

Abstract
Staphylococcus aureus is a major human pathogen that uses quorum sensing (QS) to control virulence. Its QS system is regulated by macrocyclic peptide signals (or autoinducing peptides (AIPs)) and their cognate transmembrane receptors (AgrCs). Four different specificity groups of S. aureus have been identified to date (groups I–IV), each of which uses a different AIP:AgrC pair. Non-native ligands capable of intercepting AIP:AgrC binding, and thereby QS, in S. aureus have attracted considerable interest as chemical tools to study QS pathways and as possible anti-virulence strategies for the treatment of infection. We recently reported a set of analogs of the group-III AIP that are capable of strongly modulating the activity of all four AgrC receptors. Critical to the further development of such ligands is a detailed understanding of the structural features of both native AIPs and non-native analogs that are essential for activity. Herein, we report the first three-dimensional structural analysis of the known native AIP signals (AIPs-I–IV) and several AIP-III analogs with varied biological activities using NMR spectroscopy. Integration of these NMR studies with the known agonism and antagonism profiles of these peptides in AgrC-III revealed two key structural elements that control AIP-III (and non-native peptide) activity: (1) a tri-residue hydrophobic “knob” essential for both activation and inhibition, and (2) a fourth anchor point on the exocyclic tail needed for receptor activation. These results provide strong structural support for a mechanism of AIP-mediated AgrC activation and inhibition in S. aureus, and should facilitate the design of new AgrC ligands with enhanced activities (as agonists or antagonists) and simplified chemical structures.
Introduction
Staphylococcus aureus is an opportunistic, Gram-positive bacterial pathogen that is a primary cause of human infections worldwide.1,2 The emergence of S. aureus strains resistant to last-line antibiotics,3,4 such as vancomycin, has stimulated an urgent need for the development of new antimicrobial approaches against this bacterium. Strategies that target infectivity (i.e., virulence) as opposed to growth have attracted much recent interest.5 Such anti-virulence strategies could offer reduced selection pressure for drug-resistant mutations, and represent a paradigm shift for the treatment of bacterial infection.6 Pivotal to the ability of S. aureus to initiate virulence is its capability to assess its local population density using quorum sensing (QS).7 S. aureus uses the agr (accessory gene regulator) two-component signaling system for QS, which is mediated in part by macrocyclic peptide signals (or autoinducing peptides (AIPs)) and their cognate receptors (AgrCs).8,9 The AgrCs are transmembrane, receptor histidine kinases. AIP signal concentration increases with bacterial cell density, and when a sufficient density is achieved in a given environment, binding of the AIP to the extracellular sensor domain of AgrC causes AgrC activation and autophosphorylation. AgrC then phosphorylates the response regulator, AgrA, which then goes on to directly activate expression of virulence genes.
Four different AIP:AgrC pairs have been characterized so far, resulting in the categorization of four different specificity groups of S. aureus (I–IV).7,8 The AIPs-I–IV vary in length from hepta- to nonapeptides, and share a 5-amino acid (aa) Cys→C-terminus macrocyclic thiolactone core and a 2-4-aa exocyclic tail (shown in Figure 1A). While their primary sequences differ, all four AIPs show a gradient of increasing hydrophobicity from their N to C termini, ending with bulky hydrophobic residues at the C terminal positions.10
Figure 1.

A) Structures of the native AIPs (I-IV) used by S. aureus for QS. B) Two representative AIP-III analogs identified by our research group that are potent inhibitors of AgrC receptors.
Methods to inhibit AIP:AgrC interactions represent a direct strategy to block QS, and thereby halt virulence, in S. aureus.11 Over the past decade, the development of non-native synthetic ligands (both small peptides and macromolecules) capable of AgrC inhibition has received significant attention.12-18 Intriguingly, initial studies of the agr system revealed that each of the four native AIPs are capable of cross-inhibiting the other three, non-cognate AgrC receptors.12,13,19 This cross-group interference has been suggested to potentially provide each group with a competitive advantage when establishing an infection; however, the in vivo relevance of this interference remains poorly understood, as some infection types contain specific groups of S. aureus while others contain multiple groups.9
Most past work directed toward the development of abiotic AIP:AgrC modulators has been focused on the AIP-I and AIP-II signals12-15,17,20,21 due to the prevalence of groups-I and -II S. aureus in human infections.22-24 The presence of group-III S. aureus in infections appears to be more common than previously estimated, however.22,23 We recently performed a systematic SAR study of the AIP-III signal, and identified a set of AIP-III analogs that strongly disrupt AIP:AgrC interactions and, to our knowledge, are the most potent AgrC inhibitors to be reported in all four groups of S. aureus (Figure 1B).25 These past experiments indicated that the AIP-III scaffold might provide a superior scaffold for the development of peptide-based AgrC inhibitors. Many questions remain with regards to their mechanisms of action. Critically, we lack virtually any information about the three-dimensional (3-D) solution-phase structures of any of the S. aureus AIPs or analogs thereof.26 Detailed structural analyses of these macrocyclic peptides would illuminate their modes of action and facilitate the design of new ligands with simplified chemical structures and improved properties for use both as research tools and as potential therapeutic leads.
Toward this goal, we report herein the first 3-D structural characterization of the four known native AIP signals in S. aureus and several of our AIP-III analogs using NMR spectroscopy. Comparison of the calculated structures of the native AIP-III signal to the AIP-III analogs allowed us to develop a mechanistic understanding of their modes of AgrC-III activation and inhibition that could be directly correlated to their observed biological activities in cell-based assays. Likewise, comparison of the AIP-III and analog structures to those of the native AIPs-I, -II and -IV provided us with insights into the structural features dictating their cross-group inhibitory activity of AgrC-III. While some caution must be taken when drawing conclusions about the biologically active conformations of ligands under solution-phase conditions that differ from native conditions (and in the absence of the receptor), our ability to very well correlate these structural data to the biological activities of these peptides suggest that the structures, and mechanistic rationales derived therefrom, have biological relevance. These results are significant as they represent the first structural data in support of a mechanism of AIP-mediated AgrC activation and inhibition in S. aureus, and serve to refine many prior hypotheses about AIP:AgrC interactions in general.
Experimental Section
Chemical and biological reagents
All reagents and solvents were purchased from commercial sources (Alfa-Aesar, Sigma-Aldrich, J.T. Baker, and Acros) and used without further purification. Water (18 MΩ) was purified using a Millipore Analyzer Feed System. All peptides analyzed in this study were prepared using our previously reported solid-phase synthesis and macrocyclization protocols.25 The peptides were purified to homogeneity using standard reversed-phase high performance liquid chromatography (RP-HPLC) methods.
Strain information and assay methods
The S. aureus reporter strains AH174727 and RN953214 were used for the AgrC-III antagonism and agonism assays, respectively. S. aureus AH1747 is a methicillin-resistant group-III strain harboring a P3-gfp reporter plasmid. Activation of AgrC-III results in phosphorylation of AgrA, which will then bind P3 and transcribe gfp (in addition to typical upregulation of RNAIII).27 S. aureus RN9532 is a group-I agr-null strain harboring a P3-blaZ reporter plasmid, and a chimeric AgrC receptor containing the N-terminal sensor domain of the group-III agrC coding sequence fused to the C-terminal histidine kinase domain of the group-IV agrC, followed by the agrA coding sequence. Activation of the chimeric AgrC-III-IV receptor results in phosphorylation of AgrA, which will then bind P3 and transcribe blaZ.14 [We note that Muir, Novick, and co-workers have demonstrated that the sensor domain from one AgrC receptor can be attached to the histidine kinase domain of a different AgrC without changing the signaling characteristics of that domain.28]
Antagonism of AgrC-III by peptides was measured in by monitoring GFP fluorescence using S. aureus AH1747 as previously described by our laboratory.25 Agonism of AgrC-III by peptides was measured by monitoring β-lactamase activity using S. aureus RN9532 and a modified version of the assay described by Novick and co-workers.29 Serial dilutions of peptide stock solutions were performed to obtain dose response antagonism and agonism data, and IC50 and EC50 values were calculated, respectively. See Supporting Information for full details of assay methods and dose-response data.
NMR methods and structural analyses
All NMR spectra were recorded on a Varian Inova 600 MHz spectrometer at 298 K, using 1.0–1.5 mM solutions of the peptides in 70% H2O/30% CD3CN, pH 3.65 (note, the peptides are insoluble in aqueous solutions at the concentrations needed for the NMR experiments). Spectra were processed using the Vnmr software package (v. 6.1C; Varian) and NMRPipe software.30 Chemical shifts were referenced to CD3CN at 1.94 ppm.
Two-dimensional (2-D) homonuclear experiments gcosy, wgtocsy (tocsy31,32 using DIPSI spinlock and the 3-9-19 water suppression sequence) and wgroesy (rotating frame NOE experiment with pulsed T-Roesy33 spin lock and the 3-9-19 water suppression sequence) were acquired. The gcosy experiments were collected with 1754 and 512 real data points in the direct and indirect dimensions, respectively, with 16 scans per data point. For the wgtocsy experiments, 1536 and 256 real data points were collected in the direct and indirect dimensions, respectively, with 32 scans per data point. A relaxation delay of 2 sec was used for both the gcosy and wgtocsy experiments, with a mixing time of 80 ms for the wgtocsy. For the wgroesy experiments, 3080 real data points were used in the direct dimension, with data points in the indirect dimension ranging from 256 to 400, and the number of scans ranging from 48 to 80 per data point. A relaxation delay of 3 sec and a mixing time of 300 ms were used. Presaturation water suppression was used in the 1H 1-D and 22706 real data points were acquired, with 32 scans per data point. One-dimensional 1H spectra and representative 2-D spectra for each peptide are included in the Supporting Information.
All spectra were analyzed with SPARKY.34 Assignment of resonances for each peptide (listed in Tables S-1–S-11) was achieved using standard sequential assignment methodology.35 The number of ROEs observed for each peptide are listed in Table S-12. The volumes of the ROE peaks were calculated by SPARKY and converted into a continuous distribution of interproton distance restraints, with a uniform 20% distance error applied to take into account spin diffusion. Three-dimensional structure calculations and refinements made use of the torsion angle molecular dynamics and the internal variable dynamics modules36 of Xplor-NIH (v. 2.31),37 with patches for the thioester bridge and ring closure. The target function minimized was comprised of the experimental NMR restraints (ROE-derived interproton distances and torsion angles), a repulsive van der Waals potential for the non-bonded contacts,38 a torsion angle database potential of mean force,39 and a gyration volume potential.40 PyMOL41 and Chimera42 were used for visual analysis and presentation of the peptide structures.
The pdb files for each peptide structure are included in the Supporting Information. These files have also been deposited at the BMRB SMSDep website (http://smsdep.protein.osaka-u.ac.jp/bmrb-adit/; BMRB accession numbers listed in Table S-13). Movies of the 20-structure ensembles for each peptide and the Kyte Doolittle (KD) hydrophobicities of AIP-III, AIP-III D4A, and AIP-III d-L7 are included as Web Enhanced Objects.
Results and Discussion
Biological analyses and SAR development
In our past work, we systematically altered the structure of AIP-III, evaluated the analogs for antagonism of AgrC receptors I–IV using cell-based reporter gene assays in wild-type S. aureus strains, and delineated several structure-activity relationships (SARs) for AgrC receptor antagonism.25 For the current study, we sought to connect these SARs for AgrC-III modulation with peptide 3-D structural data. We therefore further evaluated a subset of the AIP-III analogs that displayed a range of inhibitory activities against AgrC-III in our initial study and determined their relative IC50 values using the group-III reporter strain27 for a more comparative analysis of SARs (listed in Table 1). We also include the IC50 values for the native AIPs-I, -II, and –IV in Table 1 (values determined in our earlier study using the same AgrC-III antagonism assay)25 for comparison to the non-native analogs. In tandem, we evaluated the AIP-III analogs and AIPs-I, -II, and –IV for any intrinsic activity as AgrC-III agonists using an alternate mutant S. aureus reporter strain14 (see Experimental Section for full details of strains); only AIP-III d-L7 was found to display agonistic activity (with an EC50 value comparable to AIP-III, Figure S-2). Our results demonstrating the inactivity of AIPs-I, -II, and –IV in this AgrC-III agonism assay corroborate those of Novick, Muir, and co-workers.14
Table 1.
Peptides evaluated in this study and associated antagonistic activities against AgrC-III.
| Peptide | Sequence | AgrC-III antagonism IC50 value (CI)a, b |
|---|---|---|
| Native AIPs | ||
| AIP-I | Y-S-T-(C-D-F-I-M) | 0.522 nM (0.309–0.883)c |
| AIP-II | G-V-N-A-(C-S-S-L-F) | 0.532 nM (0.238–1.19)c |
| AIP-III | I-N-(C-D-F-L-L) | -- |
| AIP-IV | Y-S-T-(C-Y-F-I-M) | 0.460 nM (0.213–0.994)c |
| AIP-III analogs | ||
| AIP-III D4A | I-N-(C-A-F-L-L) | 0.0506 nM (0.0227–0.113)c |
| tAIP-III | Ac-(C-D-F-L-L) | 54.7 nM (25.9–115) |
| tAIP-III D2A | Ac-(C-A-F-L-L) | 0.329 nM (0.215–0.502)c |
| AIP-III d-F5 | I-N-(C-D-DF-L-L) | 375 nM (258–546) |
| AIP-III d-L6d | I-N-(C-D-F-DL-L) | 181 nM (98.6–333) |
| AIP-III d-L7 | I-N-(C-D-F-L-dL) | -- |
| AIP-III F5A | I-N-(C-D-A-L-L) | 182 nM (73.9–447) |
| AIP-III L6Ad | I-N-(C-D-F-A-L) | 110 nM (49.0–246) |
| AIP-III L7A | I-N-(C-D-F-L-A) | >1000 nM |
See Experimental Section for details of S. aureus reporter strain and antagonism assay.
CI = 95% confidence interval.
Value from ref. 25.
NMR structure not determined. -- indicates no observable antagonistic activity.
This collective set of AgrC-III antagonism and agonism data for the AIP-III analogs revealed chemical motifs that are critical for AIP-III activity and served to focus our selection of peptides for subsequent NMR analyses (Table 1). We summarize critical SAR here. First, replacing the endocyclic Asp4 with Ala (AIP-III D4A; structure in Figure 1B) converted AIP-III from an agonist to an extremely potent antagonist of AgrC-III (along with AgrC-I, -II, and -IV).25 Second, removal of the exocyclic tail from AIP-III yielded AgrC-III inhibitors (e.g., truncated analogs tAIP-III and tAIP-III D2A (Figure 1B)), yet maintenance of this tail was critical for AgrC-III activation. However, certain AIP-III analogs that contained the exocyclic tail were also capable of AgrC-III inhibition (e.g., AIP-III D4A). Third, the hydrophobic amino acid side chains on the AIP-III macrocycle (Phe5, Leu6, and Leu7) were important for both AgrC-III activation and inhibition, as replacing these residues with Ala or the d-isomer abolished agonistic activity (with one exception) and severely attenuated antagonistic activity (relative to AIP-III D4A). The endocyclic hydrophobic residues of AIP-I and -II have previously been shown to play a similarly important role in cognate receptor activation (residues 6–8 in AIP-I and 8–9 in AIP-II).12,15 Interestingly for AIP-III however, replacement of Leu7 with d-Leu yielded an AgrC-III agonist (AIP-III d-L7) with activity comparable to AIP-III (see above).
Turning to the AgrC-III antagonism data for AIPs-I, -II, and -IV, these native peptides each showed comparable and strong IC50 values that were only marginally higher than that for our truncated AIP-III analog, tAIP-III D2A (Table 1). Delineating precise structural features that belay these similar inhibitory activities is challenging a priori due to the divergent primary sequences of the native AIPs and tAIP-III D2A. However, two of the general SAR for AgrC-III antagonism outlined above are maintained: (1) these four peptides retain a largely hydrophobic macrocyclic core (with AIP-II the least hydrophobic overall), and (2) the presence of the exocyclic tail (and its primary structure in general) do not appear to influence their relative ability to inhibit AgrC-III.
These collective SAR for the AIP-III analogs and the native AIPs-I, -II, and -IV are largely congruent with the general hypotheses developed by Novick, Muir, and co-workers in 2004 for all AIP:AgrC interactions.10 The authors proposed that (i) a hydrophobic face of the macrocycle (or “patch”) is essential for initial receptor recognition, (ii) this binding is tuned by specific residues on the macrocycle, and (iii) the exocyclic tail along with appropriate binding contacts on the macrocycle (e.g., the thiolactone itself is essential) are required for receptor activation. These hypotheses support a mechanism by which our AIP-III analogs and the native AIPs-I, -II, and -IV can competitively bind AgrC-III but fail to activate the receptor; this model is congruent with our observation that the native AIP-III can outcompete the analogs in a dose dependent manner in the S. aureus AgrC-III reporter assay.25 The native AIPs-I and -II have also been shown to competitively inhibit AgrC-II and AgrC-I, respectively, using similar experiments.28 While there is no structural information to date on the transmembrane AgrC receptors, a hydrophobic binding pocket on the extracellular AgrC receptor distal domain is predicted that recognizes the AIP patch.10
NMR analyses
We reasoned that the modified features in our AIP-III analogs could have substantial effects on their overall 3-D structures relative to native AIP-III, and these were altering their ability to interact with AgrC-III. Further, as the native AIPs-I, -II, and -IV are each capable of inhibiting (yet not activating) AgrC-III, we reasoned that their 3-D structures could also be disparate from AIP-III and potentially share features with our AIP-III-derived AgrC-III inhibitors. To test these hypotheses, we examined the structures of the four native AIPs and seven AIP-III analogs with varied activities (listed in Table 1) using NMR spectroscopy methods. All 1-D and 2-D NMR experiments (gCOSY, wgTOCSY, and wgROESY) were recorded on a 600 MHz spectrometer (298 K, 1.0–1.5 mM solutions of peptide in 70% H2O/30% CD3CN, pH 3.65; see Experimental Section for full details of methods). ROEs were converted to distance restraints (58–100 ROEs per peptide; Table S-12), and 3-D structure calculations and refinements were performed using Xplor-NIH (see Supporting Information for overlays of 20 lowest energy structures and RMSD values for each peptide). Although the NMR conditions vary from that of our biological assays,43 we believe that the calculated structures provide important insights into the intrinsic conformational propensities of these ligands and the effects of these conformations on their observed biological activities (see below).
Comparison of the AIP-III structure to the AIP-III analog structures
The calculated structure for AIP-III is shown in Figure 2A. Both the macrocyclic and exocyclic tail portions were found to be highly structured (see also Figure S-3A). The tail backbone projects away from the macrocycle, corroborating an earlier NMR study of other native AIPs indicating their structural independence.14 Closer inspection of the AIP-III structure shows the side chains of the three endocyclic hydrophobic residues (Phe5, Leu6, and Leu7) protruding from one face of the macrocycle in a triangular arrangement that we refer to as a hydrophobic “knob” (Figure 2C). We propose that this knob represents the previously predicted hydrophobic patch that is critical for initial binding of AIP-III to AgrC receptors (see above). The side chain of Ile1 projects in the same direction as the knob, while the intervening Asn2 and Asp4 side chains project in the opposite direction. This arrangement yields a structure for AIP-III with one largely hydrophobic face and an opposing more hydrophilic face (see Figure 2C).
Figure 2.
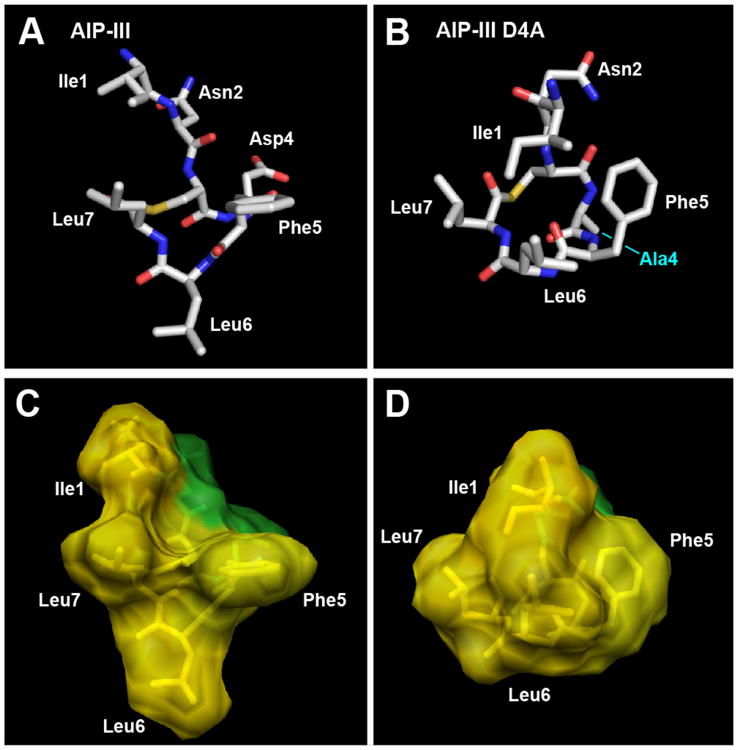
Heavy atom lowest energy structures of A) AIP-III and B) AIP-III D4A. Altered residue labeled in cyan. Carbon is shown in silver, oxygen in red, nitrogen in blue, and sulfur in yellow. Space-filling models of C) AIP-III and D) AIP-III D4A displaying hydrophobic (yellow) and hydrophilic (green) surfaces.
The structure of AIP-III D4A, our most potent AgrC-III inhibitor (IC50 value = 50.6 pM), also displays a hydrophobic face (Figure 2B), with the Phe5, Leu6, and Leu7 side chains projecting from one side. However, the Asp4→Ala modification results in a significant conformational change relative to AIP-III. The side chains of Ala4 and Phe5 now face each other in the AIP-III D4A structure, while the Asp4 and Phe5 side chains are opposed in AIP-III (Figure 2A; see Figure S-5 for an overlay). The repositioning of the Phe5 side chain results in a nearly 180° flip of the Leu6 side chain in AIP-III D4A, pulling the entire triangular hydrophobic knob towards Ile1 on the tail (Figure 2D). These structural data suggest that Asp4 plays a pivotal role in receptor activation by AIP-III, by serving to appropriately position Ile1 and arrange the triangular knob in this native AIP. In turn, the structure provides a model for understanding the inhibitory activity of AIP-III D4A. Lacking Asp4, AIP-III D4A can still use its more compressed hydrophobic knob to competitively bind to AgrC-III, yet due to the differentially positioned Ile1 residue, the analog fails to activate the receptor. Notably, this model could also explain, at least in part, the strong inhibitory activity of AIP-I D5A against AgrC-III, an AIP-I analog with the same Asp→Ala modification, as reported by Muir, Novick, and co-workers in 2002.14 However, as the native AIP-I and the truncated analog of AIP-I D5A lacking a tail, tAIP-I D2A, were also shown to inhibit AgrC-III (albeit to differing degrees; 10-fold less active and 3-fold more active, respectively),14 the mechanisms by which these three peptides inhibit AgrC-III cannot solely depend on the positioning of the exocyclic tail. We return to this point below.
The importance of the endocyclic triangular knob for receptor recognition was further underscored by the structures of the truncated versions of AIP-III and AIP-III D4A, tAIP-III and tAIP-III D2A (Figure 3). Both of these analogs lack the exocyclic tail that appears critical for AgrC-III activation. We hypothesized that tAIP-III, a ∼1000-fold weaker inhibitor than AIP-III D4A (IC50 value = 54.7 nM), would have a disrupted triangular knob. Indeed, the structure of tAIP-III reveals the Phe side chain pointing in the opposite direction as the two Leu side chains (Figure 3A). When overlaid with the structure of AIP-III (Figure 3B), the macrocycle backbone of tAIP-III is flatter than AIP-III and the Phe3 and Leu4 (but not Leu5) side chains (as numbered in tAIP-III) are splayed below and above their counterparts in AIP-III, respectively. We reason that this disrupted knob prevents tAIP-III from effectively binding AgrC-III and contributes to its weaker inhibitory activity (its poor overlay with AIP-III D4A also supports this hypothesis; see Figure S-6).
Figure 3.
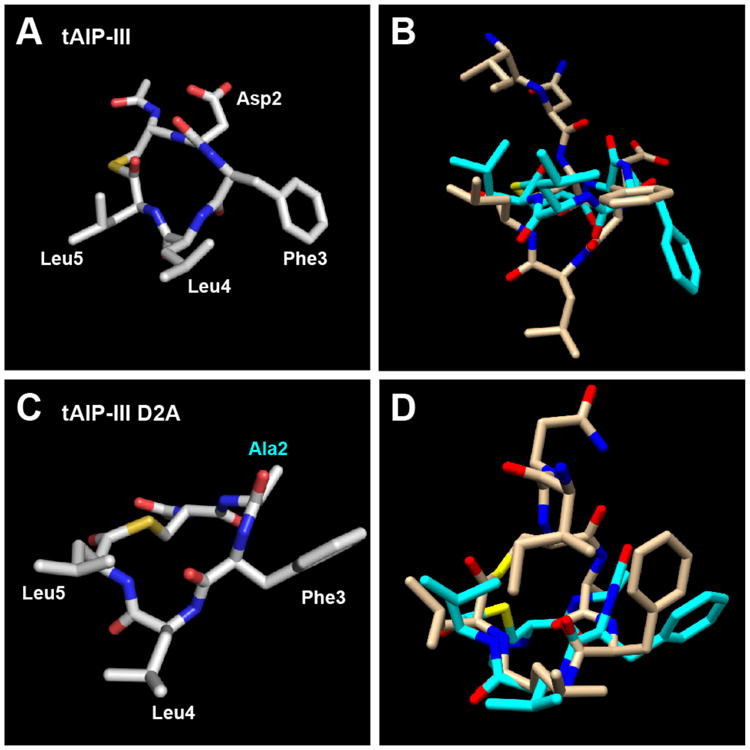
A) Heavy atom lowest energy structure of tAIP-III. B) Overlay of tAIP-III (cyan) and AIP-III (tan) structures. C) Heavy atom lowest energy structure of tAIP-III D2A. Altered residue labeled in cyan. D) Overlay of tAIP-III D2A (cyan) and AIP-III D4A (tan) structures.
tAIP-III D2A is ∼100-fold stronger inhibitor of AgrC-III relative to tAIP-III (IC50 value = 329 pM), which suggested that it would contain an appropriately oriented triangular knob. Accordingly, the NMR structure of tAIP-III D2A (Figure 3C) revealed it to adopt a conformation largely analogous to that of the full-length peptide AIP-III D4A, retaining the hydrophobic knob (see Figure 3D for an overlay; heavy atom RMSD = 2.38 Å). These data suggest that the reduced inhibitory activity of tAIP-III D2A relative to AIP-III D4A (six-fold) may be attributable to the elimination of the fourth exocyclic hydrophobic residue, Ile1, which thus plays a role in both receptor binding (as in AIP-III D4A) and, when oriented correctly, engaging in contacts for receptor activation (as in the native AIP-III; see above).
Analysis of the structures of the four AIP-III analogs with modified endocyclic hydrophobic residues further supports the importance of the triangular knob for receptor binding. The two analogs with alanine modifications, AIP-III F5A and AIP-III L7A, were weak to very weak AgrC-III inhibitors, respectively (Table 1). And, as expected for these peptides that each lacked one of the three endocyclic hydrophobic side chains, their structures lacked an obvious triangular knob motif (Figures 4A and 4B). Interestingly, the Phe5→Ala replacement promotes a more extended conformation for the tail of AIP-III F5A, while the Leu7→Ala replacement in AIP-III L7A brings the side chain of Asp4 and the amino terminus into sufficiently close proximity for a hydrogen bond (H-bond observed in 80% of the calculated structures; Figure 4B). The orientation of the Asp4 side chain and the amino terminus in AIP-III L7A (as a result of the H-bond) prevents a clear segregation of its hydrophobic and hydrophilic faces, which could further abrogate receptor binding and explain its very weak inhibitory activity against AgrC-III.
Figure 4.
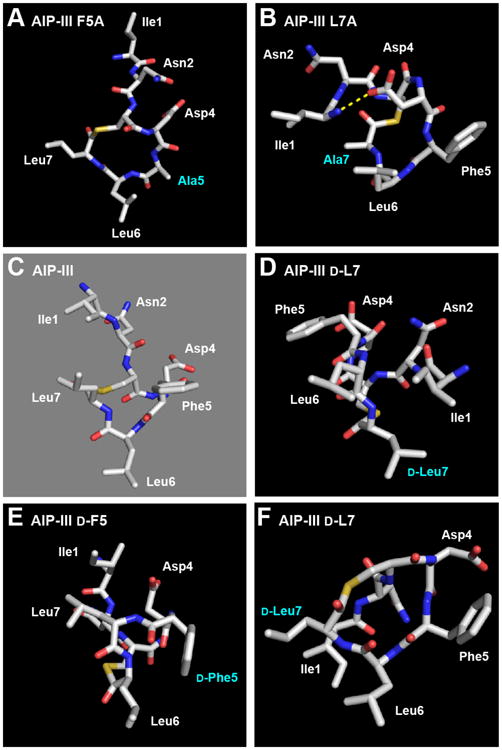
Heavy atom lowest energy structures of A) AIP-III F5A; B) AIP-III L7A (Asp4→N-terminus H-bond shown with dashed yellow line); C) AIP-III (for comparison); D) AIP-III d-L7, from an angle that shows the alternate triangular knob and activation anchor; E) AIP-III d-F5; F) AIP-III d-L7, from the same angle as the other peptides. Altered residues labeled in cyan.
AIP-III d-F5 was also a weak AgrC-III inhibitor (Table 1), suggesting that the inverse stereochemistry at Phe5 resulted in a significant conformational change compared to AIP-III. Its structure revealed two, separate hydrophobic motifs derived from the Ile1/Leu7 and d-Phe5/Leu6 side chains (Figure 4E), instead of one main triangular core. We speculate that one (or both) of these two motifs cannot compensate for the absent triangular knob, and thereby attenuates AIP-III d-F5:AgrC-III binding.
AIP-III d-L7 was the only analog evaluated in this study that was able to activate AgrC-III (with analogous activity as AIP-III; see above) and was thus predicted to possess the same structural features as AIP-III – namely the triangular knob and an activation anchor. To our surprise, the endocyclic knob was absent in its NMR structure (Figure 4F). However, closer analysis of the AIP-III d-L7 structure indicated that both the triangular knob and an activation anchor are likely still present in this analog, but in slightly different forms. We propose that the knob formed from Phe5, Leu6, and Leu7 in AIP-III is now formed instead from Ile1, d-Leu7, and Leu6 in AIP-III d-L7, whereas the activation anchor Ile1 in AIP-III is replaced by Phe5 in AIP-III d-L7 (see Figure 4C vs. 4D; space-filling models shown in Figure 5). These similar structural features may explain the ability of AIP-III d-L7 to strongly activate AgrC-III and suggest the interesting possibility that the roles of the four hydrophobic side chains in the 1, 5, 6 and 7 positions of AIP-III (as part of the knob or as the anchor) may be interchangeable. Additional AIP-III analogs could be readily constructed to test this hypothesis; such studies and related experiments to dissect steric contributions at these positions in AIP-III are ongoing.10
Figure 5.
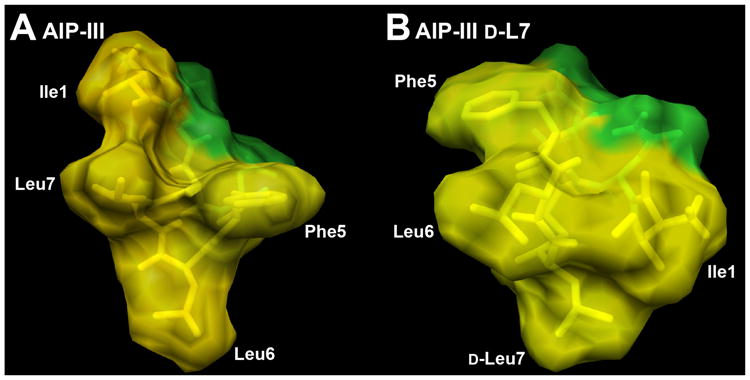
Space-filling models of A) AIP-III and B) agonist AIP-III d-L7 displaying hydrophobic (yellow) and hydrophilic (green) surfaces. Ile1 is proposed to act as the activation anchor, and Phe5, Leu6, and Leu7 as the hydrophobic knob, in the native AIP-III. In turn, Phe5 may serve as an alternate activation anchor, and Ile1, d-Leu7, and Leu6 as an alternate hydrophobic knob, in AIP-III d-L7.
Comparison of the AIP-III and analog structures to the AIP-I, -II, and –IV structures
We next turned our attention to the structures of the native AIPs-I, -II, and –IV, each of which was only a slightly less potent AgrC-III antagonist than the truncated AIP-III analog, tAIP-III D2A (IC50 values ≈ 500 pM; Table 1). We were interested to learn if they maintained the hydrophobic knob motif observed in the strong AgrC-III antagonists derived from AIP-III, as outlined above, or if they adopted alternate structures. If they did contain the knob motif, we reasoned that they likely would not have the hydrophobic anchor point in the exocyclic tail, as these three native AIPs are incapable of AgrC-III activation.
There are many interesting questions that can be addressed through a comparative analysis of the 3-D structures of the natives AIPs; however, for the purposes of the current study, we focused on examining structural features of AIPs-I, -II, and -IV that could engender inhibitory activity against AgrC-III. Nevertheless, it is useful to highlight some differences between the four native AIPs at the outset. AIP-II has a considerably different primary sequence relative to the other AIPs, containing only two bulky hydrophobic residues in its macrocycle, a longer, four-residue exocyclic tail, and a more hydrophilic sequence overall (Figure 1A). In turn, AIP-I and AIP-IV have nearly identical primary sequences, with the only difference being at the fifth residue (Asp5 in AIP-I vs. Tyr5 in AIP-IV; see Figure 1A). Both possess three bulky, hydrophobic residues in the macrocycle that resemble those in AIP-III (Phe6, Ile7, and Met8 in AIPs-I and -IV vs. Phe5, Leu6, and Leu7 in AIP-III). Turning to the cognate receptors for these AIPs, the AgrC-II N-terminal sensor domain is the most distinct (∼30% sequence similarity to the other receptors), while the sensor domains in the other three AgrC receptors are more similar (nearly 90% sequence similarity between AgrC-I and -I V, and over 50% sequence similarity between AgrC-III and -I or -IV).7 This dissimilarity suggests that AgrC-II may bind its native ligand (AIP-II) in an alternate manner relative to the other AgrC receptors, and/or that the active conformation of AIP-II is different relative to the other AIPs, even whilst maintaining some form of a hydrophobic patch (as shown to be important for activity in earlier SAR studies; see above).12 Previous investigations with a constitutively active AgrC-I receptor mutant have shown that AIP-II can behave as an inverse agonist of this mutant AgrC-I, while AIP-III and AIP-IV were determined to be neutral antagonists, further underscoring the possibility for AIP-II to engage in different interactions with AgrC receptors.44 Together, these observations suggest that AIP-II could potentially adopt a conformation that is different from that of the other native AIPs, and that of the AIP-III-derived analogs described above.
The calculated structure for AIP-I is shown in Figure 6A and, as anticipated, reveals a triangular hydrophobic knob motif comprised of the endocyclic residues Phe6, Ile7, and Met8. Similar to AIP-III and the AIP-III analogs above, the macrocycle and exocyclic tail of AIP-I were both well ordered (see Figure S-7A). This result contrasts with an earlier partial NMR study of AIP-I by Chan et al., which suggested that only the macrocycle was structured; however, these studies were performed in an alternate solvent (DMSO-D6), and NMR characterization was limited to analysis of chemical shift differences as a function of temperature.16 In our AIP-I structure, the exocyclic tail projects back and away from the macrocycle, with the Try1 side chain positioned on the opposite face of the macrocycle (Figure 6A). The presence of a hydrophobic knob in AIP-I, along with the absence of a correctly positioned hydrophobic contact on the exocyclic tail, could explain its inhibitory activity against AgrC-III. The moderately good overlap of the AIP-I macrocycle with that of the potent AgrC-III antagonist, AIP-III D4A (except for the Leu6 and Ile7 side chains; Figure 7A), and the comparable AgrC-III antagonist, tAIP-III D2A (except for the Phe3 and Phe6 side chains; Figure S-8) offers some support for this explanation. The 10-fold reduced activity of AIP-I compared to AIP-III D4A could potentially be attributed to different contacts (and side chain functionality) on the tail and knob regions in each peptide. As highlighted above, it is interesting to note that the AIP-I analog with the same modification, AIP-I D5A, has previously been shown to be a 10-fold stronger AgrC-III inhibitor relative to AIP-I.14 These data, coupled with our structural and biological data for AIP-I, AIP-III, and AIP-III D4A, suggest that this Asp→Ala modification causes an alteration in both the native AIP-I and AIP-III macrocyclic knob structures that serves to enhance AgrC-III receptor binding, yet prevents activation. We also note that the aspartate side chains in these peptides will be deprotonated at physiological pH. However, replacing the Asp5 residue in AIP-I with asparagine yielded an analog (AIP-I D5N) with almost similar agonistic activity in AgrC-I as the native AIP-I, demonstrating that a negative charge at this position is nonessential for AgrC-I binding and activation.14 Further, this analog was also capable of activating AgrC-III (with an EC50 only ∼10-fold higher than AIP-III). Additional experiments are of course warranted, but we speculate that the negative charge at Asp4 in AIP-III is also unnecessary for AgrC-III binding and activation.
Figure 6.
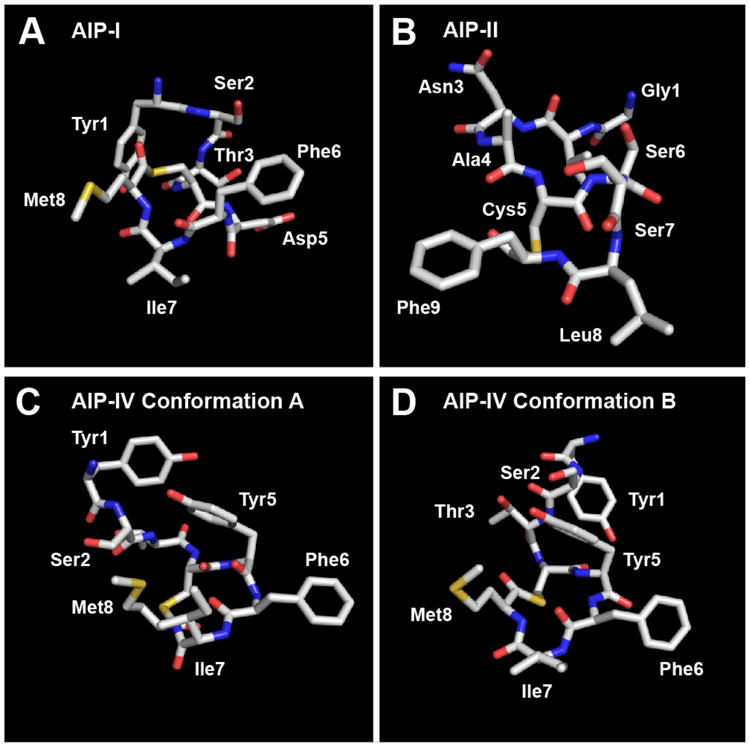
Heavy atom lowest energy structures of A) AIP-I; B) AIP-II; C) AIP-IV Conformation A; D) AIP-IV Conformation B.
Figure 7.
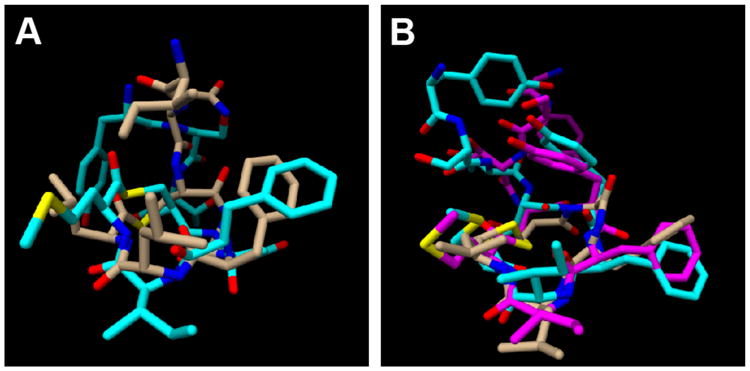
A) Overlay of AIP-I (cyan) and AIP-III D4A (tan) structures. B) Overlay of AIP-IV Conformation A (cyan), AIP-IV Conformation B (magenta) and tAIP-III D2A (tan) structures.
Structural analysis of AIP-II revealed that this peptide was relatively unstructured in solution compared to the other AIP analogs evaluated in this study (RMSD values were two-fold higher than those of the other peptides, Table S-12). This finding was perhaps not surprising, in view of the differences in its primary structure (and potentially its mode of receptor binding) as described above. The lowest energy structure for AIP-II is shown in Figure 6B; however, delineating any defined structural motif from the calculated heavy atom 20-structure ensemble for this AIP (shown in Figure S-7B) was basically impossible. Thus, we are unable to derive any mechanistic understanding into the mode by which AIP-II antagonizes AgrC-III from this structural data, other than stating that a rigidified conformation may not be essential.
Lastly, NMR structural analyses of AIP-IV also resulted in a 20-structure ensemble with relatively high RMSD values (comparable to AIP-II, Table S-12). However, further assessment of the ensemble uncovered two distinct populations, varying mainly in the orientation of the exocyclic tail, which satisfied the conformational restraints. Separation of these populations resulted in two families of structures: a 12-structure ensemble that we named “conformation A” and an 8-structure ensemble that we named “conformation B” (shown in Figures 6C, 6D, S-7C, and S-7D). The RMSD values of the two separated ensembles were very similar to those of the other AIP analogs, although these calculations were based on smaller number of structures (8 and 12 vs. 20 for the other peptides in this study; Table S-12). Consistent with our hypothesis, the two conformations of AIP-IV both possess an obvious triangular knob projecting from one side of the peptide and composed of the side chains of the three endocyclic hydrophobic residues (Figure 7A). While the orientation of the exocyclic tail was different between the two conformations, the Tyr1 side chain points away from the knob in both conformations (in analogy to AIP-I; see above). When overlaid with the structure of the potent AgrC-III inhibitor, tAIP-III D2A, the two conformations of AIP-IV had almost identical knob motifs to that of tAIP-III D2A (Figure 7B). This result may explain the comparable IC50 values of AIP-IV and tAIP-III D2A for AgrC-III inhibition.
We note that Kirchdoerfer et al. reported an X-ray crystal structure of AIP-IV bound to a monoclonal antibody (AP4-24H11 Fab) in 2011 at 2.5 Å resolution.27 This antibody can sequester AIP-IV and effectively “quench” QS in group-IV S. aureus.18 While the relevance of this structure to the actual AIP-IV:AgrC-IV receptor interaction is unknown, we were interested to compare this X-ray structure to our two calculated structures for AIP-IV. We found that the macrocycles in the X-ray structure of AIP-IV and our two calculated NMR structures (conformations A and B) show moderately good overlap when overlaid (see Figure S-9). The side chain of Try5 in the X-ray structure, however, is canted in the opposite direction relative to the NMR structures of AIP-IV. With regard to the exocyclic tail, this region was poorly ordered in the X-ray structure, and no clear overlap was observed with the tails in either calculated conformation of AIP-IV.
Conclusions
In summary, we have determined the 3-D solution-phase structures of a native AIP signal from S. aureus, AIP-III, and a set of AIP-III analogs with varied activities on the AgrC-III receptor using NMR spectroscopy. As the other native AIPs used by S. aureus (AIPs-I, -II, and -IV) are also known to inhibit AgrC-III, we simultaneously determined their 3-D solution-phase structures for direct comparison to the structures of our AIP-III analogs with strong inhibitory activity. These structural studies reveal several prominent molecular features that are shared amongst many of the peptides, which when coupled to the observed activities of these peptides in cell-based assays and the resulting SARs, support a mechanism by which these peptides modulate AgrC-III activity. The two key structural elements identified are (1) a tri-residue hydrophobic “knob” on one face of the peptide essential for both receptor activation and inhibition, and (2) a fourth hydrophobic anchor point on the exocyclic tail needed for receptor activation. In general, modifications that eliminated the knob motif significantly perturbed both the AIP-III and analog structures and strongly attenuated their agonistic or antagonistic activities. In turn, loss of the activation anchor but maintenance of the knob (in both truncated and full length peptides) yielded highly potent AgrC-III antagonists. These results represent the first structural data in support of a mechanism of AIP-mediated AgrC activation and inhibition in S. aureus, and serve to further refine several earlier hypotheses about AIP:AgrC interactions in general.
As introduced above, impetus for the development of non-native molecules capable of virulence inhibition in S. aureus is strong and growing. Several of the AIP-III analogs evaluated in this study are, to our knowledge, the most potent AgrC inhibitors to be reported. As such, we contend that the structures described herein provide 3-D scaffolds for the design of new chemical agents for the modulation of AgrC-III, and likely other AgrC receptors, with improved activities and potentially simplified, non-peptidic structures. Further structural experiments involving modified analogs of AIP-I, -II and -IV are required to obtain a more detailed understanding of their modes of action, as both cognate receptor activators and cross-receptor inhibitors. In the interim, an important next step will be to determine whether these solution-phase structures are maintained when the peptides are bound to AgrC-III; experiments toward this goal are currently underway and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the Office of Naval Research (N00014-07-1-0255), Kimberly-Clark Corporation, and Burroughs Welcome Fund (to H.E.B). The NMRFAM was supported by the NIH (P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS)). NMR facilities in the Department of Chemistry were supported in part by NIH (1 S10 RR13866-01). We thank Prof. Richard Novick and Prof. Alexander Horswill for providing S. aureus strains, and Dr. W. Milo Westler and Dr. Deborah Shalev for valuable discussions.
Footnotes
Supporting Information Available: Full bacteriological assay protocols, assay data, tables of resonance assignments and RMSD values, additional structural figures, NMR spectra, and pdb files. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Chambers HF, DeLeo FR. Nat Rev Microbiol. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein E, Smith DL, Laxminarayan R. Emerg Infect Dis. 2007;13:1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David MZ, Daum RS. Clin Microbiol Rev. 2010;23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasko DA, Sperandio V. Nat Rev Drug Disc. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 6.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nat Rev Microbiol. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novick RP, Geisinger E. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 8.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Chem Rev. 2011;111:117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.George EA, Muir TW. ChemBioChem. 2007;8:847–855. doi: 10.1002/cbic.200700023. [DOI] [PubMed] [Google Scholar]
- 10.Wright JS, Lyon GJ, George EA, Muir TW, Novick RP. Proc Natl Acad Sci U S A. 2004;101:16168–16173. doi: 10.1073/pnas.0404039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For a recent review, see:Gordon CP, Williams P, Chan WC. J Med Chem. 2013;56:1389–1404. doi: 10.1021/jm3014635.
- 12.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, Muir TW. Proc Natl Acad Sci U S A. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyon GJ, Mayville P, Muir TW, Novick RP. Proc Natl Acad Sci U S A. 2000;97:13330–13335. doi: 10.1073/pnas.97.24.13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyon GJ, Wright JS, Muir TW, Novick RP. Biochemistry. 2002;41:10095–10104. doi: 10.1021/bi026049u. [DOI] [PubMed] [Google Scholar]
- 15.McDowell P, Affas Z, Reynolds C, Holden MT, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CE, Bycroft BW, Chan WC, Williams P. Mol Microbiol. 2001;41:503–512. doi: 10.1046/j.1365-2958.2001.02539.x. [DOI] [PubMed] [Google Scholar]
- 16.Chan WC, Coyle BJ, Williams P. J Med Chem. 2004;47:4633–4641. doi: 10.1021/jm0400754. [DOI] [PubMed] [Google Scholar]
- 17.George EA, Novick RP, Muir TW. J Am Chem Soc. 2008;130:4914–4924. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- 18.Park J, Jagasia R, Kaufmann GF, Mathison JC, Ruiz DI, Moss JA, Meijler MM, Ulevitch RJ, Janda KD. Chem Biol. 2007;14:1119–1127. doi: 10.1016/j.chembiol.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji G, Beavis R, Novick RP. Science. 1997;276:2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- 20.Scott RJ, Lian LY, Muharram SH, Cockayne A, Wood SJ, Bycroft BW, Williams P, Chan WC. Bioorg Med Chem Lett. 2003;13:2449–2453. doi: 10.1016/s0960-894x(03)00497-9. [DOI] [PubMed] [Google Scholar]
- 21.Fowler SA, Stacy DM, Blackwell HE. Org Lett. 2008;10:2329–2332. doi: 10.1021/ol800908h. [DOI] [PubMed] [Google Scholar]
- 22.Jarraud S, Mougel C, Thioulouse J, Lina G, Meugnier H, Forey F, Nesme X, Etienne J, Vandenesch F. Infect Immun. 2002;70:631–641. doi: 10.1128/IAI.70.2.631-641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtfreter S, Grumann D, Schmudde M, Nguyen HTT, Eichler P, Strommenger B, Kopron K, Kolata J, Giedrys-Kalemba S, Steinmetz I, Witte W, Broker BM. J Clin Microbiol. 2007;45:2669–2680. doi: 10.1128/JCM.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Limbago B, Fosheim GE, Schoonover V, Crane CE, Nadle J, Petit S, Heltzel D, Ray SM, Harrison LH, Lynfield R, Dumyati G, Townes JM, Schaffner W, Mu Y, Fridkin SK. J Clin Microbiol. 2009;47:1344–1351. doi: 10.1128/JCM.02264-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. J Am Chem Soc. 2013;135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 26.NMR characterization has been limited to analysis of chemical shift differences in peptides. See ref. 14, 16 and 17
- 27.Kirchdoerfer RN, Garner AL, Flack CE, Mee JM, Horswill AR, Janda KD, Kaufmann GF, Wilson IA. J Biol Chem. 2011;286:17351–17358. doi: 10.1074/jbc.M111.231258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyon GJ, Wright JS, Christopoulos A, Novick RP, Muir TW. J Biol Chem. 2002;277:6247–6253. doi: 10.1074/jbc.M109989200. [DOI] [PubMed] [Google Scholar]
- 29.Ji G, Beavis RC, Novick RP. Proc Natl Acad Sci U S A. 1995;92:12055–12059. doi: 10.1073/pnas.92.26.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Bax A, Davis DG. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 32.Levitt MH, Freeman R, Frenkiel T. J Magn Reson. 1982;47:328–330. [Google Scholar]
- 33.Hwang TL, Shaka AJ. J Am Chem Soc. 1992;114:3157–3159. [Google Scholar]
- 34.Goddard TD, Kneller DG. v 3.114. University of California; San Francisco: 2007. [Google Scholar]
- 35.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York: 1986. [Google Scholar]
- 36.Schwieters CD, Clore GM. J Magn Reson. 2001;152:288–302. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- 37.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Nilges M, Clore GM, Gronenborn AM. FEBS Lett. 1988;229:317–324. doi: 10.1016/0014-5793(88)81148-7. [DOI] [PubMed] [Google Scholar]
- 39.Clore GM, Kuszewski J. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- 40.Schwieters CD, Clore GM. J Phys Chem B. 2008;112:6070–6073. doi: 10.1021/jp076244o. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, Behrmann I, Tsiaris W, Sasaki A, Schneider-Mergener J, Yoshimura A, Neel BG, Heinrich PC, Schaper F. J Biol Chem. 2003;278:661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Two differences between the NMR conditions and those of our biological assays were the pH 3.65 vs. 7.35 in biological media BHI)) and ionic strength <1 mM vs. ∼140 mM in BHI) of the aqueous solutions utilized. Direct comparisons are challenging, however, as we used a mixed aqueous/organic solvent system for our NMR experiments. This solvent system was required to dissolve the peptides at the concentrations required for the NMR studies
- 44.Geisinger E, Muir TW, Novick RP. Proc Natl Acad Sci U S A. 2009;106:1216–1221. doi: 10.1073/pnas.0807760106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.