Heart Association. Circulation. 2001;103(3):472–475. doi: 10.1161/01.cir.103.3.472. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, et al. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. Journal of Biological Chemistry. 2004;279(5):3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- Grønbæk M. Alcohol, type of alcohol, and all-cause and coronary heart disease mortality. Annals of the New York Academy of Sciences. 2002;957:16–20. doi: 10.1111/j.1749-6632.2002.tb02902.x. [DOI] [PubMed] [Google Scholar]
- Grønbæk M. Epidemiologic evidence for the cardioprotective effects associated with consumption of alcoholic beverages. Pathophysiology. 2004;10(2):83–92. doi: 10.1016/j.pathophys.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Grønbæk M, Becker U, Johansen D, et al. Type of alcohol consumed and mortality from all causes, coronary heart disease, and cancer. Annals of Internal Medicine. 2000;133(6):411–419. doi: 10.7326/0003-4819-133-6-200009190-00008. [DOI] [PubMed] [Google Scholar]
- Guiraud A, de Lorgeril M, Boucher F, et al. Cardioprotective effect of chronic low dose ethanol drinking: Insights into the concept of ethanol preconditioning. Journal of Molecular and Cellular Cardiology. 2004;36(4):561–566. doi: 10.1016/j.yjmcc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Guo D, Nguyen T, Ogbi M, et al. Protein kinase C-epsilon coimmuno-precipitates with cytochrome oxidase subunit IV and is associated with improved cytochrome-c oxidase activity and cardioprotection. American Journal of Physiology. American Journal of Physiology Heart and Circulation Physiology. 2007;293(4):H2219–H2230. doi: 10.1152/ajpheart.01306.2006. [DOI] [PubMed] [Google Scholar]
- Hagiage M, Marti C, Rigaud D. Effect of a moderate alcohol intake on the lipoproteins of normotriglyceridemic obese subjects compared with normoponderal controls. Metabolism: Clinical and Experimental. 1992;41(8):856–861. doi: 10.1016/0026-0495(92)90167-9. [DOI] [PubMed] [Google Scholar]
- Hale SL, Kloner RA. Ethanol does not exert myocardial preconditioning in an intact rabbit model of ischemia/reperfusion. Heart Disease. 2001;3(5):293–296. doi: 10.1097/00132580-200109000-00003. [DOI] [PubMed] [Google Scholar]
- Hannuksela ML, Liinamaa MJ, Kesaniemi YA, Savolainen MJ. Relation of polymorphisms in the cholesteryl ester transfer protein gene to transfer protein activity and plasma lipoprotein levels in alcohol drinkers. Atherosclerosis. 1994;110(1):35–44. doi: 10.1016/0021-9150(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Matsushita S, Imazeki H, et al. Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet. 1994;343(8899):741–742. doi: 10.1016/s0140-6736(94)91629-2. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Chen L, Ikeno F, et al. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108(19):2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Mochly-Rosen D. DeltaPKC-mediated activation of epsilonPKC in ethanol-induced cardiac protection from ischemia. Journal of Molecular and Cellular Cardiology. 2005;39(2):203–211. doi: 10.1016/j.yjmcc.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Itoya M, Morrison JD, Downey HF. Effect of ethanol on myocardial infarct size in a canine model of coronary artery occlusion-reperfusion. Molecular and Cellular Biochemistry. 1998;186(1–2):35–41. [PubMed] [Google Scholar]
- Jaburek M, Costa AD, Burton JR, et al. Mitochondrial PKC epsilon and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circulation Research. 2006;99(8):878–883. doi: 10.1161/01.RES.0000245106.80628.d3. [DOI] [PubMed] [Google Scholar]
- Jo SA, Kim EK, Park MH, et al. A Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clinica Chimica Acta. 2007;382(1–2):43–47. doi: 10.1016/j.cca.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3 beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. Journal of Clinical Investigation. 2004;113(11):1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, et al. Substrate-specific activation of sirtuins by resveratrol. Journal of Biological Chemistry. 2005;280(17):17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Karliner JS. Lessons from the besotted heart. Journal of the American College of Cardiology. 2009;54(23):2197–2198. doi: 10.1016/j.jacc.2009.06.052. [DOI] [PubMed] [Google Scholar]
- Knupfer G. Drinking for health: The daily light drinker fiction. British Journal of Addiction. 1987;82:547–555. doi: 10.1111/j.1360-0443.1987.tb01511.x. [DOI] [PubMed] [Google Scholar]
- Krenz M, Baines CP, Heusch G, et al. Acute alcohol-induced protection against infarction in rabbit hearts: Differences from and similarities to ischemic preconditioning. Journal of Molecular and Cellular Cardiology. 2001a;33(11):2015–2022. doi: 10.1006/jmcc.2001.1465. [DOI] [PubMed] [Google Scholar]
- Krenz M, Baines CP, Yang XM, et al. Acute ethanol exposure fails to elicit preconditioning-like protection in in situ rabbit hearts because of its continued presence during ischemia. Journal of the American College of Cardiology. 2001b;37(2):601–607. doi: 10.1016/s0735-1097(00)01125-6. [DOI] [PubMed] [Google Scholar]
- Krenz M, Cohen MV, Downey JM. The protective and anti-protective effects of ethanol in a myocardial infarct model. Annals of the New York Academy of Sciences. 2002a;957:103–114. doi: 10.1111/j.1749-6632.2002.tb02909.x. [DOI] [PubMed] [Google Scholar]
- Krenz M, Yang XM, Qin Q, et al. Dose-response relationships of the protective and antiprotective effects of acute ethanol exposure in isolated rabbit hearts. Heart Disease. 2002b;4(5):276–281. doi: 10.1097/00132580-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. Journal of Biological Chemistry. 1996;271(11):6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- Lakshman R, Garige M, Gong M, et al. Is alcohol beneficial or harmful for cardioprotection? Genes and Nutrition. 2009 doi: 10.1007/s12263-009-0161-2. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. Journal of Biological Chemistry. 2005;280(34):30550–30556. doi: 10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SY, Li Q, Shen JJ, et al. Attenuation of acetaldehyde-induced cell injury by overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene in human cardiac myocytes: Role of MAP kinase signaling. Journal of Molecular and Cellular Cardiology. 2006;40(2):283–294. doi: 10.1016/j.yjmcc.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Lindeman RD, Romero LJ, Allen AS, et al. Alcohol consumption is negatively associated with the prevalence of coronary heart disease in the New Mexico Elder Health Survey. Journal of the American Geriatric Society. 1999;47(4):396–401. doi: 10.1111/j.1532-5415.1999.tb07229.x. [DOI] [PubMed] [Google Scholar]
- Lucas DT, Szweda LI. Cardiac reperfusion injury: Aging, lipid peroxidation, and mitochondrial dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(2):510–514. doi: 10.1073/pnas.95.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckey SW, Tjalkens RB, Petersen DR. Mechanism of inhibition of rat liver class 2 ALDH by 4-hydroxynonenal. Advances in Experimental Medicine and Biology. 1999;463:71–77. doi: 10.1007/978-1-4615-4735-8_9. [DOI] [PubMed] [Google Scholar]
- Luu SU, Wang MF, Lin DL, et al. Ethanol and acetaldehyde metabolism in Chinese with different aldehyde dehydrogenase-2 genotypes. Proceedings of the National Science Council, Republic of China B. 1995;19(3):129–136. [PubMed] [Google Scholar]
- Mackness MI, Durrington PN, Mackness B. How high-density lipoprotein protects against the effects of lipid peroxidation. Current Opinion in Lipidology. 2000;11(4):383–388. doi: 10.1097/00041433-200008000-00007. [DOI] [PubMed] [Google Scholar]
- Markus MA, Morris BJ. Resveratrol in prevention and treatment of common clinical conditions of aging. Clinical Interventions in Aging. 2008;3(2):331–339. [PMC free article] [PubMed] [Google Scholar]
- Miyamae M, Camacho SA, Zhou HZ, et al. Alcohol consumption reduces ischemia-reperfusion injury by species-specific signaling in guinea pigs and rats. American Journal of Physiology. 1998a;275(1 Pt 2):H50–H56. doi: 10.1152/ajpheart.1998.275.1.H50. [DOI] [PubMed] [Google Scholar]
- Miyamae M, Diamond I, Weiner MW, et al. Regular alcohol consumption mimics cardiac preconditioning by protecting against ischemia-reperfusion injury. Proceedings of the National Academy of the Sciences of the USA. 1997;94(7):3235–3239. doi: 10.1073/pnas.94.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamae M, Rodriguez MM, Camacho SA, et al. Activation of epsilon protein kinase C correlates with a cardioprotective effect of regular ethanol consumption. Proceedings of the National Academy of the Sciences of the USA. 1998b;95(14):8262–8267. doi: 10.1073/pnas.95.14.8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen EL, Jensen HH, Sanders SA, Reinisch JM. Better psychological functioning and higher social status may largely explain the apparent health benefits of wine: A study of wine and beer drinking in young Danish adults. Archives of Internal Medicine. 2001;161(15):1844–1848. doi: 10.1001/archinte.161.15.1844. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Chen CM, Rao SR, Breslow RA. Alcohol consumption and cardiovascular mortality among U.S. adults, 1987 to 2002. Journal of the American College of Cardiology. 2010;55(13):1328–1335. doi: 10.1016/j.jacc.2009.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamal KJ, Maclure M, Muller JE, et al. Prior alcohol consumption and mortality following acute myocardial infarction. JAMA: Journal of the American Medical Association. 2001;285(15):1965–1970. doi: 10.1001/jama.285.15.1965. [DOI] [PubMed] [Google Scholar]
- Mukamal KJ, Mittleman MA. Acute ethanol exposure fails to elicit preconditioning-like protection in in situ rabbit hearts because of its continued presence during ischemia. Journal of the American College of Cardiology. 2001;38(4):1271. doi: 10.1016/s0735-1097(01)01531-5. [DOI] [PubMed] [Google Scholar]
- Muntwyler J, Hennekens CH, Buring JE, Gaziano JM. Mortality and light to moderate alcohol consumption after myocardial infarction. Lancet. 1998;352(9144):1882–1885. doi: 10.1016/S0140-6736(98)06351-X. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Niccoli G, Altamura L, Fabretti A, et al. Ethanol abolishes ischemic preconditioning in humans. Journal of the American College of Cardiology. 2008;51(3):271–275. doi: 10.1016/j.jacc.2007.09.042. [DOI] [PubMed] [Google Scholar]
- Opie LH, Lecour S. The red wine hypothesis: from concepts to protective signaling molecules. European Heart Journal. 2007;28:1683–1693. doi: 10.1093/eurheartj/ehm149. [DOI] [PubMed] [Google Scholar]
- Perez-Miller S, Younus H, Vanam R, et al. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nature Structural & Molecular Biology. 2010;17(2):159–164. doi: 10.1038/nsmb.1737. 20062057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher MJ, Varosy P, Kiefe CI, et al. Alcohol consumption, binge drinking, and early coronary calcification: Findings from the Coronary Artery Risk Development in Young Adults (CARDIA) Study. American Journal of Epidemiology. 2005;161(5):423–433. doi: 10.1093/aje/kwi062. [DOI] [PubMed] [Google Scholar]
- Rao MN, Marmillot P, Gong M, et al. Light, but not heavy alcohol drinking, stimulates paraoxonase by upregulating liver mRNA in rats and humans. Metabolism. 2003;52(10):1287–1294. doi: 10.1016/s0026-0495(03)00191-4. [DOI] [PubMed] [Google Scholar]
- Rehm J, Baliunas D, Borges GL, et al. The relation between different dimensions of alcohol consumption and burden of disease: An overview. Addiction. 2010;105(5):817–843. doi: 10.1111/j.1360-0443.2010.02899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard JF, Vasiliou V, Petersen DR. Characterization of 4-hydroxy-2-nonenal metabolism in stellate cell lines derived from normal and cirrhotic rat liver. Biochimica et Biophysica Acta. 2000;1487(2–3):222–232. doi: 10.1016/s1388-1981(00)00095-0. [DOI] [PubMed] [Google Scholar]
- Renaud S, de Lorgeril M. Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet. 1992;339(8808):1523–1526. doi: 10.1016/0140-6736(92)91277-f. [DOI] [PubMed] [Google Scholar]
- Renaud SC, Gueguen R, Schenker J, d’Houtaud A. Alcohol and mortality in middle-aged men from eastern France. Epidemiology. 1998;9(2):184–188. [PubMed] [Google Scholar]
- Renaud SC, Gueguen R, Siest G, Salamon R. Wine, beer, and mortality in middle-aged men from eastern France. Archives of Internal Medicine. 1999;159(16):1865–1870. doi: 10.1001/archinte.159.16.1865. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Vaughan DE, Stampfer MJ, et al. Association of moderate alcohol consumption and plasma concentration of endogenous tissue-type plasminogen activator. JAMA: Journal of the American Medical Association. 1994;272(12):929–933. doi: 10.1001/jama.1994.03520120039028. [DOI] [PubMed] [Google Scholar]
- Rimm EB, Giovannucci EL, Willett WC, et al. Prospective study of alcohol consumption and risk of coronary disease in men. Lancet. 1991;338(8765):464–468. doi: 10.1016/0140-6736(91)90542-w. [DOI] [PubMed] [Google Scholar]
- Rimm EB, Williams P, Fosher K, et al. Moderate alcohol intake and lower risk of coronary heart disease: Meta-analysis of effects on lipids and haemostatic factors. British Medical Journal. 1999;319(7224):1523–1528. doi: 10.1136/bmj.319.7224.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen MJ, Kesaniemi YA. Effects of alcohol on lipoproteins in relation to coronary heart disease. Current Opinion in Lipidology. 1995;6(4):243–250. doi: 10.1097/00041433-199508000-00009. [DOI] [PubMed] [Google Scholar]
- Sierksma A, van der Gaag MS, van Tol A, et al. Kinetics of HDL cholesterol and paraoxonase activity in moderate alcohol consumers. Alcoholism: Clinical and Experimental Research. 2002;26(9):1430–1435. doi: 10.1097/01.ALC.0000030639.57507.60. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Colditz GA, Willett WC, et al. A prospective study of moderate alcohol consumption and the risk of coronary disease and stroke in women. New England Journal of Medicine. 1988;319(5):267–273. doi: 10.1056/NEJM198808043190503. [DOI] [PubMed] [Google Scholar]
- Takeshita T, Morimoto K. Self-reported alcohol-associated symptoms and drinking behavior in three ALDH2 genotypes among Japanese university students. Alcoholism: Clinical and Experimental Research. 1999;23(6):1065–1069. [PubMed] [Google Scholar]
- Thun MJ, Peto R, Lopez AD, et al. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. New England Journal of Medicine. 1997;337(24):1705–1714. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- Tjønneland A, Grønbaek M, Stripp C, Overvad K. Wine intake and diet in a random sample of 48763 Danish men and women. American Journal of Clinical Nutrition. 1999;69(1):49–54. doi: 10.1093/ajcn/69.1.49. [DOI] [PubMed] [Google Scholar]
- Valimaki M, Kahri J, Laitinen K, et al. High density lipoprotein sub-fractions, apolipoprotein A-I containing lipoproteins, lipoprotein (a), and cholesterol ester transfer protein activity in alcoholic women before and after ethanol withdrawal. European Journal of Clinical Investigation. 1993;23(7):406–417. doi: 10.1111/j.1365-2362.1993.tb00783.x. [DOI] [PubMed] [Google Scholar]
- van de Wiel A, de Lange DW. Cardiovascular risk is more related to drinking pattern than to the type of alcoholic drinks. Netherlands Journal of Medicine. 2008;66(11):467–473. [PubMed] [Google Scholar]
- van der Gaag MS, van Tol A, Scheek LM, et al. Daily moderate alcohol consumption increases serum paraoxonase activity: a diet-controlled, randomised intervention study in middle-aged men. Atherosclerosis. 1999;147(2):405–410. doi: 10.1016/s0021-9150(99)00243-9. [DOI] [PubMed] [Google Scholar]
- van Tol A, Hendriks HF. Moderate alcohol consumption: Effects on lipids and cardiovascular disease risk. Current Opinion in Lipidology. 2001;12(1):19–23. doi: 10.1097/00041433-200102000-00004. [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Human Genomics. 2005;2(2):138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Pappa A, Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metabolism Reviews. 2004;36(2):279–99. doi: 10.1081/dmr-120034001. [DOI] [PubMed] [Google Scholar]
- Vidavalur R, Otani H, Singal PK, Maulik N. Significance of wine and resveratrol in cardiovascular disease: French paradox revisited. Experimental and Clinical Cardiology. 2006;11(3):217–225. [PMC free article] [PubMed] [Google Scholar]
- Wallerath T, Poleo D, Li H, Förstermann U. Red wine increases the expression of human endothelial nitric oxide synthase: A mechanism that may contribute to its beneficial cardiovascular effects. Journal of the American College of Cardiology. 2003;41(3):471–478. doi: 10.1016/s0735-1097(02)02826-7. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) WHO Statistical Information System (WHOSIS) 2007. Per capita recorded alcohol consumption (litres of pure alcohol) among adults (>=15 years) [Google Scholar]
- Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Kato H, Yokoyama T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and glutathione S-transferase M1 and drinking, smoking, and diet in Japanese men with esophageal squamous cell carcinoma. Carcinogenesis. 2002;23(11):1851–1859. doi: 10.1093/carcin/23.11.1851. [DOI] [PubMed] [Google Scholar]
- Zakhari S. Overview: How is alcohol metabolized by the body? Alcohol Research & Health. 2006;29(4):245–254. [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Wang Z, Chen HZ, et al. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovascular Research. 2008;80(2):191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HZ, Karliner JS, Gray MO. Moderate alcohol consumption induces sustained cardiac protection by activating PKC-epsilon and Akt. American Journal of Physiology Heart and Circulation Physiology. 2002;283(1):H165–74. doi: 10.1152/ajpheart.00408.2001. [DOI] [PubMed] [Google Scholar]