

Abstract
This report describes the use of Bayesian methods to analyze polyprotein coding region sequences (n = 217) obtained from GenBank to define the genome-wide phylogeny of foot and mouth disease virus (FMDV). The results strongly supported the monophyly of five FMDV serotypes, O, A, Asia 1, C, and SAT 3, while sequences for the two remaining FMDV serotypes, SAT 1 and SAT 2 did not separate into entirely distinct clades. The phylogenomic tree revealed three sister-group relationships, serotype O + Asia 1, A + C, and SAT 1 + 3 + 2, with a new branching pattern: {[(O, Asia 1), (A, C)], (SAT 1, 2, 3)}. Within each serotype, there was no apparent periodic, geographic, or host species influence on the evolution of global FMDVs. Analysis of the polyprotein coding region of these sequences provided evidence for the influence of purifying selection on the evolution of FMDV. Using a Bayesian coalescent approach, the evolutionary rate of FMDV isolates that circulated during the years 1932-2007 was estimated to be 1.46 × 10-3 substitutions/site/year, and the most recent common ancestor of the virus existed approximately 481 years ago. Bayesian skyline plot revealed a population expansion in the early 20th century that was followed by a rapid decline in population size from the late 20th century to the present day. These findings provide new insights into the mechanisms that impact on the evolution of this important livestock pathogen.
Keywords: foot-and-mouth disease virus, molecular evolution, phylogenomics
INTRODUCTION
Foot-and-mouth disease (FMD) caused by foot-and-mouth disease virus (FMDV) is an acute, systemic disease affecting cloven-hoofed animals, resulting in significant economic losses if not controlled. FMDV is a member of the genus Aphthovirus of the family Picornaviridae and is categorized into seven serological types (A, O, C, Asia 1, and SAT 1-3) according to the antigenic properties of the capsid proteins (Pereira, 1977). The virus has a single-stranded positive-sense RNA genome of about 8.5 kb that contains a large single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (5′ UTR and 3′ UTR, respectively). The ORF of approximately 7,000 nucleotides in full length, is translated into a single polyprotein, which is subsequently cleaved into a leader peptide (L), structural proteins P1 (1A, 1B, 1C, and 1D), and non-structural proteins P2 (2A, 2B, and 2C), P3 (3A, 3B, 3C, and 3D) (Domingo et al., 2002). Within the host cells, the leader peptide regulates host interferon responses, while the structural proteins form the viral capsid and the non-structural proteins control viral life cycle.
Many comparative studies have been performed on the basis of viral individual genes (e.g., Mohapatra et al., 2009; Schumann et al., 2008; Tully and Fares, 2008). However, genomelevel analysis of global FMDVs including all seven FMDV serotypes has not been well described. Only three studies have focused on aspects of complete genome analysis of FMDV sequences (Carrillo et al., 2005; Cooke and Westover, 2008; Lewis-Rogers et al., 2008). Carrillo et al. (2005) pioneered work in this field by determining 103 complete genomic sequences from FMDV isolates representing all seven serotypes, and then comparing them with the sequences of 30 published FMDVs using neighbor-joining, split decomposition, and puzzling methods. In their analyses, star-like topologies for sequences of 1B, 1C, 1D, or entire structural protein coding regions were presented indicating phylogenetic incongruities between the diverse FMDV serotypes. Cooke and Westover (2008) performed a phylogenomic analysis on the basis of amino acid sequences, excluding regions corresponding to antigenic regions, for 149 complete FMDV genomes. They used neighbor-joining, quartet, and minimum evolution methods. Each of five serotypes (O, Asia 1, C, A, and SAT 1+2+3) appeared as independent clades and serotype C and Asia 1 had a sister group relationship to each other. Furthermore, Lewis-Rogers et al. (2008) also analyzed 159 sequences of coding genome excluding 3B gene using Bayesian inference and maximum likelihood, and suggested that each of five serotypes (O, Asia 1, C, A, and SAT 3) were monophyletic and serotypes O and Asia 1 were most closely related to each other. However, the lack of bootstrap values in these studies did not allow the robustness of the phylogenetic relationships to be interpreted.
In relation to time scale and population dynamics of FMDVs, there was the only one study derived from only 1D gene sequences (Tully and Fares, 2008). In this report, the mean substitution rates were 2.48 × 10-3 substitutions/site/year and this virus was diversified approximately 432 years ago. Bayesian skyline plot analysis also showed that the virus grew rapidly until the 1970s, when it experienced a rapid sharp drop in population size followed by a strong population growth.
To date, most phylogenetic and evolutionary studies describing FMDV have been largely restricted to a small number of genes from a limited selection of representative viruses. The aim of this study was to undertake a comprehensive and global phylogenomic analysis of 217 coding sequences representing all seven FMDV serotypes, and utilize Bayesian coalescent analyses to estimate substitution rates, divergence times, and population size changes of the virus.
MATERIALS AND METHODS
Data collection and sequence analysis
Both nucleotide and amino acid sequences of the polyprotein regions for serotypes O (n = 101), A (n = 47), Asia 1 (n = 32), C (n = 20), SAT 1 (n = 9), SAT 2 (n = 4), and SAT 3 (n = 4) were obtained from GenBank (Supplementary Table S1). These 217 FMDV sequences were initially aligned using the CLUSTAL X 1.81 (Thompson et al., 1997), and then adjusted manually where necessary. This alignment can be obtained from the authors. The alignment of coding genome sequences was divided into the corresponding individual genes: L, 1A, 1B, 1C, 1D, 2A, 2B, 2C, 3A, 3B, 3C, and 3D. All gene positions quoted here are with respect to FMDV genome of isolate O/NY00 (GenBank accession no. AY333431).
The calculation of both nucleotide and amino acid sequence homologies of polyprotein and individual genes were conducted using BIOEDIT 7.053 (Hall, 1999). For 12 individual genes, as well as the entire polyprotein region, base frequencies, ts/tv ratios, and following numbers of sites were calculated using Modeltest 3.7 (Posada and Crandall, 1998): total sites (including gaps), conserved sites, and variable sites.
Phylogenomic tree reconstruction
Phylogenomic reconstructions were conducted on nucleotide sequences of 217 FMDV polyprotein regions using Bayesian inferences (BI). Bayesian methods were executed in MrBayes 3.1.2 (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) under the best fit model, GTR+I+G, which were selected using Akaike’s Information Criterion (AIC) in Modeltest 3.7 (Posada and Crandall, 1998). We set the parameters of nst = 6 and rates = gamma as the likelihood settings. The Monte Carlo Markov Chains (MCMC) were run for 2,000,000 generations and sampled every 100 generations: four chains were run and 20,000 initial trees were discarded (burn-in). Bayesian posterior probabilities were estimated based on the 50% majority rule consensus of the trees. In order to screen for congruent tree topologies, 12 individual protein coding gene sequences were analyzed using this Bayesian approach. We also analyzed three additional data matrices containing the viral polyprotein sequences combined with annotated information such as date of sample collection, geographical location, and host species, to examine the influence of each of these factors on the evolution of FMDV.
Selection pressure analysis
To evaluate the selection pressure driving the evolution of FMDV, the nonsynonymous/synonymous substitution ratio (ω = dN/dS) values were estimated using Clustal X 1.81 (Thompson et al., 1997), PAL2NAL (Suyama et al., 2006), and codeml program in the PAML 3.14.1 package (Nielsen and Yang, 1998; Yang, 1997).
Substitution rates, divergence times, and population size changes
Rate of nucleotide substitution, age of the most recent common ancestor (tMRCA), and changes of population size were calculated using the Bayesian Markov Chain Monte Carlo (MCMC) approach as implemented in BEAST 1.5.3 (Drummond and Rambaut, 2007). The dataset for these analyses comprised the coding genome sequences of 187 FMDVs circulating in the world during the last 76 years (from 1932 to 2008).
Preliminary analyses were performed to determine which clock and population models were most appropriate for this FMDV dataset. We employed both strict and relaxed (both uncorrelated exponential and uncorrelated lognormal) molecular clocks (Drummond et al., 2006) with different five demographic models (constant size, exponential growth, expansion growth, logistic growth, and Bayesian skyline), and the datasets were run each for 50,000,000 generations. By a comparison of Bayes factor (log10 Bayes Factors > 2 in all cases) based on the relative marginal likelihoods of the models (Suchard et al., 2001), the relaxed uncorrelated exponential clock and constant population size model were selected as the best fitting ones for the FMDV dataset. Final Bayesian coalescent analyses used the GTR+I+G substitution model, a relaxed uncorrelated exponential clock, and a constant population size. The dataset was run for 500,000,000 generations to ensure convergence of all parameters (ESSs > 100) with discarded burn-in of 10%. The changes in effective population size over the time were examined using the Bayesian skyline plot (BSP). The resulting convergence was analyzed using Tracer 1.5 (http://beast.bio.ed.ac. uk/Tracer) and the statistical uncertainties were summarized in the 95% highest probability density (HPD) intervals.
RESULTS
Sequence analyses
The characteristics of the 217 FMDV polyprotein region (ORF) and individual gene sequences are summarized in Table 1. The overall length of the polyprotein region alignment (including gaps) was 7,066 base pairs, and the deduced amino acid sequences were 2,355 residues in length. The complete polyprotein region sequences of the FMDVs revealed a very low degree of genetic similarities; 2,904 (41.1%) of the nucleotide positions and 1,083 (46.0%) of the amino acid positions were conserved. Pairwise comparisons demonstrated that the average identities among the complete polyprotein region sequences were 85.1% for the nucleotide sequences and 90.8% for the amino acid sequences, respectively. Of the 12 individual genes, 1D was most variable (average sequence identities of 69.0% and 72.2% for nucleotides and amino acids, respectively), while 3D was most conserved (average sequence identities of 91.5% and 97.4% for nucleotides and amino acid, respectively). The transition/transversion ratio estimated from the entire polyprotein region sequences was 3.31. The transition/ transversion ratio of 3D gene was highest (5.34), while 1D had the lowest ratio (2.30).
Table 1.
Summary of the genomic regions of FMDV for phylogenomic analysis
| Genomic region | Total sites including gaps, nt/ aa | Conserved sites (%), nt/ aa | Average identities (%), nt/ aa | Ts/Tv rates | Base frequencies, A, C, G, T (%) | ω value |
|---|---|---|---|---|---|---|
| ORF | 7,066/ 2,355 | 2,904 (41.1)/ 1,083 (46.0) | 85.1/ 90.8 | 3.31 | 25.3, 28.0, 25.7, 21.1 | 0.0675 |
| L | 658/ 219 | 216 (32.8)/ 82 (37.4) | 83.0/ 87.7 | 3.92 | 25.4, 27.1, 25.2, 22.2 | 0.0601 |
| 1A | 207/ 69 | 99 (47.8)/ 39 (56.5) | 85.7/ 97.2 | 3.47 | 29.2, 31.1, 19.3, 20.4 | 0.0194 |
| 1B | 657/ 221 | 226 (34.4)/ 84 (38.0) | 76.8/ 84.9 | 2.65 | 25.3, 28.9, 25.6, 20.2 | 0.0146 |
| 1C | 673/ 223 | 221 (32.8)/ 72 (32.3) | 76.6/ 82.9 | 2.60 | 22.4, 30.2, 26.2, 21.2 | 0.0263 |
| 1D | 677/ 224 | 130 (19.2)/ 45 (20.1) | 69.0/ 72.2 | 2.30 | 25.3, 30.8, 25.0, 18.8 | 0.0488 |
| 2A | 48/ 16 | 18 (37.5)/ 8 (50) | 90.2/ 94.4 | 2.83 | 22.3, 24.9, 29.5, 23.3 | 0.0532 |
| 2B | 462/ 154 | 232 (50.2)/ 84 (54.5) | 90.6/ 96.6 | 4.54 | 21.7, 29.4, 25.9, 22.9 | 0.0393 |
| 2C | 954/ 318 | 469 (49.2)/ 181 (56.9) | 90.7/ 96.6 | 4.98 | 26.9, 28.4, 24.5, 20.2 | 0.0323 |
| 3A | 459/ 153 | 134 (29.2)/ 45 (29.4) | 86.8/ 90.0 | 4.15 | 29.2, 24.8, 25.3, 20.6 | 0.0932 |
| 3B | 213/ 72 | 82 (38.5)/ 28 (38.9) | 90.5/ 94.3 | 3.29 | 30.8, 25.0, 29.4, 14.8 | 0.0959 |
| 3C | 639/ 214 | 337 (52.7)/ 130 (60.7) | 90.3/ 96.8 | 4.13 | 24.4, 26.2, 27.8, 21.6 | 0.0282 |
| 3D | 1410/ 470 | 741 (52.6)/ 285 (60.6) | 91.5/ 97.4 | 5.34 | 24.3, 26.8, 26.1, 22.9 | 0.0357 |
Genome variability of the polyprotein region for each FMDV serotype is presented in Tables 2, 3, and 4. Average identities of intra-serotypic sequences were more than 83.7% for nucleotide and 90.3% for amino acid (within SAT 2 isolates) (Table 2), whereas inter-serotypic identities ranged from 73.3 (between O and SAT 3) to 85.3 (between A and C) % for the nucleotide sequences and from 77.7 (between C and SAT 3) to 91.3% (between O and Asia 1) for the amino acid sequences (Table 3), respectively. The transition/transversion ratio of serotype C was highest (5.26) while that of SAT 2 was lowest (1.25). The presence of amino acid residues in the FMDV polyprotein regions that were conserved between the sequences of each serotype was revealed in the multiple alignment (data not shown). There were 8 conserved amino acid residues for O, 8 for A, 17 for Asia 1, 12 for C, and 49 for SATs (Table 4) that were largely distributed through three structural genes, 1B, 1C, and 1D, although for the SATs these were spread more widely over additional non-structural genes, 2B, 2C, 3C, and 3D.
Table 2.
Summary of the polyprotien region sequences of the seven different FMDV serotypes
| Serotype | Total sites including gaps, nt/ aa | Conserved sites (%), nt/ aa | Average identities (%), nt/ aa | Ts/Tv rates | Base frequencies, A, C, G, T (%) | ω value |
|---|---|---|---|---|---|---|
| O | 7,066/ 2,355 | 4,196 (59.4)/ 1,714 (72.8) | 91.5/ 96.5 | 4.71 | 25.2, 27.9, 25.8, 21.1 | 0.0706 |
| A | 7,066/ 2,355 | 4,356 (61.6)/ 1,812 (76.9) | 89.5/ 95.3 | 4.41 | 25.4, 27.9, 25.6, 21.0 | 0.0532 |
| Asia1 | 7,066/ 2,355 | 4,613 (65.3)/ 1,805 (76.6) | 91.4/ 96.5 | 4.40 | 25.1, 27.9, 25.8, 21.3 | 0.0497 |
| C | 7,066/ 2,355 | 5,538 (78.4)/ 1,714 (72.8) | 94.4/ 97.2 | 5.26 | 25.2, 28.1, 25.6, 21.1 | 0.2205 |
| SAT1 | 7,066/ 2,355 | 4,817 (68.0)/ 1,897 (80.6) | 88.3/ 93.8 | 2.14 | 25.7, 28.7, 24.9, 20.7 | 0.0533 |
| SAT2 | 7,066/ 2,355 | 5,065 (71.7)/ 1,937 (82.3) | 83.7/ 90.3 | 1.25 | 25.5, 28.0, 25.5, 21.1 | 0.0650 |
| SAT3 | 7,066/ 2,355 | 6,021 (85.2)/ 2,191 (93.0) | 92.0/ 96.4 | 2.36 | 25.4, 28.3, 25.2, 21.1 | 0.0983 |
Table 3.
Average identities (%) among the polyprotien region sequences of the seven different FMDV serotypes (above, nucleotide; below, amino acid).
| Serotype | O | A | Asia1 | C | SAT1 | SAT2 | SAT3 |
|---|---|---|---|---|---|---|---|
| O | 84.7 | 84.9 | 84.4 | 74.1 | 76.1 | 73.9 | |
| A | 91.2 | 84.5 | 85.3 | 74.3 | 76.3 | 73.8 | |
| Asia1 | 91.3 | 90.5 | 84.3 | 74.2 | 76.1 | 73.6 | |
| C | 91.0 | 90.9 | 90.6 | 74.0 | 75.7 | 73.3 | |
| SAT1 | 78.8 | 78.8 | 78.6 | 78.3 | 78.5 | 84.7 | |
| SAT2 | 80.6 | 80.5 | 80.5 | 80.0 | 83.3 | 77.7 | |
| SAT3 | 78.5 | 78.2 | 78.2 | 77.7 | 88.7 | 82.3 | |
Table 4.
Conserved serotype specific amino acid sequences
| Genomic region | Serotypes | ||||
|---|---|---|---|---|---|
| O (n = 101) | A (n = 47) | Asia1 (n = 32) | C (n = 20) | SAT (n = 17) | |
| 1A | |||||
| 1B | T-64a, D-73, R-77, E-192 | S-37, H-42, H-93, W-129, S-185 | V-38, A-42, L-73, Y-80, M-95, Q-136, L-173, K-190, G-192, M-201, A-203 | M78, V-81, V-82, P-87, K-96 | H-19, T-21, D-40, T-112, T-114, T-154, H-168, E-198 |
| 1C | F-187, L-202 | F-63, G-84 | Y-44, A-132, N-135, A-196 | F-14, Q-15, N-16, P-61, Y-62, V-63, K-72, A-80, F-81, H-83, K-87, Y-111, Y-143, P-175, A-178, D-191, T-192 | |
| 1D | Q-149, A-199 | V-79, E-195, V-196 | Q-49, K-95, T-134, D-195 | K-85, T-131, Y-139 | G-7, D-14, H-18, T-40, G-82, D-102, F-115, T-162, Y-177, Y-195, H-197 |
| 2B | T-44, K-51, D-52, V-114 | ||||
| 2C | S-35, S-206, D-298 | ||||
| 3C | A-14, F-67, Q-201 | ||||
| 3D | N-42, K-44 | ||||
aAll positions of each gene were referred to FMDV coding genome sequences of strain O/NY00 (GenBank accession no. AY333431).
Phylogenomic analyses
Ambiguous positions were removed from the nucleotide alignment so that a total of 7,066 nucleotide positions were included for the phylogenomic analysis. The Bayesian inference tree (Figs. 1 and 2, - lnL = 177878.36) showed the monophyly of each of five serotypes (O, A, Asia 1, C, and SAT 3), with very high posterior probabilities (all, PP = 1.00). However, the remaining two serotypes (SAT 1 a nd SAT 2) were not c learly discriminated by this analysis. The FMDV sequences had a branching order of {[(O, Asia 1), (A, C)], (SAT 1 + 3 + 2)}; O + Asia 1 and A + C appeared as a two pairs of sister-group relationships, while three remaining serotypes SAT 1, 2, and 3 were clearly depicted as a single independent clade (PP = 1.00) in which all of the SAT 1 members, excluding only one isolate (AY593844), were closely related to SAT 3 members.
Fig. 1. Bayesian inference tree (-lnL = 177878.36) showing phylogenetic relationships of the 217 global FMDV isolates obtained from the complete polyprotein sequences under the GTR+I+G model of sequence evolution. Posterior probabilities (≥ 0.80) are indicated above the branches. Base frequencies: A = 0.35, C = 0.16, G = 0.20, T = 0.29.

Fig. 2. Topology of each serotype within the Bayesian inference tree showing phylogenetic relationships of the 217 global FMDV isolates obtained from the complete polyprotein sequences.

In addition to analysis at the level of complete polyprotein sequences, we performed Bayesian analyses on each viral gene of the 217 isolates. The resulting 12 individual BI trees were compared to the polyprotein region-based phylogenomic tree and showed that the 1D gene tree which had a branching pattern of {[(O, Asia 1), (A, C)], [(SAT 1, 2), 3]} most closely resembled the polyprotein region tree. This 1D gene tree (Fig. 3, - lnL = 24368.62) conformed to all major aspects of the polyprotein tree except for minor differences in topology: both SAT 1 and SAT 2 serotypes were represented as a monophyletic group (all, PP = 1.00), rather than exhibiting a paraphyletic relationship; and the SAT 1 sequences had sister-group relationship with SAT 2 (PP = 1.00) rather than SAT 3.
Fig. 3. Bayesian inference tree (-lnL = 24344.28) showing phylogenetic relationships of the 217 global FMDV isolates obtained from the complete 1D gene sequences under the TIM+I+G model of sequence evolution. Posterior probabilities (≥ 0.80) are indicated above the branches. Base frequencies: A = 0.24, C = 0.33, G = 0.25, T = 0.18.

Further analysis examined the influences of time periods, geographic locations, and host species on evolution within each of the FMDV serotypes (Figs. 1 and 2). At the top level, viruses of the serotype O were either divided into one of three groups or were unclassified (one isolate). Here, the viruses included in Group 1 were collected during 1958-2002 from 15 countries (Korea, China, Japan, Taiwan, Philippines, India, Israel, Iran, UAE, Turkey, UK, Ireland, France, South Africa, and Uganda). Their hosts were a mixture of three different susceptible species, bovine, porcine, and sheep. Isolates belonged to Group 2 were selected from China, Taiwan, and Hong Kong during the same periods (1958-2002) as Group 1. Their hosts were bovine and porcine. The remaining cluster, Group 3 contained FMD viruses from 8 nations, UK, Belgium, Poland, Italy, Argentina, Brazil, Uruguay, and Venezuela during 1939-2007. And they had bovine and sheep as their host species. Similar configurations appeared within each serotype (A, Asia1, C, and SATs), as well. Accordingly, the branching pattern of the FMDV tree within each serotype did not appear to be influenced by the time, place, or hosts from where the samples were collected.
Selection pressure analysis
The nonsynonymous/synonymous substitution ratio (ω = dN/ dS) values based on the entire data set were calculated and are presented in Tables 1 and 2. The dN/dS value of coding sequences within the genome was 0.0675, and all the values for each component gene within each of the FMDV serotypes were also lower than 1. In particular, the dN/dS value of 3B gene had the highest values (0.0959), while the corresponding values for 1B was lowest (0.0146) of the 12 individual FMDV genes. Of the 7 FMDV serotypes, C had the highest overall dN/dS values (0.2205), whereas Asia 1 exhibited the lowest ratio (0.0497). Accordingly, these analyses provide an indication that purifying selection is acting on these FMDV coding sequences.
Substitution rates, divergence times, and population size changes
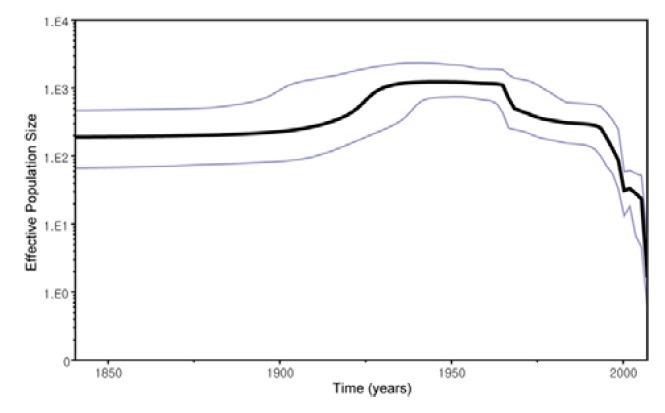
The polyprotein coding region sequences of FMDVs collected during the past 76 years (1932-2007) were also analyzed using a Bayesian coalescent approach. The relaxed uncorrelated exponential clock and constant population size model were selected at the best fit for our FMDV dataset using a Bayesian coalescent approach. The evolutionary rate of FMDV isolates was estimated to be 1.46 × 10-3 (95% HPD = 1.06 × 10-3 - 1.86 × 10-3) substitutions/site/year, projecting the most recent common ancestor back to the 481 (95% HPD = 208-851) years ago. SAT 1 diverged first 397 (95% HPD = 144-685) years ago, followed by sequential divergence of serotype SAT 2 (396 years ago; 95% HPD = 145-687), A (147 years ago; 95% HPD = 103-202), O (121 years ago; 95% HPD = 92-155), Asia 1 (89 years ago; 95% HPD = 64-117), C (86 years ago; 95% HPD = 62-125), and SAT 3 (83 years ago; 95% HPD = 62-113). Although limited historical sequences are available, the Bayesian skyline plot analysis of FMDV isolates (Fig. 4) indicates that the viruses appeared to have been evolving under almost constant population size until the early 20th century, when they experienced a population expansion followed by a rapid sharp decline in population size from the late 20th century to the present day.
Fig. 4. Bayesian skyline plot estimated from the 187 FMDVs sampled between 1932 and 2008. The bold line represents the median estimate of the effective number of infections through time. The blue lines indicate the upper and lower bounds of 95% highest posterior density.
DISCUSSION
This study revealed a high degree of genetic diversity between the 217 FMDV coding sequences available on GenBank. These results are in concordance with the views of Drake and Holland (1999) who stated that FMDV was characterized by high mutation rates of 10-3 to 10-5 per replication cycle. This rapid evolutionary rate of FMDV has generated more than 65 strains among the seven serotypes (Domingo et al., 2002). Of the 12 individual polyprotein genes, our analyses showed that 1D was most variable, while 3D was most conserved. This finding is consistent with the resolution described by Carrillo et al. (2005), especially for the 1D gene that encodes the structural protein, VP1. This capsid protein is a immunogenic target for neutralizing antibodies and variation at specific amino acid sites within the G-H loop (residues 140-160) and C-terminus (residues 200-213) contributes to the antigenic variability of the virus (Sobrino et al., 2001). An additional finding of this study is that within each of the serotypes, there were a number of amino acid residues that were conserved. These serotype specific amino acid residues might provide useful markers for the classification and diagnosis of FMDV.
Although the phylogeny of FMDV has been studied in recent years using individual gene (predominantly 1D) or genome sequences, the present study provides further details on the genome-wide phylogeny. Our results indicated that five FMDV serotypes, O, A, Asia 1, C, and SAT 3, were each monophyletic but the remaining two serotypes SAT 1 and SAT 2 were not so clearly differentiated. This configuration is consistent with a previous study based on coding sequences of the genome excluding the 3B gene (Lewis-Rogers et al., 2008). Tully and Fares (2008), however, have claimed that both SAT 1 and SAT 2, as well as above five serotypes, were also monophyletic on the basis of 1D gene sequences.
In common with the results from previous studies (Carrillo et al., 2005; Cooke and Westover, 2008; Lewis-Rogers, 2008; Tully and Fares, 2008), our findings revealed that there were two clearly defined top-level clades for FMDV; one containing the Eurasian serotypes and the other comprising the SATs serotypes. Within the Eurasian clade, the present study also showed two sister-group relationships, Asia 1 + O and A + C. The first sister-group relationship of serotypes Asia 1 and O supported the conclusions of Lewis-Rogers et al. (2008) and Tully and Fares (2008). In contrast to these reports, Cooke and Westover (2008) suggested that the serotype Asia 1 had a closer relationship with serotype C than for other serotypes, based on the amino acid sequences of FMDV genomes excluding antigenic regions. Our 1D gene trees were in agreement with analysis undertaken for complete polyprotein sequences except for minor differences in topology. Namely, in the 1D gene tree, the serotype SAT 1 and SAT 2 were represented as a monophyletic group respectively; isolate AY593844 was more closely related to SAT2 in coding gene tree and was grouped with SAT1 isolates in 1D gene tree, and isolate AY593847 more closely related to SAT 3 viruses in coding gene tree was also combined with SAT 2 viruses 1D gene tree. These tree incongruities may reflect recombination events outside of the 1D region between isolates from different serotypes. Serotypes SAT 1 and SAT 2 had a sister-group relationship to each other. This point was in accordance with the idea of Tully and Fares (2008). In addition, our two BI trees (data not shown) based on the 1B and 1C gene sequences also revealed that all the seven serotypes were monophyletic, but with differences of phylogenetic relationships among serotypes: {[(O, C), (A, Asia 1)], [(SAT 1, 3) 2]} in the 1B tree and [(C, Asia 1), O], A, [(SAT 1, 3), 2] in the 1C tree.
Subsequently, we found no apparent correlation between the time, place of collection, or host species and the evolution of global FMDVs within each serotype. These findings were substantiated by additional results derived from three data matrices consisting of all the viral sequences with additional annotation information included in the GenBank file (Supplementary Figs. S1, S2, and S3). The lack of structure in these analyses is consistent with other investigators in the field (Cottam et al., 2006; Lewis-Rogers et al., 2008) and presumably reflects the transboundary nature of FMD, and arises as a consequence of rapid spread of the virus via frequent international trade of livestock (or livestock products).
The influence of purifying selection acting upon FMDV has been studied from both individual gene and complete genome perspectives (Cooke and Westover, 2008; Lewis-Rogers et al., 2008; Tully and Fares, 2006). Across short time periods Cottam et al. (2006), observed that the highest dN/dS value (1.0) appeared in 1A gene, in contrast to low values (0.1) in 2C based on coding genome sequences of serotype O viruses, In this study, we used more sequence information than previous data sets and our results supported the strong purifying selection of overall FMDV polyprotein region.
Only one previous study (Tully and Fares, 2008) has used 1D gene sequences to define the historical origin and epidemiological dynamics of FMDV serotypes. In contrast to the single gene based research, our study utilsed coding genome data to can elucidate evolutionary mechanisms of FMDV. We estimated a mean evolutionary rate of 1.46 × 10-3 substitutions/ site/year for polyprotein, which is higher than the value (2.48 × 10-3) previously reported for the 1D gene. On the basis of our analyses, the divergence of the common ancestor of FMDV was relatively recent event that occurred no earlier than approximately 481 years ago, which is similar to the value (approximately 432 years ago) reported by Tully and Fares (2008). Of the 7 serotypes, SAT 1 diverged first (397 years ago), followed by sequential segregation of serotype SAT 2 (396 years ago), A (147 years ago), O (121 years ago), Asia 1 (89 years ago), C (86 years ago), SAT 3 C (83 years ago). This result, although not in accordance with the previous study (Tully and Fares, 2008) that suggested the sequential divergence of serotypes fits more closely with the likely ancestral source of FMDV as the Cape buffalo (Syncerus caffer) in Africa.
In terms of dynamics in the population size of FMDV, our Bayesian skyline plot analysis of FMDV isolates showed that the virus population size remained constant until an expansion in the early 20th century after which the population size underwent a rapid sharp decline from the late 20th century to the present day. This pattern is in contrast with the result derived from 1D gene sequence analysis (Tully and Fares, 2008); where the virus grew rapidly until the 1970s, when it experienced a rapid sharp drop in population size followed by a strong population growth. Based on the widespread distribution of FMDV, it is difficult to correlate this pattern with discrete changes that have influenced the circulation of FMDV at the global level. However, we postulate that the sharp decrease of FMDV effective population size since the late 20th century might be due to the increased use of vaccines across the world (Balamurugan, 2004; Barteling, 2002).
FMDV is still one of the most notorious pathogens in global livestock industry; therefore, prevention-and-control strategies to control or limit the spread of disease are in great demand. The expanding information of genome-wide phylogeny and evolutionary mechanism of the viruses obtained from this study could be very useful for our understanding of its emergence and epidemiology and aid in development of new vaccines and diagnostic tests. This further phylogenomic study is the first detailed investigation of evolutionary mechanisms on the basis of the coding genome sequences representing all 7 FMDV serotypes.
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
Acknowledgments
This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ006471)” Rural Development Administration, Republic of Korea, and with the FMD Reference Laboratory in UK (DEFRA [project SE2938], UK support to DK).
References
- 1.Barteling S.J. Development and performance of inactivated vaccines against foot and mouth disease. Rev. Sci. Tech. (2002);21:577–588. doi: 10.20506/rst.21.3.1361. [DOI] [PubMed] [Google Scholar]
- 2.Balamurugan V., Kumar R.M., Suryanarayana V.V. Past and present vaccine development strategies for the control of foot-and-mouth disease. Acta Virol. (2004);48:201–214. [PubMed] [Google Scholar]
- 3.Carrillo C., Tulman E.R., Delhon G., Lu Z., Carreno A., Vagnozzi A., Kutish G.F., Rock D.L. Comparative genomics of foot-and-mouth disease virus. J. Virol. (2005);79:6487–6504. doi: 10.1128/JVI.79.10.6487-6504.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooke J.N., Westover K.M. Serotype-specific differences in antigenic regions of foot-and-mouth disease virus (FMDV): a comprehensive statistical analysis. Infect. Genet. Evol. (2008);8:855–863. doi: 10.1016/j.meegid.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Cottam E.M., Haydon D.T., Paton D.J., Gloster J., Wilesmith J.W., Ferris N.P., Hutchings G.H., King D.P. Molecular epidemiology of the foot-and-mouth disease virus outbreak in the United Kingdom in 2001. J. Virol. (2006);80:11274–11282. doi: 10.1128/JVI.01236-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingo E., Baranowski E., Escarmis C., Sobrino F. Foot and-mouth disease virus. Comp. Immunol. Microbiol. Infect. Dis. (2002);25:297–308. doi: 10.1016/s0147-9571(02)00027-9. [DOI] [PubMed] [Google Scholar]
- 7.Drake J.W., Holland J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA. (1999);96:13910–13913. doi: 10.1073/pnas.96.24.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. (2007);7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drummond A.J., Ho S.Y.W., Phillips M.J., Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. (2006);4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall T.A. BIOEDIT: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acid Symp. Ser. (1999);41:95–98. [Google Scholar]
- 11.Huelsenbeck J.P., Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. (2001);17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 12.Lewis-Rogers N., McClellan D.A., Crandall K.A. The evolution of foot-and-mouth disease virus: impacts of recombination and selection. Infect. Genet. Evol. (2008);8:786–798. doi: 10.1016/j.meegid.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Mohapatra J.K., Priyadarshini P., Pandey L., Subramaniam S., Sanyal A., Hemadri D., Pattnaik B. Analysis of the leader proteinase (L(pro)) region of type A foot-and-mouth disease virus with due emphasis on phylogeny and evolution of the emerging VP3(59)-deletion lineage from India. Virus Res. (2009);141:34–46. doi: 10.1016/j.virusres.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen R., Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV- 1 envelope gene. Genetics. (1998);148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira H.G. Subtyping of foot-and-mouth disease virus. Dev. Biol. Stand. (1977);35:167–174. [PubMed] [Google Scholar]
- 16.Posada D., Crandall K.A. Modeltest: testing the model of DNA substitution. Bioinformatics. (1998);14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 17.Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. (2003);19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 18.Schumann K.R., Knowles N.J., Davies P.R., Midgley R.J., Valarcher J.F., Raoufi A.Q., McKenna T.S., Hurtle W., Burans J.P., Martin B.M., et al. Genetic characterization and molecular epidemiology of foot-and-mouth disease viruses isolated from Afghanistan in 2003-2005. Virus Genes. (2008);36:401–413. doi: 10.1007/s11262-008-0206-4. [DOI] [PubMed] [Google Scholar]
- 19.Sobrino F., Saiz M., Jimenez-Clavero M.A., Nunez J.I., Rosas M.F., Baranowski E., Ley V. Foot-and-mouth disease virus: a long known virus, but a current threat. Vet. Res. (2001);32:1–30. doi: 10.1051/vetres:2001106. [DOI] [PubMed] [Google Scholar]
- 20.Suchard M.A., Weiss R.E., Sinsheimer J.S. Bayesian selection of continuous- time Markov chain evolutionary models. Mol. Biol. Evol. (2001);18:1001–1013. doi: 10.1093/oxfordjournals.molbev.a003872. [DOI] [PubMed] [Google Scholar]
- 21.Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acid Res. (2006);34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G. The Clustal-windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res. (1997);22:4673–4680. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tully D.C., Fares M.A. Unravelling selection shifts among foot-and-mouth disease virus (FMDV) serotypes. Evol. Bioinform. (2006);2:237–251. [PMC free article] [PubMed] [Google Scholar]
- 24.Tully D.C., Fares M.A. The tale of a modern animal plague: tracing the evolutionary history and determining the time-scale for foot and mouth disease virus. Virology. (2008);382:250–256. doi: 10.1016/j.virol.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. (1997);13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]