

Abstract
Lysophosphatidic acid (LPA) is a lipid growth factor that exerts diverse biological effects, including rapid neurite retraction and cell migration. Alterations in cell morphology, including neurite retraction, in neurodegenerative disorders such as Alzheimer’s disease involve hyperphosphorylation of the cytoskeletal protein tau. Since LPA has been shown to induce neurite retraction in various cultured neural cells and the detailed underlying molecular mechanisms have not yet been elucidated, we investigated whether LPA induced neurite retraction through taumediated signaling pathways in differentiated neuroblastoma cells. When Neuro2a cells differentiated with retinoic acid (RA) were exposed to LPA, cells exhibited neurite retraction in a time-dependent manner. The retraction of neurites was accompanied by the phosphorylation of tau. The LPA-induced neurite retraction and tau phosphorylation in differentiated Neuro2a cells were significantly abolished by the glycogen synthase kinase-3β (GSK-3β) inhibitor lithium chloride. Interestingly, the LPA-stimulated tau phosphorylation and neurite retraction were markedly prevented by the administration of H89, an inhibitor of both cyclic-AMP dependent protein kinase (PKA) and cyclic- AMP response element-binding protein (CREB). Transfection of the dominant-negative CREBs, K-CREB and ACREB, failed to prevent LPA-induced tau phosphorylation and neurite retraction in differentiated Neuro2a cells. Taken together, these results suggest that GSK-3β and PKA, rather than CREB, play important roles in tau phosphorylation and neurite retraction in LPA-stimulated differentiated Neuro2a cells.
Keywords: CREB, GSK-3β, lysophosphatidic acid, neurite retraction, Tau phosphorylation
INTRODUCTION
Normal development of the nervous system requires remarkable alterations in cell morphology that are dependent upon dynamic changes in cytoskeletal proteins. Differentiating neurons actively extend and retract cytoplasmic processes, known as neurites, during the course of brain development (Sayas et al., 2002a). Rearrangements of the cytoskeleton in response to extracellular stimulations play a key role in the developmental process through which undifferentiated neuroblasts become differentiated functional neurons. Two major components of the cytoskeleton are involved in axonogenesis: actin microfilaments and microtubules. Microfilaments appear to regulate the initial events that are part of the growth cone tip, filopodia, and lamellipodia progression, and in a second step, microtubules stabilize the extended neurite (Tanaka et al., 1995).
In particular, tau, one of the most abundant axonal microtubule- associated proteins (MAPs), may play a key role in axonogenesis (Mandell and Banker, 1996). It can be phosphorylated by several protein kinases (Steiner et al., 1990). Phosphorylation by tau protein kinase II at serine 404 is primarily responsible for the functional loss of tau-mediated tubulin polymerization. In addition, phosphorylation of microtubule-associated tau results in the dissociation of tau from the microtubules and tubulin depolymerization (Evans et al., 2000; Steiner et al., 1990). Tau phosphorylation is developmentally regulated, and is lower in the adult than in the fetal brain. Additionally, tau hyperphosphorylation occurs in the aberrant structures known as paired helical filaments that are a neuropathological hallmark in neuronal disorders such as Alzheimer’s disease (Greenberg and Davies, 1990).
Tau phosphorylation and neurite retraction by lysophosphatidic acid (LPA; 1-acyl-sn-glycerol-3-phosphate) stimulation have been shown in neuroblastoma cell lines (Jalink et al., 1993) and in PC-12 cells (Tigyi and Miledi, 1992). In addition, Saito showed that LPA promoted growth cone collapse in some primary neurons (Saito, 1997). LPA, one of the simplest natural phospholipids, acts on cognate G protein-coupled receptors (GPCRs) to induce a host of cellular responses, including cell proliferation and differentiation, platelet aggregation, protection from apoptosis, promotion of cell survival, stress fiber formation, tumor cell invasion, and cell morphological change (Contos et al., 2000; Fukushima et al., 2002; Moolenaar, 1999; Nam et al., 2010). LPA signals act through at least five specific cell membrane- bound GPCRs. Several members (Edg-2/Vzg-1, Edg-4, and Edg-7, which are also known as LPA1, LPA2, and LPA3, respectively) of the endothelial cell differentiation gene family have been identified as high-affinity receptors for LPA (Fukushima and Chun, 2001; Goetzl and An, 1999). In contrast to the three highly related LPA receptors, LPA4/GPR23 and LPA5/GPR92 are more closely related to nucleotide receptors of the P2Y GPCR family (Anliker and Chun, 2004; Lee et al., 2006).
LPA-induced tau phosphorylation and neurite retraction have been shown in neuroblastoma cells. However, the signaling pathways of LPA-regulated tau phosphorylation and neurite retraction are not very clear. Thus, this study was undertaken in order to determine signaling pathways responsible for the activation of glycogen synthase kinase-3β (GSK-3β), cyclic-AMP dependent protein kinase (PKA), and cyclic-AMP response element-binding protein (CREB). Herein, we provide evidence that LPA-induced neurite retraction is caused by the activation of GSK-3β and PKA, rather than CREB.
MATERIALS AND METHODS
Materials
All chemicals used were of analytical grade unless stated otherwise. LPA (1-oleoyl-sn-glycerol 3-phosphate), lithium chloride (LiCl), all-trans retinoic acid (RA), anti-β-Tubulin, Y27632, and pertussis toxin (PTX) were purchased from Sigma-Aldrich Co. (USA). LPA was dissolved in 0.1% fatty-free bovine serum albumin at 5 mM in stock solution. Antibodies to phospho-GSK- 3β, GSK-3β, CREB, and β-actin were obtained from Cell Signaling Technology, Inc. (USA). Antibodies to phosphor-tauser396 and phosphor-tauser404 were purchased from Santa Cruz Biotechnology, Inc. (USA). PD98059 and SB203580 were from Tocris Bioscience (UK). Modified Eagle’s Medium (MEM), fetal bovine serum (FBS), and Lipofectamine 2000 were obtained from Invitrogen Corporation (USA). The dominant-negative CREBs, K-CREB and A-CREB, were gifts from Dr. David Ginty and Dr. Hiroshi Ishiguro (Johns Hopkins Univ., USA).
Cell culture
Neuroblastoma neuro2a cells were grown at 37℃ under a humidified atmosphere of 5% CO2. Cells were cultured in MEM containing 10% FBS, penicillin (50 U/ml), and streptomycin (50 μg/ml). For differentiation, cells were maintained in MEM containing 2% FBS and 10 μM RA for 24 h. For LPA-stimulation, differentiated cells were treated with various inhibitors, as described in the figure legends, prior to LPA stimulation.
RNA extraction and reverse transcriptase (RT)-polymerase chain reaction (PCR)
Total RNA was isolated from cells and tissue by the guanidine isothiocyanate method (Chomczynski and Sacchi, 1987), and cDNA was made using 1 μg of total RNA and AMV reverse transcriptase (Promega Corporation, USA) in 20 μl reaction mixtures in the presence of 2.5 μM oligo(dT) primer and 20 μM dNTP mixture for 60 min at 42℃. For PCR amplification, specific oligonucleotide primer pairs (0.5 μM each) were incubated with 200 ng cDNA, 2 units of Taq polymerase (PerkinElmer, Inc., USA), 1× Taq buffer [10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2] and 10 μM dNTP in 20 μl reaction mixtures. The sequences of primers used in this study were as follows: LPA1 forward primer, 5′-TCTTCTGGGCCATTTTCAAC-3′; LPA1 reverse primer, 5′-TGCCTGAAGGTGGCGCTCAT-3′; LPA2 forward primer, 5′-CCTACCTCTTCCTCATGTTC-3′; LPA2 reverse primer, 5′-TAAAGGGTGGAGTCCATCAG-3′; LPA3 forward primer, 5′-AGTGTCACTATGACAAGC-3′; LPA3 reverse primer, 5′-GAGATGTTGCAGAGGC-3′; LPA4 forward primer, 5′-TGAAGGCTTCTCCAAACGTGTCTG-3′; LPA4 reverse primer, 5′-GTTCAGAGTTGCAAGGCACAAGGT-3′; β-actin forward primer, 5′-TGGAATCCTGTGGCATCCATGAAA-3′; and β-actin reverse primer, 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. PCR cycling conditions were 95℃ for 30 s, 55-60℃ for 45 s, and 72℃ for 60 s, for a total of 30-33 cycles. The PCR products were applied to a 1.5% agarose gel and visualized with ethidium bromide.
Isolation of total protein and Western blot analysis
Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 1 mM Na3VO4, 5 mM NaF, and protease inhibitor cocktail). After incubation on ice for 30 min with vortexing every 10 min, the lysates were centrifuged (15,000 × g, 15 min). Supernatants were collected and protein concentrations were determined by a Bradford assay (Bio-Rad Laboratories, Inc., USA). Equal amounts of protein were separated by SDS-PAGE (10% or 12% reducing gels), transferred to polyvinylidene difluoride membranes (Millipore, USA), and blocked with 5% non-fat milk. Membranes were incubated in primary antibodies overnight at 4℃. Membranes were then washed in TBST (10 mM Tris, 140 mM NaCl, 0.1% Tween 20, pH 7.6), incubated with appropriate secondary antibodies, and washed again in TBST. Bands were visualized by enhanced chemiluminescence and exposed to X-ray film.
Transient transfection with K-CREB and A-CREB
Neuro2a cells were plated in 6-well cell culture dishes to 80% confluence (Corning, Inc., USA). Twenty-four h later, they were incubated with OPTI-MEM media containing 10% FBS without antibiotics. DNA and the Lipofectamine 2000 reagent were diluted separately in 250 μl of serum-free OPTI-MEN media without antibiotics, mixed together, and incubated at room temperature for 20 min. The culture plates were washed with phosphate- buffered saline (PBS, pH 7.4), and 1.5 ml of antibioticfree OPTI-MEM medium containing 10% FBS was added. Then, 500 μl of the plasmid/Lipofectamine 2000 reagent mixture was gently added to each well, and the plates were incubated at 37℃ for 24 h.
Immunocytochemistry
Cells were plated onto four-well chamber slides at a density of 60-70% confluency. After chemical stimulation, cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton X- 100, each for 10 min at room temperature. Following three rinses in PBS, cells were blocked with 10% normal goat serum in PBS for 1 h at room temperature. Cells were incubated overnight with the primary antibody (β-tubulin at 1:800 dilution) at 4℃. Cells were washed three times with PBS and then stained using the ABC Kit (Vector Laboratories, Inc., USA) and 0.05% diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich Co.). Following three rinses in PBS, the stained coverslips were mounted and observed under a microscope (200X; Olympus PROVIS AX-70, Olympus Corporation, Japan).
Statistical analysis
Results are presented as the mean ± S.E. from more than three independent experiments. Differences between the groups were assessed with paired t-tests in Prism 4 (GraphPad Software, Inc., USA).
RESULTS
LPA induced neurite retraction in differentiated Neuro2a cells
In order to examine the expression of the LPA receptors in Neuro2a cells, we utilized RT-PCR (Fig. 1G). Primers were designed from sequences conserved in the mouse, as well as with mouse tissues, including hippocampus, cortex, ovary, and testis (Chun et al., 2002). The RT-PCR reaction showed the expression of LPA1 and LPA2 receptor and a little LPA4 receptor mRNA. However, the LPA3 receptor mRNA expression was not detectable in both undifferentiated and differentiated Neuro2a cells. In addition, there were no differences in the levels of expression of the mRNA of all LPA receptors between Neuro2a cells and differentiated Neuro2a cells.
Fig. 1. Lysophosphatidic acid (LPA)-induced neurite retraction of differentiated neuroblastoma cells, Neuro2a. Neuro2a cells were cultured on a chamber slide. Cells were incubated with (B-F) or without (A) 10 μM retinoic acid (RA) for 24 h. After incubating with RA, cells were treated without (B) or with 5 μM LPA for 5 min (C), 15 min (D), 30 min (E), or 60 min (F), respectively. Cells were immunostained with an antibody against β-tubulin. Scale bar: 50 μm. In order to examine the levels of expression of the LPA receptor genes in Neuro2a cells, we analyzed RT-PCR. Primers were designed according to conserved mouse sequences. Amplification of β- actin was used as a relative loading control for cDNA quantity (G).
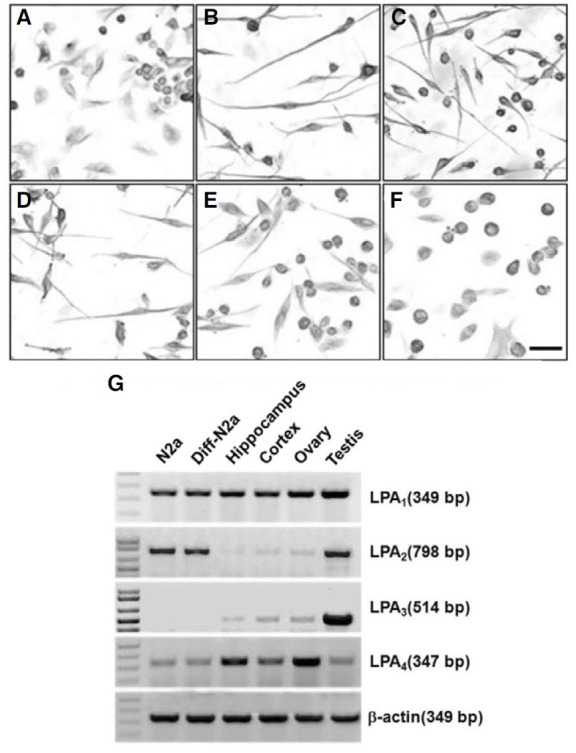
We characterized the neuronal-like phenotype of Neuro2a cells after RA treatment (Mao et al., 2000). The differentiated Neuro2a cells were obtained after addition of 10 μM RA in the MEM culture media containing 2% FBS. After 24 h incubation with RA, most of the Neuro2a cells exhibited a neuron-like phenotype with long neurites (Fig. 1B). Before the LPA treatment, more than 90% of the differentiated cells showed processes. After a 5 min treatment with LPA, differentiated cells showed growth cone collapse and membrane ruffling. Neurite retraction was time-dependent, and the extended processes were almost completely reduced after 1 h with LPA addition (Figs. 1B-1F). In addition, LPA-induced neurite retraction was maintained 12 h after LPA treatment (data not shown).
Activation of the GSK-3β cascade was involved in the LPA-induced neurite retraction
Recent evidence suggests that GSK-3β may be involved in neurite retraction in certain cell types (Bhat et al., 2000; Hartigan and Johnson, 1999; Mandell and Banker, 1996). Additionally, these previous reports prompted us to test the involvement of the GSK-3β cascade in the LPA-stimulated neurite retraction in differentiated Neuro2a cells. To test this possibility, we performed immunoblot analyses. As shown in Fig. 2A, treatment of differentiated Neuro2a cells with LPA resulted in a time-dependent increase of the level of tyrosine-phosphorylated GSK-3β. In addition, the GSK-3β phosphorylated in serine did not change with the LPA treatment (Fig. 2A). The results from the immunoblot analysis demonstrated that GSK-3β is activated by LPA in differentiated Neuro2a cells.
Fig. 2. Phosphorylation of GSK-3β and tau in differentiated Neuro2a cells by LPA. After 24 h differentiation, Neuro2a cells were treated with 5 μM LPA in a time-dependent manner. Whole cell lysates were subjected to 10% SDS-PAGE and immunoblotted with phosphospecific antibodies GSK-3β (A) and tau (B). The amplification intensity of total-GSK-3β and β-actin were used as a loading control.
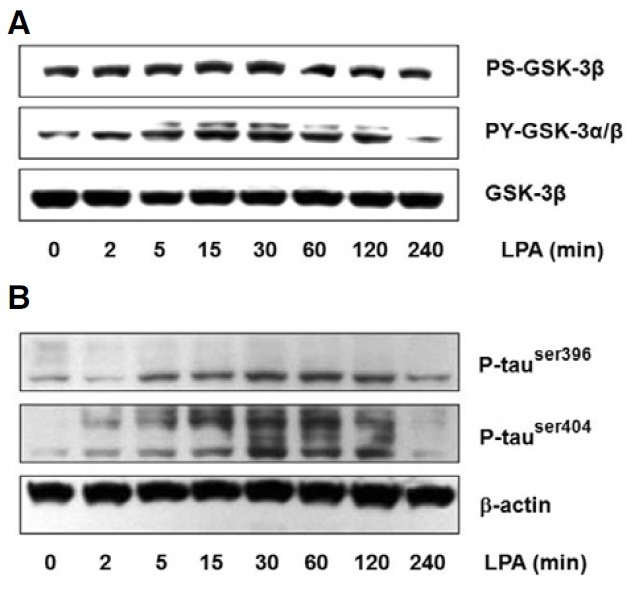
During neurite retraction, one of the most important elements that mediates the stability of microtubules is the phosphorylation level of MAPs (Mandell and Banker, 1996). A recent study reported that GSK-3β phosphorylates tau at sites of serine (Ser) 396 and Ser404, which is involved in neurite retraction, a key step in the development of neurofibrillary pathology in the Alzheimer’s disease brain (Li et al., 2006; Tatebayashi et al., 2006). Considering these facts, we investigated the possibility of LPA-stimulated tau phosphorylation at the Ser396 and Ser404 sites in differentiated Neuro2a cells. Phosphorylation of tau was assessed by immunoblot analysis with phosphospecific antibodies against Ser396 and Ser404 of tau. The level of phosphorylated tau rapidly increased at sites Ser396 and Ser404 after treatment for 5 min with LPA, and the levels were the highest 30 min after LPA addition and then decreased slowly (Fig. 2B).
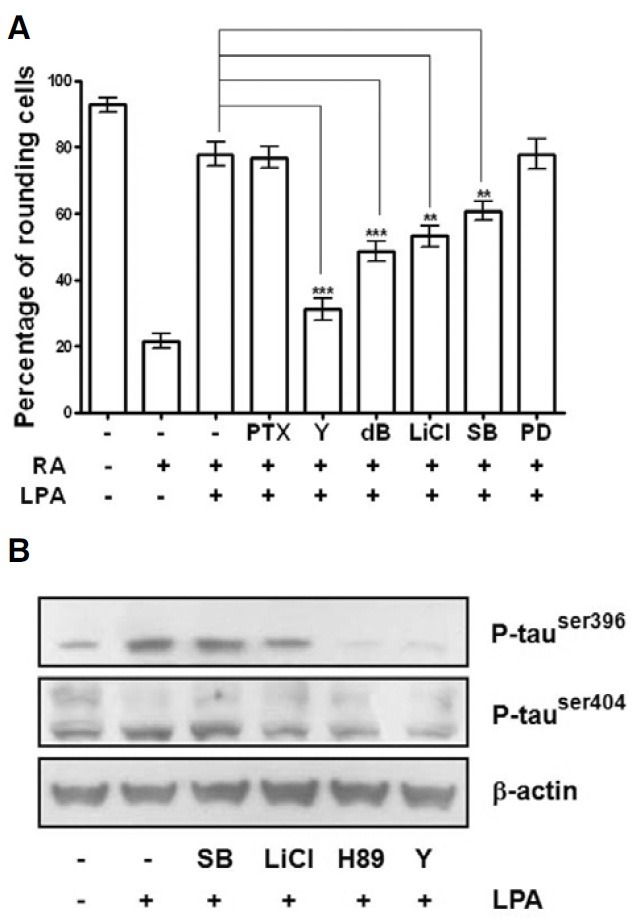
To further test which signaling pathway was involved in LPA induced neurite retraction in differentiated Neuro2a cells, the effects of various pharmacological inhibitors of protein kinases on the neurite retraction and phosphorylation of tau were examined. As shown in Fig. 3A, LPA-induced neurite retraction was significantly reduced by Y27632, a Rho-associated coiled-coil forming protein serine/threonine kinase (ROCK) inhibitor. Furthermore, Y27632 also significantly prevented tau phosphorylation in differentiated Neuro2a cells (Fig. 3B). In order to confirm that GSK-3β is the kinase responsible for tau phosphorylation by LPA in differentiated Neuro2a cells, we treated cells with LiCl, which is an inhibitor of GSK-3β (Klein and Melton, 1996; Stambolic et al., 1996). Pretreatment with 20 mM LiCl partially prevented LPA-induced neurite retraction (Fig. 3A) and decreased the tau phosphorylation (Fig. 3B). Interestingly, although LPA-induced neurite retraction in differentiated Neuro2a cells was partially reduced by pretreatment with SB203580, a p38 MAPK inhibitor, it failed to prevent LPA-induced tau phosphorylation (Fig. 3). These data suggest that p38 MAPK-mediated neurite retraction by LPA did not involve tau phosphorylation. Moreover, PTX (a Gi inhibitor) and PD98059 (a MAPK inhibitor) did not regulate LPA-induced neurite retraction in differentiated Neuro2a cells (Fig. 3A). Taken together, these results demonstrate that the activation of ROCK, GSK-3β, and p38 MAPK were involved in LPA-induced neurite retraction of differentiated Neuro2a cells.
Fig. 3. Involvement of GSK-3β in LPA-induced neurite retraction of differentiated Neuro2a cells. Differentiated Neuro2a cells were pretreated with 100 ng/ml pertussis toxin (PTX; 6 h), 4 μM Y27632 (Y; 30 min), 2 mM db-cAMP (db; 1 h), 20 mM lithium chloride (LiCl; 2 h), 20 μM SB203580 (SB; 1 h), and 50 μM PD98059 (PD; 1 h), followed by incubation with 5 μM LPA for 1 h. The cells were then photographed at 200X magnification under a phase-contrast microscope. With these photographs, the numbers of cells without or with the neurite shape were counted in four randomly selected fields. Results are expressed as the percentages of rounding cells among the total cells (A). Differentiated Neuro2a cells were pretreated with 20 μM SB203580, 20 mM LiCl, 10 μM H89 or 4 μM Y27632 before exposure to 5 μM LPA for 30 min. The cytoplasmic extracts were used to study the levels of tau phosphorylation by western blot analysis probed with phospho-specific tau antibodies as described in “Materials and Methods”. (B). Results of the mean ± S.E. of four different rectangular areas of three independent experiments are shown. **P < 0.01; ***P < 0.001.
CREB activation is not required for LPA-induced differentiated Neuro2a cell neurite retraction
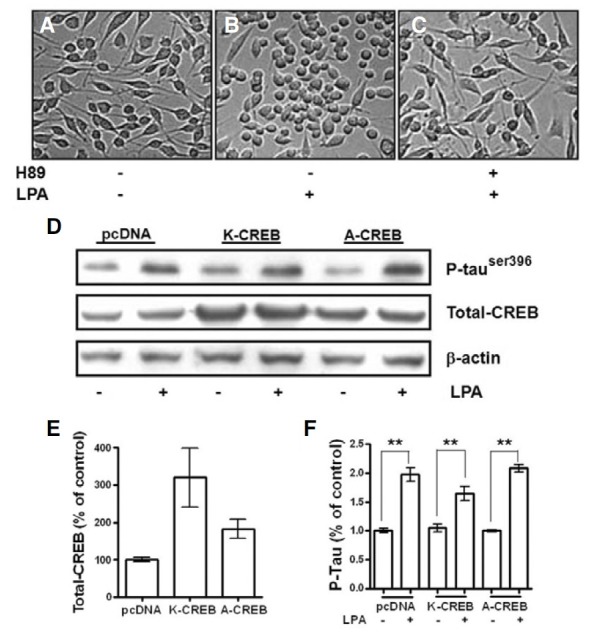
We found that H89 dramatically blocked tau phosphorylation (Fig. 3B) and significantly prevented neurite retraction in differentiated Neuro2a cells (Figs. 4A-4C). H89, an inhibitor of PKA, has recently been shown to inhibit MSK-1 activity (Lee et al., 2003; Thomson et al., 1999; Zhong et al., 2001). Previous studies have demonstrated that CREB is downstream of the GSK- 3β signaling pathway (Grimes and Jope, 2001). To further investigate the mechanisms of LPA-induced neurite retraction in differentiated Neuro2a cells, we examined the role of CREB in LPA-stimulated neurite retraction by using the dominant-negative CREBs, K-CREB and A-CREB. The protein levels of CREB were significantly increased when Neuro2a cells were transfected with A-CREB and K-CREB (Figs. 4D and 4E). However, LPA-induced tau phosphorylation was not significantly different between the dominant-negative CREB-transfected cells and pcDNA-transfected cells (Figs. 4D and 4F). These findings indicate that LPA-stimulated activation of CREB may not result in LPA-stimulated neurite retraction and tau phosphorylation in differentiated Neuro2a cells.
Fig. 4. CREB is not a component of the signaling cascade of LPA-induced neurite retraction of differentiated Neuro2a cells. Neuro2a cells were differentiated in Modified Eagle’s Medium containing 2% FBS and 10 μM RA for 24 h (A). They were pretreated with 10 μM H89 (C) for 1 h before being incubated with 5 μM LPA (B and C) for 1 h. Then, the cells were photographed at 200X magnification under a phase-contrast microscope. Neuro2a cells were transfected with pcDNA, K-CREB, or A-CREB. After differentiation, cells were incubated with 5 μM LPA for 30 min. Phospho- tau and total-CREB were detected by immunoblot analyses using phospho-specific antibodies that recognize phospho-tau and CREB-1 antibodies, respectively (D). The amplification intensity of β-actin was used as a loading control. The intensities of the total-CREB bands (E) and the phospho- Tau bands (F) were determined by densitometric scanning and analyzed by Bio-Profil software. Results are mean ± S.E. and representatives of three independent experiments are shown. **P < 0.01.
DISCUSSION
The question addressed in this study was how LPA induced neurite retraction and tau phosphorylation in differentiated neuroblastoma Neuro2a cells. Our main finding was that GSK- 3β and PKA signaling pathways partially influenced the induction of neurite retraction and tau phosphorylation in LPAstimulated differentiated Neuro2a cells. In addition, we demonstrated that phosho-p38 MAPK, which was activated by LPA, was partially involved in LPA-induced neurite retraction, but did not regulate tau phosphorylation in differentiated Neuro2a cells. Interestingly, although LPA-induced CREB phosphorylation is involved in various biological progresses, CREB seemed to be incapable of regulating neurite retraction in differentiated Neuro2a cells. Our results, incorporated with the findings of previous studies (Fukushima et al., 2002; Sayas et al., 1999), add new observations that are leading to a more detailed understanding of the role of CREB in LPA-induced neurite retraction and tau phosphorylation in differentiated neuroblastoma cells.
A number of studies have shown that LPA mediates morphological changes in neuronal cells through the regulation of microtubule-binding proteins (Chun, 2005; Sayas et al., 2006). It has been proposed that these effects are mediated by LPA and LPA receptors, including LPA1, LPA2, and LPA4 (Fukushima, 2004; Yanagida et al., 2007; Ye et al., 2002). GSK-3β is a key regulatory component of a large number of cellular processes, and aberrant control of GSK-3-regulated pathways plays a role in a number of human diseases, such as diabetes, Alzheimer’s disease, and cancer. More than 40 proteins are phosphorylated by GSK-3β, and these substrates include metabolic proteins, cytoskeleton proteins, and transcription factors (Cai et al., 2006). Here, we found that LPA increased GSK-3β activity, which induced tau phosphorylation and neurite retraction in differentiated Neuro2a cells. Neurite retraction is a significant process not only during development (neurogenesis and neuritogenesis), but also in some pathological circumstances such as neurodegeneration (Sayas et al., 1999; 2002b). Previous studies have demonstrated that GSK-3β regulated AP-1, CREB activation, and CRE/CREB-dependent gene transcription (Boer et al., 2007; Hu et al., 2007). Recently, we demonstrated that LPA-induced CREB phosphorylation played an important role in the induction of c-fos expression, cyclin D1 accumulation, cell proliferation, and so on (Kwon et al., 2009). Thus, we investigated the possibility of the involvement of CREB in LPA induced neurite retraction in differentiated Neuro2a cells. As shown in Fig. 3C, LPA-induced tau phosphorylation was notably reduced by the protein kinase inhibitor, H89, which is a CREB inhibitor. Furthermore, H89 significantly prevented LPA induced neurite retraction in differentiated Neuro2a cells (Fig. 4A). H89 has also been reported to inhibit PKA (Ginty et al., 1991). In order to investigate whether PKA or CREB was involved in LPA-induced neurite retraction in our study, we used the dominant-negative CREBs, K-CREB and A-CREB, as the selective inhibitors of CREB. As a result, we found that both KCREB and A-CREB failed to prevent LPA-stimulated tau phosphorylation in differentiated Neuro2a cells. Thus, we conclude that CREB is not involved in LPA-stimulated Neuro2a cell neurite retraction. Previous studies have demonstrated that cAMPmediated neuritogenesis requires the activation of PKA and PI- 3K in order to initiate neurite elongation (Sanchez et al., 2001). The addition of db-cAMP, an activator of PKA, before LPA addition is known to prevent growth cone collapse and neurite retraction by maintaining the neuron-like phenotype (Sayas et al., 1999). Our biochemical data confirm that the addition of dbcAMP before LPA treatment partially blocked growth cone collapse and neurite retraction in differentiated Neuro2a cells (Fig. 3A). Taken together, LPA-induced neurite retraction in differentiated Neuro2a cells occurred through the tau-mediated signaling pathway, which involved an increase of GSK-3β and PKA activity, rather than CREB activation.
Moreover, we found that LPA-induced neurite retraction in differentiated Neuro2a cells was partially reduced by pretreatment with SB203580, a p38 MAPK inhibitor (Fig. 3A). Although SB203580 partially prevented LPA-stimulated neurite retraction, it failed to regulate tau phosphorylation (Fig. 3B). Thus, p38 MAPK activation is required in LPA-induced neurite retraction, but it is not dependent on tau phosphorylation in differentiated Neuro2a cells.
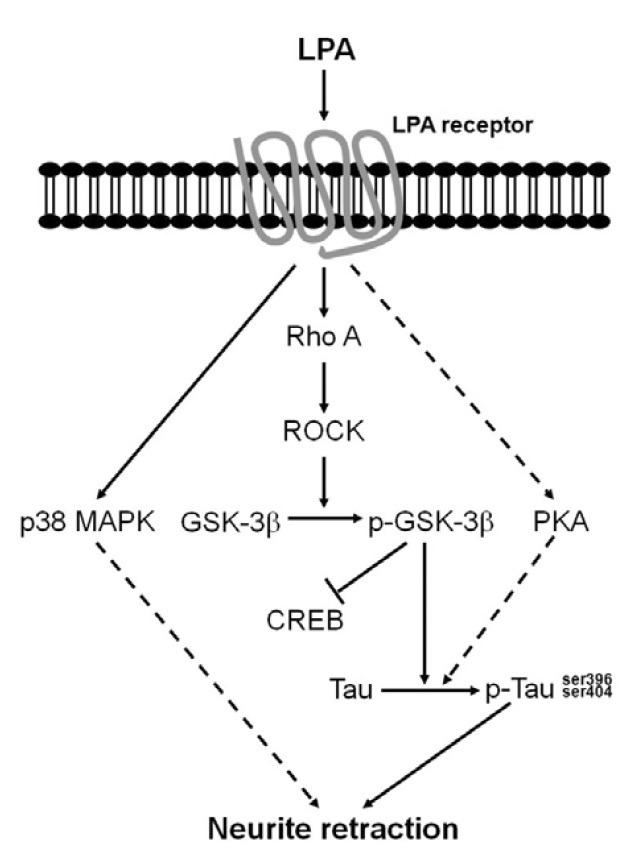
In summary, as shown in Fig. 5, we demonstrated tau phosphorylation and neurite retraction by extracellular LPA in differentiated Neuro2a cells. The LPA-induced neurite retraction in differentiated Neuro2a cells occurred through a tau-mediated signaling pathway, which involved an increase of GSK-3β and PKA rather than CREB activation. Moreover, the p38 MAPK signaling pathway-induced neurite retraction by LPA was not dependent on tau phosphorylation. Tau protein phosphorylation and neurite retraction have been implicated in the cascade of events associated with Alzheimer’s disease. An increased understanding of tau phosphorylation and neurite retraction may open up possibilities for Alzheimer’s disease therapeutic processes.
Fig. 5. A proposed model for the tau signaling cascade in LPA induced neurite retraction in differentiated Neuro2a cells. LPA increased tau phosphorylation, which was followed by activation of the GSK-3β signaling pathway. The activated GSK-3β and PKA signaling pathway mediated the hyperphosphorylation of tau, which subsequently resulted in differentiated Neuro2a cell neurite retraction.
Acknowledgments
We thank Dr. David Ginty and Dr. Hiroshi Ishiguro (Johns Hopkins Univ., USA) for providing us with the dominant-negative CREBs, K-CREB and A-CREB. We are grateful to Prof. Mark Curry for critical reading of the manuscript. This study was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A090792).
References
- 1.Anliker B., Chun J. Cell surface receptors in lysophospholipid signaling. Semin. Cell Dev. Biol. (2004);15:457–465. doi: 10.1016/j.semcdb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Bhat R.V., Shanley J., Correll M., Fieles W.E., Keith R.A., Scott C.W., Lee C.M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA. (2000);97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boer U., Eglins J., Krause D., Schnell S., Schofl C., Knepel W. Enhancement by lithium of cAMP-induced CRE/ CREB-directed gene transcription conferred by TORC on the CREB basic leucine zipper domain. Biochem. J. (2007);408:69–77. doi: 10.1042/BJ20070796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai X., Li M., Vrana J., Schaller M.D. Glycogen synthase kinase 3- and extracellular signal-regulated kinasedependent phosphorylation of paxillin regulates cytoskeletal rearrangement. Mol. Cell. Biol. (2006);26:2857–2868. doi: 10.1128/MCB.26.7.2857-2868.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. (1987);162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 6.Chun J. Lysophospholipids in the nervous system. Prostaglandins Other Lipid Mediat. (2005);77:46–51. doi: 10.1016/j.prostaglandins.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Chun J., Goetzl E.J., Hla T., Igarashi Y., Lynch K.R., Moolenaar W., Pyne S., Tigyi G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. (2002);54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- 8.Contos J.J., Ishii I., Chun J. Lysophosphatidic acid receptors. Mol. Pharmacol. (2000);58:1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 9.Evans D.B., Rank K.B., Bhattacharya K., Thomsen D.R., Gurney M.E., Sharma S.K. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. (2000);275:24977–24983. doi: 10.1074/jbc.M000808200. [DOI] [PubMed] [Google Scholar]
- 10.Fukushima N. LPA in neural cell development. J. Cell Biochem. (2004);92:993–1003. doi: 10.1002/jcb.20093. [DOI] [PubMed] [Google Scholar]
- 11.Fukushima N., Chun J. The LPA receptors. Prostaglandins. (2001);64:21–32. doi: 10.1016/s0090-6980(01)00105-8. [DOI] [PubMed] [Google Scholar]
- 12.Fukushima N., Weiner J.A., Kaushal D., Contos J.J., Rehen S.K., Kingsbury M.A., Kim K.Y., Chun J. Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Mol. Cell. Neurosci. (2002);20:271–282. doi: 10.1006/mcne.2002.1123. [DOI] [PubMed] [Google Scholar]
- 13.Ginty D.D., Glowacka D., Bader D.S., Hidaka H., Wagner J.A. Induction of immediate early genes by Ca2+ influx requires cAMP-dependent protein kinase in PC12 cells. J. Biol. Chem. (1991);266:17454–17458. [PubMed] [Google Scholar]
- 14.Goetzl E.J., An S. A subfamily of G protein-coupled cellular receptors for lysophospholipids and lysosphingolipids. Adv. Exp. Med. Biol. (1999);469:259–264. doi: 10.1007/978-1-4615-4793-8_38. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg S.G., Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA. (1990);87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimes C.A., Jope R.S. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J. Neurochem. (2001);78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartigan J.A., Johnson G.V. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J. Biol. Chem. (1999);274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 18.Hu X., Chen J., Wang L., Ivashkiv L.B. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J. Leukoc. Biol. (2007);82:237–243. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- 19.Jalink K., Eichholtz T., Postma F.R., van Corven E.J., Moolenaar W.H. Lysophosphatidic acid induces neuronal shape changes via a novel, receptor-mediated signaling pathway: similarity to thrombin action. Cell Growth Differ. (1993);4:247–255. [PubMed] [Google Scholar]
- 20.Klein P.S., Melton D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA. (1996);93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwon Y.J., Sun Y., Kim N.H., Huh S.O. Phosphorylation of CREB, a cyclic AMP responsive element binding protein, contributes partially to lysophosphatidic acid-induced fibroblast cell proliferation. Biochem. Biophys. Res. Commun. (2009);380:655–659. doi: 10.1016/j.bbrc.2009.01.159. [DOI] [PubMed] [Google Scholar]
- 22.Lee C.W., Nam J.S., Park Y.K., Choi H.K., Lee J.H., Kim N.H., Cho J., Song D.K., Suh H.W., Lee J., et al. Lysophosphatidic acid stimulates CREB through mitogen- and stressactivated protein kinase-1. Biochem. Biophys. Res. Commun. (2003);305:455–461. doi: 10.1016/s0006-291x(03)00790-3. [DOI] [PubMed] [Google Scholar]
- 23.Lee C.W., Rivera R., Gardell S., Dubin A.E., Chun J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. (2006);281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 24.Li X., Lu F., Tian Q., Yang Y., Wang Q., Wang J.Z. Activation of glycogen synthase kinase-3 induces Alzheimer-like tau hyperphosphorylation in rat hippocampus slices in culture. J. Neural Transm. (2006);113:93–102. doi: 10.1007/s00702-005-0303-7. [DOI] [PubMed] [Google Scholar]
- 25.Mandell J.W., Banker G.A. Microtubule-associated proteins, phosphorylation gradients, and the establishment of neuronal polarity. Perspect. Dev. Neurobiol. (1996);4:125–135. [PubMed] [Google Scholar]
- 26.Mao A.J., Bechberger J., Lidington D., Galipeau J., Laird D.W., Naus C.C. Neuronal differentiation and growth control of neuro-2a cells after retroviral gene delivery of connexin43. J. Biol. Chem. (2000);275:34407–34414. doi: 10.1074/jbc.M003917200. [DOI] [PubMed] [Google Scholar]
- 27.Moolenaar W.H. Bioactive lysophospholipids and their G protein-coupled receptors. Exp. Cell Res. (1999);253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- 28.Nam J.H., Shin D.H., Min J.E., Ye S.K., Jeon J.H., Kim S.J. Ca2+ signaling induced by sphingosine 1-phosphate and lysophosphatidic acid in mouse B cells. Mol. Cells. (2010);29:85–91. doi: 10.1007/s10059-010-0020-4. [DOI] [PubMed] [Google Scholar]
- 29.Saito S. Effects of lysophosphatidic acid on primary cultured chick neurons. Neurosci. Lett. (1997);229:73–76. doi: 10.1016/s0304-3940(97)00397-2. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez S., Sayas C.L., Lim F., Diaz-Nido J., Avila J., Wandosell F. The inhibition of phosphatidylinositol-3-kinase induces neurite retraction and activates GSK3. J. Neurochem. (2001);78:468–481. doi: 10.1046/j.1471-4159.2001.00453.x. [DOI] [PubMed] [Google Scholar]
- 31.Sayas C.L., Moreno-Flores M.T., Avila J., Wandosell F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J. Biol. Chem. (1999);274:37046–37052. doi: 10.1074/jbc.274.52.37046. [DOI] [PubMed] [Google Scholar]
- 32.Sayas C.L., Avila J., Wandosell F. Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim. Biophys. Acta. (2002a);1582:144–153. doi: 10.1016/s1388-1981(02)00149-x. [DOI] [PubMed] [Google Scholar]
- 33.Sayas C.L., Avila J., Wandosell F. Glycogen synthase kinase-3 is activated in neuronal cells by Galpha12 and Galpha13 by Rho-independent and Rho-dependent mechanisms. J. Neurosci. (2002b);22:6863–6875. doi: 10.1523/JNEUROSCI.22-16-06863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sayas C.L., Ariaens A., Ponsioen B., Moolenaar W.H. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1- mediated neurite retraction. Mol. Biol. Cell. (2006);17:1834–1844. doi: 10.1091/mbc.E05-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stambolic V., Ruel L., Woodgett J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. (1996);6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 36.Steiner B., Mandelkow E.M., Biernat J., Gustke N., Meyer H.E., Schmidt B., Mieskes G., Soling H.D., Drechsel D., Kirschner M.W., et al. Phosphorylation of microtubule-associated protein tau: identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. (1990);9:3539–3544. doi: 10.1002/j.1460-2075.1990.tb07563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka T., Iqbal K., Trenkner E., Liu D.J., Grundke-Iqbal I. Abnormally phosphorylated tau in SY5Y human neuroblastoma cells. FEBS Lett. (1995);360:5–9. doi: 10.1016/0014-5793(95)00061-d. [DOI] [PubMed] [Google Scholar]
- 38.Tatebayashi Y., Planel E., Chui D.H., Sato S., Miyasaka T., Sahara N., Murayama M., Kikuchi N., Yoshioka K., Rivka R., et al. c-jun N-terminal kinase hyperphos-phorylates R406W tau at the PHF-1 site during mitosis. FASEB J. (2006);20:762–764. doi: 10.1096/fj.05-4362fje. [DOI] [PubMed] [Google Scholar]
- 39.Thomson S., Clayton A.L., Hazzalin C.A., Rose S., Barratt M.J., Mahadevan L.C. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. (1999);18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tigyi G., Miledi R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. (1992);267:21360–21367. [PubMed] [Google Scholar]
- 41.Yanagida K., Ishii S., Hamano F., Noguchi K., Shimizu T. LPA4/p2y9/GPR23 mediates rho-dependent morphological changes in a rat neuronal cell line. J. Biol. Chem. (2007);282:5814–5824. doi: 10.1074/jbc.M610767200. [DOI] [PubMed] [Google Scholar]
- 42.Ye X., Ishii I., Kingsbury M.A., Chun J. Lysophosphatidic acid as a novel cell survival/apoptotic factor. Biochim. Biophys. Acta. (2002);1585:108–113. doi: 10.1016/s1388-1981(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 43.Zhong S., Jansen C., She Q.B., Goto H., Inagaki M., Bode A.M., Ma W.Y., Dong Z. Ultraviolet B-induced phosphorylation of histone H3 at serine 28 is mediated by MSK1. J. Biol. Chem. (2001);276:33213–33219. doi: 10.1074/jbc.M103973200. [DOI] [PubMed] [Google Scholar]