

Abstract
Introduction
A recent genome-wide association study in European systemic sclerosis (SSc) patients identified three loci (PSORS1C1, TNIP1 and RHOB) as novel genetic risk factors for the disease. The aim of this study was to replicate the previously mentioned findings in a large multicentre independent SSc cohort of Caucasian ancestry.
Methods
4389 SSc patients and 7611 healthy controls from different European countries and the USA were included in the study. Six single nucleotide polymorphisms (SNP): rs342070, rs13021401 (RHOB), rs2233287, rs4958881, rs3792783 (TNIP1) and rs3130573 (PSORS1C1) were analysed. Overall significance was calculated by pooled analysis of all the cohorts. Haplotype analyses and conditional logistic regression analyses were carried out to explore further the genetic structure of the tested loci.
Results
Pooled analyses of all the analysed SNPs in TNIP1 revealed significant association with the whole disease (rs2233287 pMH=1.94×10−4, OR 1.19; rs4958881 pMH=3.26×10−5, OR 1.19; rs3792783 pMH=2.16×10−4, OR 1.19). These associations were maintained in all the subgroups considered. PSORS1C1 comparison showed association with the complete set of patients and all the subsets except for the anti-centromere-positive patients. However, the association was dependent on different HLA class II alleles. The variants in the RHOB gene were not associated with SSc or any of its subsets.
Conclusions
These data confirmed the influence of TNIP1 on an increased susceptibility to SSc and reinforced this locus as a common autoimmunity risk factor.
INTRODUCTION
Systemic sclerosis or scleroderma (SSc) is a complex autoimmune disorder that affects the connective tissue causing fibrosis in the skin and different internal organs.1 The contribution of different genetic factors to the development and prognosis of the disease is now widely accepted.2 Over the past few years, genome-wide association studies (GWAS) have been a useful tool in the genetic dissection of autoimmune pathologies and other complex diseases.3 Radstake et al4 performed the first SSc GWAS in Caucasian populations, which represented the first large-scale GWAS in SSc. This work reinforced the association within the HLA region, especially with the HLA-DQB1 gene, which was also reported in a comprehensive multiethnic SSc HLA study.5 It also confirmed the associations found in STAT4 and IRF5 and identified CD247 as a new SSc risk locus. It is worth mentioning that the role of CD247 in SSc has recently been independently replicated.6 This GWAS has led to three follow-up studies, which have described several novel SSc susceptibility factors, ie, IRF8, GRB10, SOX5, NOTCH4, IL12RB2, CSK, PSD3 and NFKB1.7-9 Interestingly, SOX5 and NOTCH4 are directly related to the fibrotic process, which is a main hallmark of SSc.
A GWAS has recently been performed in a French Caucasian SSc discovery cohort.10 In this GWAS, 17 single-nucleotide polymorphisms (SNP) showing tier two associations were selected for follow-up in independent cohorts. Three of the selected SNP were located within the HLA region corresponding to the HLA-DQB1 and PSORS1C1 genes; and the remaining SNP were located in six independent non-HLA loci. After the replication step, the associations of HLA-DQB1, CD247, STAT4 and IRF5 were confirmed, and six SNP located in three loci (TNIP1, RHOB, PSORS1C1) were proposed as novel SSc risk factors.
It has been observed that associations identified from a single GWAS, even passing the established statistical significance thresholds, tend to have inflated effect sizes.11 This effect size is called the winner’s curse, and it also affects the predictive ability of the discovered associations and the estimate of the risk variance explained by the associations.11 Replication in independent comparable populations is thus essential for firmly establishing a genotype–phenotype association.11,12 Therefore, we aimed to perform a large-scale replication study of the novel SSc genetic risk factors identified by GWAS strategy in an independent white European and US SSc population.
PATIENTS AND METHODS
Subjects
4389 SSc patients and 7611 controls of Caucasian ancestry (Spain, The Netherlands, USA, Italy, Sweden, UK and Norway) were included in this study. Patients were classified as having limited or diffuse SSc, as defined by LeRoy et al.13 The following clinical data were collected for ascertainment of the clinical phenotype of the patients with SSc: age, gender, disease duration and presence of SSc-associated autoantibodies, anti-topoisomerase (ATA) and anti-centromere (ACA). Supplementary table S1 (available online only) shows the cohort-specific SSc patient data. The control population consisted of unrelated healthy individuals recruited in the same geographical regions as SSc patients and matched by age, sex and ethnicity with the SSc patient groups. Local ethics committees from all the participating centres approved the study. Both patients and controls were included in the study after written informed consent.
In the meta-analysis with previously published data by Allanore et al,10 which includes 2246 SSc patients and 5702 healthy controls from France, Italy, Germany and Eastern Europe, the total cohort size reached 6635 patients and 13 313 controls (except for rs13021401 and rs3792783, which were not available for The Netherlands and US GWAS cohorts, respectively).
Genotyping
Genotype data of six SNP (rs342070, rs13021401 (RHOB), rs2233287, rs4958881, rs3792783 (TNIP1) and rs3130573 (PSORS1C1)) was obtained from both available GWAS genotyping platforms and SNP genotyping assays. When possible, genotypes from the Spanish, Dutch and US cohorts from Radstake et al4 were used (Spain I, The Netherlands I and US I cohorts). In addition, additional Spanish SSc patients and controls were genotyped using the Illumina HumanCytoSNP-12 DNA Analysis BeadChip and Illumina Human1M-Duo DNA Analysis BeadChip ((Illumina Inc., San Diego, CA, USA), respectively, this information was thus included in the Spain I set when available. The remaining European cohorts (Spain II, The Netherlands II, Italy, Sweden, UK and Norway) were analysed using TaqMan SNP genotyping assays in a 7900HT Real-Time PCR System from Applied Biosystems following the manufacturer’s suggestions (Applied Biosystems, Foster City, California, USA). The differences in the number of samples included in the analyses of each polymorphism correspond to the availability of the genotype data in each platform (see supplementary table S2, available online only). Stringent quality control filters and principal component analysis were applied to the GWAS and the HLA imputed data as described in Radstake et al.4 The genotyping call-rate for the individuals genotyped using TaqMan assays reached: rs342070 93.77%, rs13021401 94.98%, rs2233287 96.08%, rs4958881 93.75%, rs3792783 93.29% and rs3130573 95.19%.
Statistical analysis
PLINK (V.1.07) software (http://pngu.mgh.harvard.edu/purcell/plink/) was used for individual population association tests (significance was calculated by 2×2 contingency tables and Fisher’s exact test or χ2 when necessary, and in the case of the haplotypes each haplotype was tested against all others), logistic regression and conditional logistic regression analyses. The different cohorts were considered covariables in the logistic regression analyses. OR and their 95% CI were reported. TNIP1 haplotypes were constructed using PLINK (V.1.07) and HaploView 4.2 (http://www.broadinstitute.org/haploview/haploview) only with those individuals successfully genotyped for the three included variants (2432 SSc patients and 3496 healthy controls). The Breslow–Day test was performed as implemented in PLINK and StatsDirect to assess the homogeneity of the association among populations. Pooled analyses and meta-analyses were carried out using a Mantel–Haenszel test under a fixed effects by PLINK (V.1.07), METAL14 (http://www.sph.umich.edu/csg/abecasis/metal/) and StatsDirect (V.2.6.6 StatsDirect Ltd) in the case of haplotypes. Significant heterogeneity among populations was found in the meta-analysis of RHOB locus polymorphisms; consequently, in this case a random effects model was applied using StatsDirect. Genotypic frequency distributions for the meta-analysis were kindly provided by Allanore et al10 for the meta-analysis by personal communication. All cohorts were in Hardy–Weinberg equilibrium at a significance level of 0.01 for all the included SNP. Power was calculated using the software Power Calculator for Genetic Studies 2006 and assuming an additive model at the 5% significance level and previously reported OR (rs342070 minor allele (A) frequency (MAF) 0.226, OR 1.20; rs13021401 MAF 0.225, OR 1.21; rs2233287 MAF 0.096, OR 1.31; rs4958881 MAF 0.115, OR 1.29; rs3792783 MAF 0.152, OR 1.29; rs3130573 MAF 0.321, OR 1.25).15
RESULTS
Non-HLA loci analysis
In this study we analysed five SNP located in two non-HLA loci, TNIP1 and RHOB. Regarding the TNIP1 locus, we replicated the previously described associations, with rs4958881 showing the most significant relationship, pMH=3.26×10−5, OR 1.19, 95% CI 1.09 to 1.29 (table 1). We also observed that the associations were consistent through the different clinical and serological subsets. The Breslow–Day test showed no evidence of interpopulation heterogeneity either in the whole disease analyses or in the stratified groups (supplementary table S3, available online only, shows individual cohort analyses). Statistical power was over 99% for the three SNP. Moreover, all the TNIP1 genetic variants showed significant association at GWAS level in the meta-analysis with the initial report (rs2233287 pMH=1.7×10−9, OR 1.23 95%, CI 1.15 to 1.32; rs4958881 pMH=2.88×10−11, OR 1.24, 95% CI 1.16 to 1.32; rs3792783 pMH=9.11×10−16, OR 1.31, 95% CI 1.23 to 1.40; figure 1).
Table 1.
Pooled analysis of the novel SSc non-HLA susceptibility loci
| CHR | BP | SNP | Locus | 1/2 | Subgroup (N) | Genotype, N (%)
|
2/2 | MAF N (%) | Allele test
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/1 | 1/2 | pMH* | OR (95% CI) | pBD | ||||||||
| 2 | 20548952 | rs342070 | RHOB | C/T | Controls (n=7193) | 448 (6.23) | 2713 (37.72) | 4032 (56.05) | 3609 (25.09) | |||
| SSc (n=4249) | 292 (6.87) | 1515 (35.66) | 2442 (57.47) | 2099 (24.70) | 0.76 | 1.01 (0.95 to 1.08) | 0.08 | |||||
| lcSSc (n=2649) | 184 (6.95) | 953 (35.98) | 1512 (57.08) | 1321 (24.93) | 0.75 | 1.01 (0.94 to 1.09) | 0.24 | |||||
| dcSSc (n=1227) | 80 (6.52) | 444 (36.19) | 703 (57.29) | 604 (24.61) | 0.95 | 1.00 (0.90 to 1.10) | 0.21 | |||||
| ACA+ (n=1492) | 92 (6.17) | 529 (35.46) | 871 (58.38) | 713 (23.89) | 0.52 | 0.97 (0.88 to 1.07) | 0.52 | |||||
| ATA+ (n=864) | 71 (8.22) | 303 (35.07) | 490 (56.71) | 445 (25.75) | 0.22 | 1.08 (0.96 to 1.21) | 0.04 | |||||
| 2 | 20552000 | rs13021401 | RHOB | T/C | Controls (n=6557) | 405 (6.18) | 2416 (36.85) | 3736 (56.98) | 3226 (24.60) | |||
| SSc (n=4259) | 280 (6.57) | 1524 (35.78) | 2455 (57.64) | 2084 (24.47) | 0.61 | 1.02 (0.95 to 1.09) | 0.06 | |||||
| lcSSc (n=2644) | 174 (6.58) | 966 (36.54) | 1504 (56.88) | 1314 (24.85) | 0.49 | 1.03 (0.95 to 1.11) | 0.11 | |||||
| dcSSc (n=1239) | 79 (6.38) | 444 (35.84) | 716 (57.79) | 602 (24.29) | 0.96 | 1.00 (0.90 to 1.10) | 0.38 | |||||
| ACA+ (n=1487) | 91 (6.12) | 530 (35.64) | 866 (58.24) | 712 (23.94) | 0.91 | 0.99 (0.90 to 1.09) | 0.39 | |||||
| ATA+ (n=872) | 65 (7.45) | 316 (36.24) | 491 (56.31) | 446 (25.57) | 0.17 | 1.09 (0.97 to 1.22) | 0.11 | |||||
| 5 | 150420290 | rs2233287 | TNIP1 | T/C | Controls (n=7164) | 93 (1.30) | 1226 (17.11) | 5845 (81.59) | 1412 (9.85) | |||
| SSc (n=4152) | 52 (1.25) | 859 (20.69) | 3241 (78.06) | 963 (11.60) | 1.94E-04 | 1.19 (1.08 to 1.30) | 0.72 | |||||
| lcSSc (n=2586) | 33 (1.28) | 528 (20.42) | 2025 (78.31) | 594 (11.48) | 1.69E-03 | 1.18 (1.07 to 1.31) | 0.70 | |||||
| dcSSc (n=1194) | 15 (1.26) | 253 (21.19) | 926 (77.55) | 283 (11.85) | 2.81E-03 | 1.23 (1.07 to 1.41) | 0.76 | |||||
| ACA+ (n=1442) | 19 (1.32) | 309 (21.43) | 1114 (77.25) | 347 (12.03) | 2.27E-03 | 1.22 (1.07 to 1.39) | 0.86 | |||||
| ATA+ (n=849) | 14 (1.65) | 186 (21.91) | 649 (76.44) | 214 (12.60) | 4.94E-03 | 1.26 (1.07 to 1.47) | 0.73 | |||||
| 5 | 150430429 | rs4958881 | TNIP1 | C/T | Controls (n=7182) | 131 (1.82) | 1512 (21.05) | 5539 (77.12) | 1774 (12.35) | |||
| SSc (n=4226) | 98 (2.32) | 1035 (24.49) | 3093 (73.19) | 1231 (14.56) | 3.26E-05 | 1.19 (1.09 to 1.29) | 0.33 | |||||
| lcSSc (n=2637) | 57 (2.16) | 637 (24.16) | 1943 (73.68) | 751 (14.24) | 1.23E-03 | 1.17 (1.06 to 1.28) | 0.58 | |||||
| dcSSc (n=1213) | 31 (2.56) | 302 (24.90) | 880 (72.55) | 364 (15.00) | 5.57E-04 | 1.24 (1.10 to 1.41) | 0.29 | |||||
| ACA+ (n=1490) | 31 (2.08) | 378 (25.37) | 1081 (72.55) | 440 (14.77) | 3.29E-03 | 1.19 (1.06 to 1.34) | 0.14 | |||||
| ATA+ (n=868) | 29 (3.34) | 232 (26.73) | 607 (69.93) | 290 (16.71) | 1.31E-05 | 1.36 (1.19 to 1.57) | 0.57 | |||||
| 5 | 150435925 | rs3792783 | TNIP1 | C/T | Controls (n=3704) | 113 (3.05) | 995 (26.86) | 2596 (70.09) | 1221 (16.48) | |||
| SSc (n=2704) | 108 (3.99) | 829 (30.66) | 1767 (65.35) | 1045 (19.32) | 2.16E-04 | 1.19 (1.09 to 1.31) | 0.70 | |||||
| lcSSc (n=1708) | 65 (3.81) | 518 (30.33) | 1125 (65.87) | 648 (18.97) | 4.44E-03 | 1.17 (1.05 to 1.30) | 0.56 | |||||
| dcSSc (n=715) | 30 (4.20) | 222 (31.05) | 463 (64.76) | 282 (19.72) | 5.41E-03 | 1.23 (1.06 to 1.42) | 0.59 | |||||
| ACA+ (n=1041) | 40 (3.84) | 326 (31.32) | 675 (64.84) | 406 (19.50) | 6.09E-03 | 1.20 (1.05 to 1.36) | 0.18 | |||||
| ATA+ (n=623) | 24 (3.85) | 208 (33.39) | 391 (62.76) | 256 (20.55) | 3.36E-03 | 1.26 (1.08 to 1.47) | 0.70 | |||||
Controls are used as reference for all comparisons.
All p values have been calculated for the allelic model.
ACA+, anti-centromere autoantibody-positive patients; ATA+, anti-topoisomerase autoantibody-positive patients; BP, base pair; CHR, chromosome; CTRL, healthy controls; dcSSc, diffuse cutaneous systemic sclerosis; lcSSc, limited cutaneous systemic sclerosis; MAF, minor allele (A) frequency; pMH, Mantel–Haenszel test under fixed effect; pBD, homogeneity Breslow–Day test; 1/2, minor allele/major allele; SNP, single nucleotide polymorphism.
Figure 1.
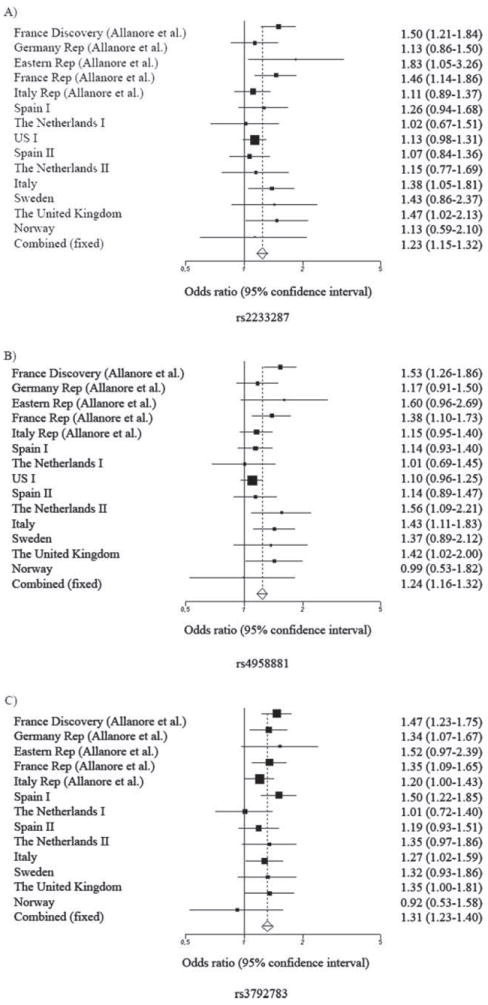
(A) Forest plot for the meta-analysis of the rs2233287 TNIP1 genetic variant. (B) Forest plot for the meta-analysis of the rs4958881 TNIP1 genetic variant. (C) Forest plot for the meta-analysis of the rs3792783 TNIP1 genetic variant.
As previously described, the three SNP examined belong to the same haplotype block.10 As reported by Allanore et al,10 the polymorphisms studied in the TNIP1 region showed moderate to high linkage disequilibrium (see supplementary figures S1 and S2, available online only). Therefore, haplotype analysis was performed. The Breslow–Day test show homogeneity in the association of the haplotypes among populations. Haplotype block analysis revealed the association of two haplotypes with the disease. Haplotypes CTT and TCC (SNP order rs2233287–rs4958881–rs3792783) represent the combinations of the major and minor alleles of each SNP, respectively, and subsequently show a protective or a susceptibility role that is concordant with the individual SNP associations, ie, major alleles are protective while minor alleles are risk variants (see supplementary table S4, available online only). However, haplotype block analysis did not show more significant p values than individual SNP analyses, and no additive or multiplicative effect of the SNP was observed. With the aim of clarifying possible underlying dependence among the SNP, we performed conditional logistic regression analysis. Nevertheless, due to the linkage disequilibrium between the analysed SNP, this approach did not enable us to identify an independent association signal (see supplementary table S5, available online only).
As shown in table 1, none of the tested polymorphisms in RHOB showed significant associations with SSc, or any of the examined subgroups. Only weak association signals could be detected in the Italian cohort. The power for the analyses of the rs342070 and rs13021401 RHOB genetic variants in the overall cohort was of 99% in both cases. Meta-analyses with the previous report showed significant OR heterogeneity in the Breslow–Day tests and no significant association under a random effects model for both polymorphisms (rs342070 Prandom=0.19; rs13021401 Prandom=0.13).
PSORS1C1 analysis
The study of the PSORS1C1 reported variant, rs3130573, showed a suggestive but heterogeneous association of this polymorphism with increased SSc susceptibility (table 2). Moreover, the association was maintained in all the subgroups (including the ACA-negative and ATA-negative subsets) except for the ACA-positive patients. Considering that the association of the HLA region with SSc is influenced primary by the autoantibody profile of the patients,5,7 we aimed to test for an uncovered influence of the HLA genes. We thus carried out a step-wise logistic regression conditional analysis of the analysed PSORS1C1 variant with all the independent signals from the most significant to the lowest observed p values in the HLA region. The considered conditions included SNPs, imputed aminoacidic positions and imputed classic HLA-alleles (as described in Raychaudhuri et al)16 in the analysed GWAS cohorts (Spain I, The Netherlands I and USA I) (unpublished data). Standard logistic regression analyses in the GWAS cohorts showed evidence of association only in the whole disease versus controls and in the ACA-negative patients versus controls comparisons (Plog=0.034, OR 1.09; Plog=0.01, OR 1.12, respectively). However, the previously mentioned association with the whole set of SSc patients lost its significance when it was conditioned to the HLA-DPB1*1301 allele (p value conditioning on DPB1*13 : 01=0.06; OR conditioning on DPB1*13 : 01=1.08), HLA-DRB1*11 : 04 (p value conditioning on DRB1*11 : 04=0.08; OR conditioning on DRB1*11 : 04=1.07) and the HLA-DQA1*05 : 01 (p value conditioning on DQA1*05 : 01=0.36; OR conditioning on DQA1*05 : 01=1.04) alleles. Moreover, the association with the ACA-negative subset of patients was also shown to be dependent on the HLA-DQA1*05 : 01 allele (p value conditioning on DQA1*05 : 01=0.27; OR add to DQA1*05 : 01=1.05). Therefore, our data suggest that the association of PSORS1C1 with SSc is not independent from the HLA region.
Table 2.
Pooled-analysis of the rs3130573 PSORS1C1 HLA-region genetic variant
| CHR | BP | SNP | 1/2 | Subgroup (N) | Genotype, N (%)
|
MAF N (%) | Allele test
|
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/1 | 1/2 | 2/2 | pMH* | OR (95% CI) | pBD | ||||||
| 6 | 31214247 | rs3130573 | G/A | Controls (n=7139) | 892 (12.49) | 3220 (45.10) | 3027 (42.40) | 5004 (35.05) | |||
| SSc (n=4130) | 574 (13.90) | 1953 (47.29) | 1603 (38.81) | 3101 (37.54) | 1.17E-05 | 1.14 (1.07 to 1.21) | 0.02 | ||||
| lcSSc (n=2575) | 350 (13.59) | 1221 (47.42) | 1004 (38.99) | 1921 (37.30) | 1.16E-03 | 1.12 (1.05 to 1.20) | 0.07 | ||||
| dcSSc (n=1187) | 177 (14.91) | 561 (47.26) | 449 (37.83) | 915 (38.54) | 3.09E-04 | 1.18 (1.08 to 1.29) | 0.07 | ||||
| ACA+ (n=1446) | 181 (12.52) | 666 (46.06) | 599 (41.42) | 1028 (35.55) | 0.28 | 1.05 (0.96 to 1.14) | 0.12 | ||||
| ACA− (n=2511) | 356 (15.07) | 1143 (48.37) | 864 (36.56) | 1855 (39.25) | 1.01E-07 | 1.21 (1.13 to 1.29) | 0.05 | ||||
| ATA+ (n=845) | 123 (14.56) | 411 (48.64) | 311 (36.80) | 657 (38.88) | 6.48E-03 | 1.16 (1.04 to 1.29) | 0.51 | ||||
| ATA− (n=3147) | 413 (13.92) | 1395 (47.00) | 1160 (39.08) | 2221 (37.42) | 1.03E-04 | 1.13 (1.07 to 1.21) | 0.03 | ||||
Controls are used as reference for all comparisons.
All p values have been calculated for the allelic model.
ACA±, anti-centromere autoantibody-positive/negative patients; ATA±, anti-topoisomerase autoantibody-positive/negative patients; BP, base pair; CHR, chromosome; CTRL, healthy controls; dcSSc, diffuse cutaneous systemic sclerosis; lcSSc, limited cutaneous systemic sclerosis; MAF, minor allele (A) frequency; pMH, Mantel–Haenszel test under fixed effect; pBD, homogeneity Breslow–Day test; 1/2, minor allele/major allele; SNP, single nucleotide polymorphism.
DISCUSSION
In this study we conducted a large multicentre replication of the novel SSc risk variants identified by Allanore et al,10 and we confirmed the association of the TNIP1 locus with SSc. However, the associations observed in the RHOB gene and the independence of PSORS1C1 from the HLA region were not supported by our data.
Due to the lack of association observed in the RHOB locus, we suggest that the initial association reported in this gene might have been a false positive finding. It is worth mentioning that RHOB has never been robustly associated with an autoimmune disease, and the previously reported association with SSc in this gene did not reach the GWAS significance level even after replication.10
PSORS1C1 was proposed as an HLA-independent SSc risk factor.10 The authors performed a dependence analysis controlling for the described association in the HLA-DQB1 gene and reported the independence of both loci. Nevertheless, HLA-DQB1 has been specifically related with the ACA-positive subset of patients,5,7 and both in our data and in the previous study no association of PSORS1C1 with ACA positivity has been shown.10 Therefore, a deeper analysis of this locus was needed, and we performed for the first time conditional logistic regression including all the independent signals in the HLA region. In our initial approach we found a signal in the PSORS1C1 gene that was comparable to the one described in the previous work;10 however, a comprehensive analysis showed that the PSORS1C1 association is dependent from the HLA-DPB1*13 : 01, HLA-DQA1*05 : 01 and HLA-DRB1*11 : 04 alleles (especially the HLA-DQA1*0501). These HLA loci have previously been described as ATA positivity risk factors,5,7 which is consistent with the lack of association in the ACA-positive subgroup. Altogether, our data do not confirm PSORS1C1 as an independent player in the SSc genetic susceptibility network.
Regarding the association of TNFAIP3 interacting protein 1 (TNIP1), our data clearly support TNIP1 as a SSc risk factor. Our replication study confirmed that the association of the three SNPs tested is maintained in all the subsets, indicating that this association peak corresponds to the whole disease. Therefore, TNIP1 might be implicated in the development of the disease but may not act as a disease modifier. Remarkably, TNIP1 is involved in HIV replication, acts as a negative regulator of the nuclear factor κB pathway (a key regulator of the immune response, which has also recently been associated with SSc),9 and also represses agonist-bound retinoic acid receptors and peroxisome proliferator-activated receptors.17-19 Furthermore, recent studies focused on the control of TNIP1 transcription have reported a complex mechanism behind TNIP1 expression that combines constitutive transcription factors and inducible factors (nuclear factor κB and peroxisome proliferator-activated receptors).20 Interestingly, Allanore et al10 showed that the transcription and expression of TNIP1 is decreased both in the skin of SSc patients and SSc cultured fibroblasts, thus the anti-inflammatory effect of this molecule may be reduced in SSc patients, providing evidence for a relevant role of TNIP1 in the disease. TNIP1 is also a well-established risk factor for different autoimmune diseases, such as psoriasis, psoriatic arthritis and systemic lupus erythematosus. 21-23 Furthermore, TNIP1 association with psoriasis has also been reported in Asian populations,24 suggesting that the role of TNIP1 in autoimmune diseases is consistent through different ethnicities. Therefore, this locus can be considered a common autoimmune disease risk factor that can be used as a new therapeutic target.
To conclude, our replication study has reinforced the influence of TNIP1 in an increased susceptibility to SSc and its role as a new player in the autoimmunity genetic background. Future research will identify the causal variant for the TNIP1 association and its implication in SSc pathophysiology.
Supplementary Material
Acknowledgments
The authors would like to thank Sofia Vargas, Sonia García and Gema Robledo for excellent technical assistance and all the patients and control donors for their essential collaboration. They thank Banco Nacional de ADN (University of Salamanca, Spain) and the Norwegian Bone Marrow Donor Registry who supplied part of the control DNA samples. They are also grateful to EUSTAR (the EULAR Scleroderma Trials and Research group) for the facilitation of this project.
Funding This work was supported by the following grants: JM was funded by GEN-FER from the Spanish Society of Rheumatology, SAF2009–11110 from the Spanish Ministry of Science, CTS-4977 from Junta de Andalucía, Spain, in part by Redes Temáticas de Investigación Cooperativa Sanitaria Programme, RD08/0075 (RIER) from Instituto de Salud Carlos III (ISCIII), Spain and by Fondo Europeo de Desarrollo Regional (FEDER). TRDJR was funded by the VIDI laureate from the Dutch Association of Research (NWO) and Dutch Arthritis Foundation (National Reumafonds). JM and TRDJR were sponsored by the Orphan Disease Programme grant from the European League Against Rheumatism (EULAR). BPCK is supported by the Dutch Diabetes Research Foundation (grant 2008.40.001) and the Dutch Arthritis Foundation (Reumafonds, grant NR 09-1-408). TW was granted by DFG WI 1031/6.1. This study was also funded by PI-0590-2010, Consejería de Salud, Junta de Andalucía, Spain. The USA studies were supported by NIH/NIAMS Scleroderma Registry and DNA Repository (N01-AR-0-2251), NIH/NIAMS-RO1-AR055258 and NIH/NIAMS Center of Research Translation in Scleroderma (1P50AR054144), and the Department of Defense Congressionally Directed Medical Research Programmes (W81XWH-07-01-0111).
Footnotes
Contributors LBC and JEM contributed to the analysis and interpretation of data and in drafting the article. JB, CPS, LB, OG, MCV, NOC, GE, PC, PGdlP, NO, JARI, MJC, MAGG, LSC, IC, AJS, AEV, AMHV, RH, AN, CL, RS, JMvL, PS, AH, JW, CF, CD, FKT, FCA and SA participated in analysis and interpretation of data and critically revised the manuscript draft. BPK, MDM, TRDJR and JM were involved in the conception and design of the study and critically revised the submitted version of the manuscript. All authors approved the final version to be published.
Additional supplementary data are published online only. To view these files please visit the journal online (http://dx.doi.org/10.1136/annrheumdis-2012-201888.bmj.com).
Competing interests None.
Patient consent Obtained.
Ethics approval Local ethics committees from all the participating centres approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 2.Martin JE, Bossini-Castillo L, Martin J. Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012;131:1023–37. doi: 10.1007/s00439-011-1137-z. [DOI] [PubMed] [Google Scholar]
- 3.Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radstake TR, Gorlova O, Rueda B, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42:426–9. doi: 10.1038/ng.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnett FC, Gourh P, Shete S, et al. Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann Rheum Dis. 2010;69:822–7. doi: 10.1136/ard.2009.111906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieude P, Boileau C, Guedj M, et al. Independent replication establishes the CD247 gene as a genetic systemic sclerosis susceptibility factor. Ann Rheum Dis. 2011;70:1695–6. doi: 10.1136/ard.2010.147009. [DOI] [PubMed] [Google Scholar]
- 7.Gorlova O, Martin JE, Rueda B, et al. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet. 2011;7:e1002178. doi: 10.1371/journal.pgen.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossini-Castillo L, Martin JE, Broen J, et al. A GWAS follow-up study reveals the association of the IL12RB2 gene with systemic sclerosis in Caucasian populations. Hum Mol Genet. 2012;21:926–33. doi: 10.1093/hmg/ddr522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin JE, Broen JC, Carmona FD, et al. Identification of CSK as a systemic sclerosis genetic risk factor through genome wide association study follow-up. Hum Mol Genet. 2012;21:2825–35. doi: 10.1093/hmg/dds099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allanore Y, Saad M, Dieude P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7:e1002091. doi: 10.1371/journal.pgen.1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–29. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chanock SJ, Manolio T, Boehnke M, et al. Replicating genotype–phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 13.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 14.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skol AD, Scott LJ, Abecasis GR, et al. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–13. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 16.Raychaudhuri S, Sandor C, Stahl EA, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–6. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta K, Ott D, Hope TJ, et al. A human nuclear shuttling protein that interacts with human immunodeficiency virus type 1 matrix is packaged into virions. J Virol. 2000;74:11811–24. doi: 10.1128/jvi.74.24.11811-11824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mauro C, Pacifico F, Lavorgna A, et al. ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–8. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 19.Gurevich I, Aneskievich BJ. Liganded RARalpha and RARgamma interact with but are repressed by TNIP1. Biochem Biophys Res Commun. 2009;389:409–14. doi: 10.1016/j.bbrc.2009.08.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurevich I, Zhang C, Encarnacao PC, et al. PPARgamma and NF-kappaB regulate the gene promoter activity of their shared repressor, TNIP1. Biochim Biophys Acta. 2012;1819:1–15. doi: 10.1016/j.bbagrm.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowes J, Orozco G, Flynn E, et al. Confirmation of TNIP1 and IL23A as susceptibility loci for psoriatic arthritis. Ann Rheum Dis. 2011;70:1641–4. doi: 10.1136/ard.2011.150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun LD, Cheng H, Wang ZX, et al. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat Genet. 2010;42:1005–9. doi: 10.1038/ng.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.