

Abstract
Hypoxia of skin is an important physiopathological process in many diseases, such as pressure ulcer, diabetic ulcer, and varicose ulcer. Although cellular injury and inflammation have been involved in hypoxia-induced dermatic injury, the underlying mechanisms remain largely unknown. This study was conducted to investigate the effects of cobalt chloride (CoCl2), a hypoxia-mimicking agent, on human skin keratinocytes (HaCaT cells) and to explore the possible molecular mechanisms. Exposure of HaCaT cells to CoCl2 reduced cell viability and caused overproduction of reactive oxygen species (ROS) and oversecretion of interleukin-6 (IL-6) and interleukin-8 (IL-8). Importantly, CoCl2 exposure elicited overexpression of cyclooxygenase-2 (COX-2) and phosphorylation of nuclear factor-kappa B (NF-κB) p65 subunit. Inhibition of COX-2 by NS-398, a selective inhibitor of COX-2, significantly repressed the cytotoxicity, as well as secretion of IL-6 and IL-8 induced by CoCl2. Inhibition of NF-κB by PDTC (a selective inhibitor of NF-κB) or genetic silencing of p65 by RNAi (Si-p65), attenuated not only the cytotoxicity and secretion of IL-6 and IL-8, but also overexpression of COX- 2 in CoCl2-treated HaCaT cells. Neutralizing anti-IL-6 or anti-IL-8 antibody statistically alleviated CoCl2-induced cytotoxicity in HaCaT cells. N-acetyl-L-cysteine (NAC), a well characterized ROS scavenger, obviously suppressed CoCl2-induced cytotoxicity in HaCaT cells, as well as secretion of IL-6 and IL-8. Additionally, NAC also repressed overexpression of COX-2 and phosphorylation of NF-κB p65 subunit induced by CoCl2 in HaCaT cells. In conclusion, our results demonstrated that oxidative stress mediates chemical hypoxia-induced injury and inflammatory response through activation of NF-κB–COX-2 pathway in HaCaT cells.
Keywords: chemical hypoxia, cyclooxygenase-2, inflammation, keratinocytes, nuclear factor-kappa B, oxidative stress
INTRODUCTION
Hypoxic injury of skin is one of common clinical events, such as pressure ulcer, diabetic ulcer, and varicose ulcer (Barcelos et al., 2009; Lazarides and Giannoukas, 2007; Mustoe et al., 2006). Previous studies showed that hypoxic injury of skin was intimately associated with oxidative stress and inflammatory responses (Houwing et al., 2000; Peirce et al., 2001). Mounting evidence suggested that cyclooxygenase-2 (COX-2) and its product prostaglandin E2 (PGE2) played a pivotal role in inflammatory response (Birnbaum et al., 2005; Chi and Kim, 2005). The roles of COX-2 overexpression are complicated and ambivalent in different models, mediating either cytotoxicity or cytoprotection (Booth et al., 2008; Carnieto et al., 2009; Ito et al., 2003; Kwak et al., 2010; Oh et al., 2010; Tu et al., 2009; Yun et al., 2009). Some studies indicated that inhibition of COX-2 activation attenuated ischemic neuronal injury, characterized by a decrease in cerebral infarction and CA1 hippocampal neuron death (Dore et al., 2003; Nakayama et al., 1998), while other reports showed that during cardiotoxicity induced by ischemia-reperfusion (I/R), up-regulation of COX-2 expression obviously possessed protective effects (Booth et al., 2008; Kwak et al., 2010). However, the role of COX-2 in hypoxic injury of skin remains unknown.
COX-2 induction is mainly due to activation of a transcriptional factor, nuclear factor-kappa B (NF-κB). NF-κB family is composed of five proteins: Rel A (p65), Rel B, c-Rel, NF-κB1 (p50), and NF-κB2 (p52), each of which may form homo- or heterodimers. NF-κB signaling pathway plays potent roles in general inflammatory response and cell death. In quiescent cells, NF-κB in cytosol is bound to its inhibitory molecule, IκBα protein. Upon stimulation, IκBα is phosphorylated by the upstream kinase, IκB kinase (IKK), which induces the polyubiquitination and degradation of IκB by proteases, subsequently leading to the phosphorylation and translocation of NF-κB complex into the nucleus (Chen et al., 1999). Activated NF-κB binds to specific DNA sequences and regulates the expression of its target genes. Reactive oxygen species (ROS) are important triggers to induce NF-κB activation in many in vivo and in vitro experiments, and antioxidants are consequently used to suppress NF-κB activation effectively (Huang et al., 2001; Pieper et al., 2001; Shrivastava and Aggarwal, 1999).
Numerous studies have demonstrated that cobalt chloride (CoCl2), as a chemical hypoxia agent, can mimic the hypoxic/ ischemic condition and induce the generation of ROS and cytotoxicity (Chen et al., 2010; Jung and Kim, 2004; Jung et al., 2007; Zou et al., 2001). Accumulating evidence showed that CoCl2 exposure induced inflammatory response, characterized by enhanced secretion of interleukin-6 (IL-6) and interleukin-8 (IL-8) in endotheliocyte and astrocytes (Kim et al., 2006; Montopoli et al., 2007; Schmalz et al., 2000). Keratinocytes, which represent the major component of skin, function as a protective barrier against exogenous insults. Upon activated by stimuli, keratinocytes produce a variety of cytokines that can affect the immune response and cell growth. Hence, keratinocytes are widely used to establish models of skin injury (Eidsmo et al., 2007).
To date, although oxidative stress and inflammatory response have been implicated in hypoxia-stimulated insults, the roles of both and the underlying mechanisms in chemical hypoxia- induced dermatic injury remain largely unknown. We therefore tested the hypothesis that NF-κB-COX-2 pathway mediated CoCl2-induced injury and inflammation and roles of the NF-κB-COX-2 pathway in CoCl2-induced injuries was ROSdependent.
MATERIALS AND METHODS
Materials
CoCl2, N-acetyl-L-cysteine (NAC), NS-398 (a selective inhibitor of COX-2), PDTC (a selective inhibitor of NF-κB) and 2′,7′- dichlorfluorescein-diacetate (DCFH-DA) were purchased from Sigma-Aldrich Corporation (USA). Cell Counter Kit-8 (CCK-8) was bought from Dojindo Laboratory (Japan). Dulbecco’s modified Eagle’s medium F12 (DMEM-F12 medium) and fetal bovine serum (FBS) were supplied by Gibco BRL (USA).
Cell culture
HaCaT cell, an immortalized keratinocyte, is derived from spontaneous transformation of human adult keratinocyte. It was generously provided by Professor Zeng (Department of Dermatology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, People’s Republic of China) and maintained in DMEM-F12 medium supplemented with 10% FBS at 37℃ under an atmosphere of 5% CO2 and 95% air.
Gene knockdown
Small interfering RNA (Si-RNA) against human NF-κB p65 subunit mRNA (GenBank accession no. L19067.1) was synthesized by GenePharma Co., Ltd (People’s Republic of China). The Si-RNA of p65 (Si-p65) and random non-coding RNA (Si- NC) were transfected into HaCaT cells using Lipofectamine 2000, according to the manufacturer’s instruction (Invitrogen, USA). Si-p65 and Si-NC (20 nmol/L) were incubated with the cells for 6 h in order to transfect into the cells. Efficiency of genetic silencing by Si-RNA was evaluated by Western blot assay.
Cell viability assay
Cell viability assay was performed according to the manufacturer’s instruction of CCK-8 kit. HaCaT cells were plated in 96- well plates at a density of 5,000 cells/well. When cells were grown to approximately 70-80% confluence, indicated conditioned mediums were added into 4 wells of each group and cocultured with HaCaT cells for 24 h. CCK-8 solution at a dilution of 1/10 with FBS-free DMEM/F12 was added to each well and the cells were incubated for 3 h at 37℃. Absorbance at 450 nm was measured with a microplate reader (Molecular Devices, USA). The mean optical density (OD) of four wells in each group was used to calculate percentage of cell viability as follows:
Cell viability (%) = (ODtreatment group- ODBlank) / (ODcontrol group- ODBlank) × 100
Experiments were performed in triplicate.
Cytokine ELISA
HaCaT cells were plated in 96-well plates. After the indicated treatments, levels of both IL-6 and IL-8 in the culture media were determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instruction (Boster biotech, LTD, People’s Republic of China). The amount of IL-6 and IL-8 in the culture medium was normalized with cell viability. The experiment was performed at least three times with similar outcomes.
Western blot assay
At the end of treatments, HaCaT cells were harvested and resuspended in ice-cold cell lysis solution and the homogenate was centrifuged at 10,000 g for 15 min at 4℃. After quantitated with BCA protein assay kit (Kangchen bio-tech, People’s Republic of China), proteins were separated by 12% SDS-PAGE. The proteins in the gel were transferred into polyvinylidene difluoride (PVDF) membranes. Blocked with 5% fat-free dry milk in TBS-T for 1 h at room temperature, the membranes were incubated with the primary antibodies specific to COX-2 (1:1,000 dilution; Bioworld Technology, USA), p65 (1: 1,000 dilution; Cell Signaling Technology, USA), phosphorylated (p)- p65 (1:500 dilution; Cell Signaling Technology, USA) or horseradish peroxidase (HRP)-conjugated β-actin (1:8,000 dilution; Kangchen Biotech, People’s Republic of China) with gentle agitation at 4℃ overnight followed by further incubation with HRP-conjugated secondary antibodies (1:4,000 dilution; Boster bio-tech, LTD, People’s Republic of China) for 1.5 h at room temperature. The immunoreactive signals were visualized using an enhanced chemiluminescence (ECL) detection system (Applygen Technologies, People’s Republic of China). For quantifying the protein expression, the X-ray films were scanned and analyzed with IMAGEJ 1.41o software [National Institutes of Health (NIH), USA].
Measurement of intracellular ROS generation
Intracellular ROS level was determined by fluorescent DCF derived from cell-permeable DCFH-DA. After treatment with indicated conditioned mediums, HaCaT cells were incubated with 10 μmol/L DCFH-DA solution at 37℃ for 30 min in the dark. DCF fluorescence was measured over the entire field of vision with a fluorescent microscope connected to an imaging system (BX50-FLA; Olympus, Japan). Mean fluorescence intensity of DCF from three random fields was analyzed with IMAGEJ 1.41o software.
Data analysis
Data are representatives of triplicate experiments and are expressed as the mean ± standard error (SE). Differences between groups were analyzed by one-way analysis of variance (ANOVA) and least significant difference (LSD)-t test with SPSS 13.0 (SPSS, USA). A P value of less than 0.05 was considered statistically significant.
RESULTS
Roles of oxidative stress in CoCl2-induced cytotoxicity in HaCaT cells
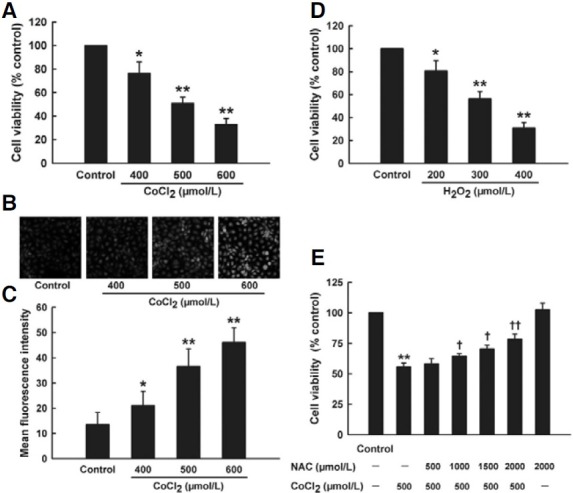
HaCaT cells were treated with chemical hypoxia-mimicking agent CoCl2 to set up an in vitro model of skin hypoxia. At the concentrations from 400 to 600 μmol/L, exposure of HaCaT cells to CoCl2 for 24 h significantly deceased cell viability in a dose-dependent manner (Fig. 1A). Notably, CoCl2-induced cytotoxicity was accompanied with overproduction of intracellular ROS. As shown in Fig. 1B, low amount of ROS was observed in quiescent cells (Fig. 1B control group). Exposure of HaCaT cells to CoCl2 from 400 to 600 μmol/L for 2 h concentration- dependently enhanced intracellular ROS content (Figs. 1B and 1C). Additionally, to examine the sensitivity of HaCaT cells to direct oxidative stress, HaCaT cells were treated with H2O2. Data of Fig. 1D showed that treatment with H2O2 (200-400 μmol/L) for 24 h also obviously reduced cell viability in HaCaT cells. In order to confirm whether intracellular ROS accumulation was involved in CoCl2-induced cytotoxicity, HaCaT cells were exposed to NAC (a ROS scavenger) prior to CoCl2 challenge. As shown in Fig. 1E, pretreatment with NAC (500-2000 μmol/L) for 1 h before exposure of HaCaT cells to 500 μmol/L CoCl2 concentration-dependently inhibited CoCl2-induced decrease in cell viability. These results suggest that CoCl2-induced cytotoxicity is associated with ROS overproduction in HaCaT cells.
Fig. 1. Effect of oxidative stress on cytotoxicity induced by CoCl2 in HaCaT cells. (A) Ha- CaT cells were treated with increasing concentrations of CoCl2 (400-600 μmol/L) for 24 h. Cell viability was measured by CCK-8 assay. (B) After treatment of HaCaT cells with indicated concentrations of CoCl2, intracellular ROS content was observed by DCFH-DA staining followed by photofluorography. (Control) quiescent HaCaT cells, (400, 500, and 600 μmol/L) HaCaT cells treated with CoCl2 at 400, 500, and 600 μmol/L for 2 h, respectively. (C) Quantitative analysis of the mean fluorescence intensity in B by IMAGEJ 1.41o software. (D) HaCaT cells were treated with increasing concentrations of H2O2 (200-400 μmol/L) for 24 h. (E) Pretreatment of HaCaT cells with NAC (500-2000 μmol/L) for 1 h before exposure to 500 μmol/L CoCl2 for 24 h or treatment with 2000 μmol/L NAC for 1 h followed by 24 h culture. Cell viability was measured by CCK-8 assay. Data are the mean ± SE (n = 3). *P < 0.05, **P < 0.01 compared with control group. + P < 0.05, ++ P < 0.01 compared with CoCl2 treatment group.
Roles of oxidative stress in inflammation induced by CoCl2 in HaCaT cells
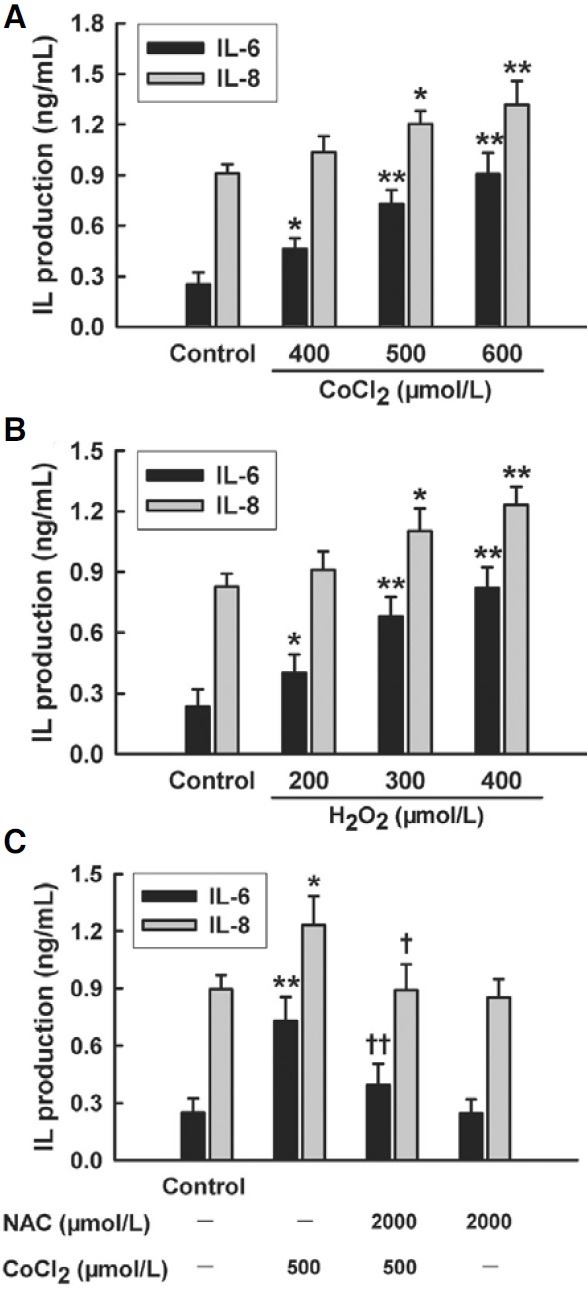
As shown in Fig. 2, treatment of HaCaT cells with CoCl2 (400- 600 μmol/L) for 24 h dose-dependently enhanced secretion of both IL-6 and IL-8 (Fig. 2A), revealing that CoCl2 elicited inflammatory response in HaCaT cells. Additionally, treatment of HaCaT cells with H2O2 (200-400 μmol/L) for 24 h also dosedependently enhanced secretion of both IL-6 and IL-8 (Fig. 2B). To further assess the role of oxidative stress in CoCl2-induced inflammatory response, HaCaT cells were preconditioned with 2000 μmol/L NAC for 1 h prior to exposure of cells to 500 μmol/L CoCl2. The results showed that the preconditioning with NAC obviously inhibited CoCl2-induced increase in both IL-6 and IL-8 secretion (Fig. 2C). These findings indicate that oxidative stress is involved in CoCl2-induced inflammatory response in HaCaT cells.
Fig. 2. Effect of oxidative stress on inflammation induced by CoCl2 in HaCaT cells. (A) HaCaT cells were treated with CoCl2 a t 400 μmol/L to 600 μmol/L for 24 h. (B) HaCaT cells were treated with H2O2 at 200 μmol/L to 400 μmol/L for 24 h. (C) HaCaT cells were pretreated with 2000 μmol/L NAC for 1 h before challenge of 500 μmol/L CoCl2 for 24 h. The levels of IL-6 and IL-8 in cell medium were measured by ELISA. Data are the mean ± SE (n = 3). *P < 0.05, **P < 0.01 compared with control group. + P < 0.05, ++ P < 0.01 compared with CoCl2 treatment group.
Roles of COX-2 in CoCl2-induced cytotoxicity and inflammation in HaCaT cells
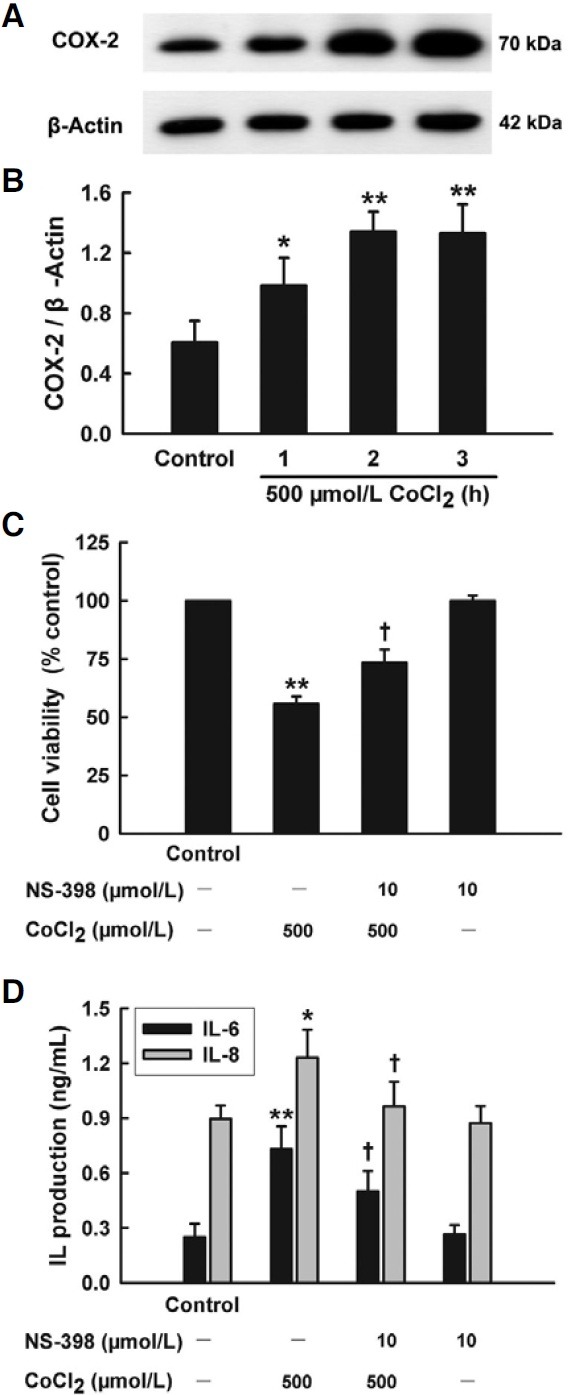
COX-2 is an inducible gene in process of oxidative stress and inflammatory response. As shown in Figs. 3A and 3B, treatment of HaCaT cells with 500 μmol/L CoCl2 for 1 to 3 h upregulated expression of COX-2 protein in a time-dependent manner. Pretreatment for 30 min with NS-398 (10 μmol/L), a selective inhibitor of COX-2, which alone did not affect on cell viability, reduced CoCl2-induced cytotoxicity in HaCaT cells (Fig. 3C). Furthermore, pretreatment with 10 μmol/L NS-398 for 30 min also attenuated both IL-6 and IL-8 secretion induced by 500 μmol/L CoCl2 (Fig. 3D).
Fig. 3. Roles of COX-2 in cytotoxicity and inflammation induced by CoCl2 in HaCaT cells. (A) HaCaT cells were treated with 500 μmol/L CoCl2 for the indicated time (1-3 h). COX-2 expression was detected by Western blot assay. (B) Densitometric analysis results from A. (C, D) HaCaT cells were treated with 500 μmol/L CoCl2 for 24 h in the presence or absence of pretreatment with 10 μmol/L NS-398 for 30 min. CCK-8 assay was applied to measure cell viability (C). ELISA was used to detect the levels of IL-6 and IL-8 in cell culture medium (D). Data are the mean ± SE (n = 3). *P < 0.05, **P < 0.01 compared with control group. + P < 0.05 compared with CoCl2 treatment group.
Roles of NF-κB p65 phosphorylation in COX-2-mediated cytotoxicity and inflammation
The transcription factor NF-κB plays an important role in regulating genes that govern the onset of oxidative stress and inflammatory response. To investigate whether phosphorylation of NF-κB p65 subunit (an essential step of NF-κB activation) participated in COX-2-mediated cytotoxicity and inflammation induced by CoCl2, the effect of CoCl2 on NF-κB p65 phosphorylation was observed. As shown in Figs. 4A and 4B, exposure of HaCaT cells to 500 μmol/L CoCl2 for 0.5 to 2 h time-dependently augmented expression of p-p65.
Fig. 4. NF-κB activation mediated CoCl2- induced COX-2 overexpression and injury in HaCaT cells. The expression of p-p65, p65 or COX-2 was tested by Western blot assay. (A) HaCaT cells were treated with 500 μmol/L CoCl2 for indicated time (0.5-2 h). (C) HaCat cells were co-cultured with small interfering RNA (Si-p65) or random non-coding RNA (Si-NC) for 6 h. (E) Ha- CaT cells were treated with 500 μmol/L CoCl2 for 2 h in the presence or absence of pretreatment with 10 μmol/L PDTC for 30 min or co-incubation with Si-p65 for 6 h. (B, D, and F) Densitometric analysis of the results from A, C, and E, respectively. (G, H) HaCaT cells were treated with 500 μmol/L CoCl2 for 24 h in the presence or absence of pretreatment with 10 μmol/L PDTC for 30 min or co-incubation with Sip65 for 6 h. CCK-8 assay was used to measure cell viability (G). ELISA was employed to detect the levels of IL-6 and IL-8 in cell culture medium (H). Data are the mean ± SE (n = 3). *P < 0.05, **P < 0.01 compared with control group. + P < 0.05 compared with CoCl2 treatment group.
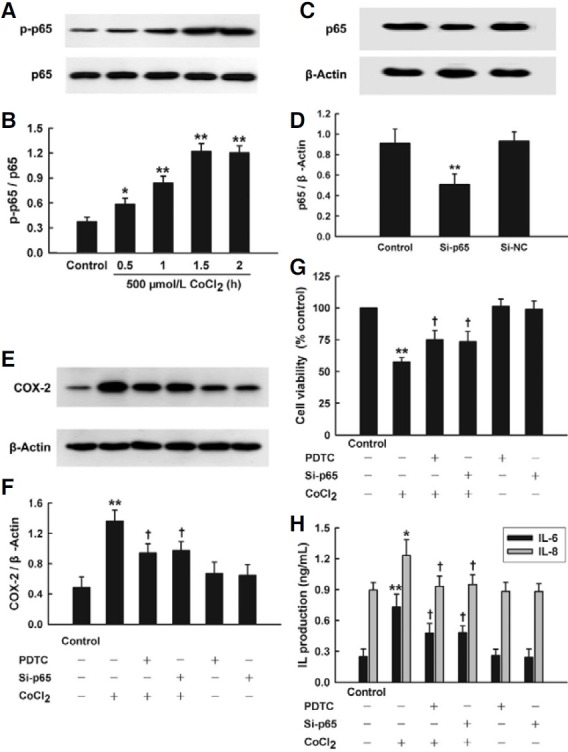
Importantly, pretreatment of HaCaT cells with 10 μmol/L PDTC (a selective inhibitor of NF-κB) for 30 min before CoCl2 treatment, significantly repressed CoCl2-induced COX-2 overexpression, which alone had no obvious effect on COX-2 expression (Figs. 4E and 4F). Additionally, after p65 expression was suppressed by small interfering RNA si-p65 (Figs. 4C and 4D), CoCl2-induced increase in COX-2 expression was markedly inhibited (Figs. 4E and 4F). The above findings of present study suggest that the activation of NF-κB at least partly mediates CoCl2-induced overexpression of COX-2.
Interestingly, pretreatment with 10 μmol/L PDTC for 30 min or si-p65 considerably protected HaCaT cells against CoCl2- induced injury, specifically enhancing cell viability (Fig. 4G), reducing both IL-6 and IL-8 secretion (Fig. 4H).
Roles of NF-κB-COX-2 pathway-mediated cytokine secretion in CoCl2-induced cytotoxicity in HaCaT cells
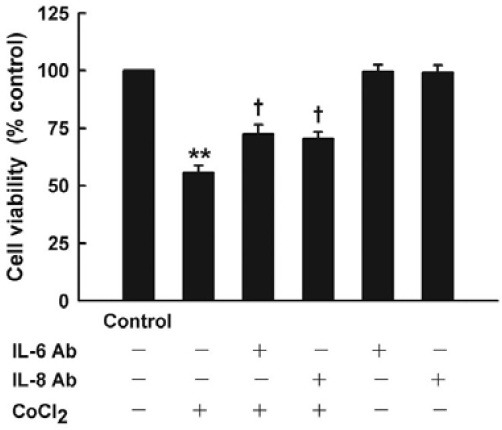
To clarify the role of CoCl2-induced inflammation in survival of cells, neutralizing anti-IL-6 or anti-IL-8 antibody was used to be co-incubated with CoCl2-treated HaCaT cells. Results (Fig. 5) of cell viability showed that co-incubation of neutralizing anti-IL-6 or anti-IL-8 antibody at 100 ng/ml for 24 h significantly protected HaCaT cells against CoCl2-induced cytotoxicity, increasing cell viability by 16.7 percent or 14.7 percent, respectively.
Fig. 5. Effect of neutralizing antibody to IL-6 or IL-8 on CoCl2- induced cytotoxicity in HaCaT cells. HaCaT cells were treated with 500 μmol/L CoCl2 for 24 h in the presence or absence of coincubation with neutralizing antibody (100 ng/ml) to IL-6 or IL-8. Cell viability was measured by CCK-8 assay. Data are the mean ± SE (n = 3). **P < 0.01 compared with control group. + P < 0.05 compared with CoCl2 treatment group.
Roles of oxidative stress in CoCl2-induced activation of NF-κB-COX-2 pathway
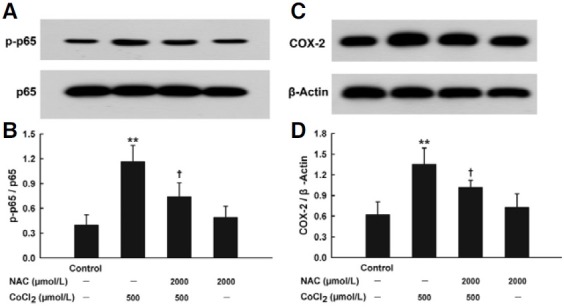
To explore whether CoCl2-induced activation of NF-κB-COX-2 pathway was ROS-dependent, the effects of NAC on expressions of both NF-κB p-p65 and COX-2 were detected. As shown in Figs. 6A and 6B, pretreatment with NAC (2000 μmol/L) for 1 h significantly blocked overexpression of NF-κB pp65 induced by 500 μmol/L CoCl2 treatment for 1.5 h. Similarly, exposure of HaCaT cells to 2000 μmol/L NAC for 1 h before CoCl2 treatment antagonized CoCl2-induced COX-2 overexpression (Figs. 6C and 6D). These results suggest that CoCl2- induced ROS trigger the overexpression of NF-κB p-p65 and COX-2.
Fig. 6. Roles of oxidative stress in CoCl2- inudced overexpression of NF-κB p-p65 and COX-2 in HaCaT cells. HaCaT cells were pretreated with 2000 μmol/L NAC for 1 h before exposure to 500 μmol/L CoCl2 for 1.5 h (A) or 2 h (C). Western blot assay was used to detect the expression of p-p65 or COX-2, respectively. (B, D) Densitometric analysis of the results from A and C, respectively. Data are the mean ± SE (n = 3). **P < 0.01 compared with control group. +P < 0.05 compared with CoCl2 treatment group.
DISCUSSION
At present, in order to mimic hypoxia, two methods are mainly used to produce experiment hypoxia as cellular hypoxia model. The effects of hypoxia are induced by either physical hypoxia, such as 1% O2, 5% CO2, and 94% N2 using the modular incubator chamber method, or a chemical hypoxia, hypoxia-mimicking agent CoCl2. In this study, we used CoCl2-treated HaCaT cells as a model of chemical hypoxia to explore the potential mechanisms underlying hypoxia-induced dermatic injury and inflammatory response.
In agreement with the previous study (Ermolli et al., 2001), exposure of HaCaT cells to CoCl2 dose-dependently attenuated cell viability. Interestingly, we found that treatment of HaCaT cells with CoCl2 enhanced generation of ROS and oversecretion of both IL-6 and IL-8, which was not reported by Ermolli and his coworkers (Ermolli et al., 2001). Similarly, CoCl2 induced overproduction of ROS in PC12 cells (Jung and Kim, 2004; Zou et al., 2001) and H9c2 cells (Chen et al., 2010). In human umbilical vein endothelial cells (HUVECs), CoCl2 also induced inflammatory response by increasing expression of vascular cell adhesion molecule-1 (VCAM-1) and decreasing expression of platelet endothelial cell adhesion molecule-1 (PECAM-1) (Montopoli et al., 2007). Furthermore, Wagner and his colleagues demonstrated that treatment with CoCl2 promoted IL-6 and IL-8 transcriptions in HUVECs (Wagner et al., 1998). These studies (Chen et al., 2010; Jung and Kim, 2004; Montopoli et al., 2007; Wagner et al., 1998; Zou et al., 2001) provide support for our research. Since both oxidative stress and inflammatory response are main pathological processes in the dermatic injury under ischemia or hypoxia condition, our results mentioned above suggest that CoCl2-treated HaCaT cells may serve as a simple and useful in vitro model for exploring the mechanisms underlying the hypoxia-linked damages of skin.
Accumulating evidence verified that COX-2 is responsible for the development of skin necrosis and inflammatory responses. Some reports showed that pressure or I/R treatment increased expression of COX-2 (Gomez-Pinilla et al., 2010; Tsutakawa et al., 2009). Additionally, in ultraviolet light B (UVB)-irradiated HaCaT cells COX-2 expression was obviously enhanced (Fernau et al., 2010). In accordance with previous data (Chi and Kim, 2005; Fernau et al., 2010; Tsutakawa et al., 2009), we observed that exposure of HaCaT cells to CoCl2 upregulated COX-2 expression. Furthermore, inhibition of COX-2 by NS-398, a selective inhibitor of COX-2, significantly attenuated CoCl2- induced cytotoxicity and secretion of both IL-6 and IL-8, indicating involvement of COX-2 in CoCl2-induced cytotoxicity and inflammation in HaCaT cells. Consistent with our results, it was reported that wogonin derived from traditional Chinese medicine, Huang-Qin, attenuated I/R-induced dermatic injury partly by inhibition of COX-2 (Chi and Kim, 2005). Inhibition of COX-2 by NS-398 ameliorated cytokine immunologic imbalances and decreased liver injury in septic rats (Li et al., 2009). However, a study showed that activation of COX-2/PG signaling pathway was involved in atorvastatin-induced cardioprotection (Birnbaum et al., 2005). The reasons underlying the detrimental versus cytoprotective effects of COX-2 may be complicated. One explanation for the different roles of COX-2 may be associated with tissue-specific regulatory mechanisms.
NF-κB is an inducible transcription factor and a positive regulator of COX-2 expression in response to various cytokines and growth factors (Huang et al., 2001; Kang et al., 2006; Liu et al., 2003). NF-κB exists in a homo- or heterodimeric form of members of Rel protein family. The most abundant form is composed of p50 and p65. It has been shown that CoCl2 activated the translocation of NF-κB into the cell nucleus and enhanced its binding to a NF-κB consensus sequence in endothelial cells (Wagner et al., 1997). In agreement with the previous studies (Huang et al., 2001; Kang et al., 2006; Liu et al., 2003; Wagner et al., 1997), we found that exposure of HaCaT cells to CoCl2 elevated phosphorylation of p65 subunit, which is a key step of NF-κB activation. Notably, inhibition of NF-κB by PDTC (a selective inhibitor of NF-κB) or genetic silencing of p65 by RNAi (Si-p65) significantly blocked CoCl2-induced cytotoxicity and oversecretion of both IL-6 and IL-8, revealing that NF-κB activation plays an important role in CoCl2-induced cytotoxicity and inflammatory response. Further study demonstrated that NF-κB had a positive effect on COX-2 expression in response to exposure of HaCaT cells to CoCl2. Our findings showed that both PDTC and Si-p65 markedly inhibited overexpression of COX-2 induced by CoCl2 treatment. Based on the above results of present study, we provide novel evidence that NF-κB-COX-2 pathway plays a critical role in CoCl2-induced injury and inflammatory response in HaCaT cells.
In addition, we observed that neutralizing anti-IL-6 or anti-IL-8 antibody obviously alleviated CoCl2-induced cytotoxicity, leading to an increase in cell viability, revealing involvement of both IL-6 and IL-8 in cytotoxicity induced by CoCl2 in HaCaT cells. Similarly, a more recent study showed that sulforaphane (4- methylsulfinylbutyl isothiocyanate, SF), which is found in cruciferous vegetables and has anti-inflammatory effects (Talalay et al., 2007), can suppress UVB-induced reduction of cell viability and production of IL-6 in HaCaT cells (Shibata et al., 2010).
Recently, it was reported that ROS generated by physical hypoxia or CoCl2 enhanced expression of vasoactive intestinal contractor (VIC) / endothelin-2 (ET-2), one of hypoxia-induced gene (Grimshaw et al., 2002; Kotake-Nara and Saida, 2007; Kotake-Nara et al., 2005). To explore the roles of ROS in NF- κB-COX-2 pathway-mediated inflammatory response and cytotoxicity induced by CoCl2 in HaCaT cells, we observed the effects of NAC, a well characterized ROS scavenger. The findings of present study showed that pretreatment with NAC protected HaCaT cells against CoCl2-induced cytotoxicity and inflammation, evidenced by increased cell viability and reduced secretion of IL-6 and IL-8. Additionally, exposure of HaCaT cells to H2O2 also induced cytotoxicity and oversecretion of IL-6 and IL-8. Taken together, these findings suggest that oxidative stress plays pivotal roles in cellular injuries and inflammatory response induced by CoCl2. Importantly, NAC pretreatment significantly inhibited phosphorylation of NF-κB p65 subunit and overexpression of COX-2, indicating that ROS mediate CoCl2- induced injury and inflammatory response at least partly through activation of NF-κB-COX-2 pathway in HaCaT cells.
In summary, the present study provided novel evidence that NF-κB-COX-2 pathway mediates CoCl2-induced injury and inflammatory response and that activation of NF-κB-COX-2 pathway is ROS-dependent in HaCaT cells. Knowledge of the signal mechanism of NF-κB-COX-2 pathway might provide clue for the development of new therapeutic strategy for hypoxiainduced dermatic insults.
Acknowledgments
This work was supported by Science and Technology Planning Project of Guangdong Province in China (2010B080701035, 2008B080703053).
References
- 1.Barcelos L.S., Duplaa C., Krankel N., Graiani G., Invernici G., Katare R., Siragusa M., Meloni M., Campesi I., Monica M., et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ. Res. (2009);104:1095–1102. doi: 10.1161/CIRCRESAHA.108.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birnbaum Y., Ye Y., Rosanio S., Tavackoli S., Hu Z.Y., Schwarz E.R., Uretsky B.F. Prostaglandins mediate the cardioprotective effects of atorvastatin against ischemia-reperfusion injury. Cardiovasc. Res. (2005);65:345–355. doi: 10.1016/j.cardiores.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Booth E.A., Flint R.R., Lucas K.L., Knittel A.K., Lucchesi B.R. Estrogen protects the heart from ischemia-reperfusion injury via COX-2-derived PGI2. J. Cardiovasc. Pharmacol. (2008);52:228–235. doi: 10.1097/FJC.0b013e3181824d59. [DOI] [PubMed] [Google Scholar]
- 4.Carnieto A., Jr., Dourado P.M., Luz P.L., Chagas A.C. Selective cyclooxygenase-2 inhibition protects against myocardial damage in experimental acute ischemia. Clinics (Sao Paulo) (2009);64:245–252. doi: 10.1590/S1807-59322009000300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F., Castranova V., Shi X., Demers L.M. New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin. Chem. (1999);45:7–17. [PubMed] [Google Scholar]
- 6.Chen S.L., Yang C.T., Yang Z.L., Guo R.X., Meng J.L., Cui Y., Lan A.P., Chen P.X., Feng J.Q. Hydrogen sulphide protects H9c2 cells against chemical hypoxia-induced injury. Clin. Exp. Pharmacol. Physiol. (2010);37:316–321. doi: 10.1111/j.1440-1681.2009.05289.x. [DOI] [PubMed] [Google Scholar]
- 7.Chi Y.S., Kim H.P. Suppression of cyclooxygenase-2 expression of skin fibroblasts by wogonin, a plant flavone from Scutellaria radix. Prostaglandins Leukot. Essent. Fatty Acids. (2005);72:59–66. doi: 10.1016/j.plefa.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Dore S., Otsuka T., Mito T., Sugo N., Hand T., Wu L., Hurn P. D., Traystman R.J., Andreasson K. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann. Neurol. (2003);54:155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- 9.Eidsmo L., Fluur C., Rethi B., Eriksson Ygberg S., Ruffin N., De Milito A., Akuffo H., Chiodi F. FasL and TRAIL induce epidermal apoptosis and skin ulceration upon exposure to Leishmania major. Am. J. Pathol. (2007);170:227–239. doi: 10.2353/ajpath.2007.060068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ermolli M., Menne C., Pozzi G., Serra M.A., Clerici L.A. Nickel, cobalt and chromium-induced cytotoxicity and intracellular accumulation in human HaCaT keratinocytes. Toxicology. (2001);159:23–31. doi: 10.1016/s0300-483x(00)00373-5. [DOI] [PubMed] [Google Scholar]
- 11.Fernau N.S., Fugmann D., Leyendecker M., Reimann K., Grether- Beck S., Galban S., Ale-Agha N., Krutmann J., Klotz L.O. Role of HuR and p38MAPK in ultraviolet B-induced posttranscriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT. J. Biol. Chem. (2010);285:3896–3904. doi: 10.1074/jbc.M109.081430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez-Pinilla P.J., Camello P.J., Tresguerres J.A., Pozo M.J. Tempol protects the gallbladder against ischemia/ reperfusion. J. Physiol. Biochem. (2010);66:161–172. doi: 10.1007/s13105-010-0021-y. [DOI] [PubMed] [Google Scholar]
- 13.Grimshaw M.J., Naylor S., Balkwill F.R. Endothelin-2 is a hypoxia-induced autocrine survival factor for breast tumor cells. Mol. Cancer Ther. (2002);1:1273–1281. [PubMed] [Google Scholar]
- 14.Houwing R., Overgoor M., Kon M., Jansen G., van Asbeck B.S., Haalboom J.R. Pressure-induced skin lesions in pigs: reperfusion injury and the effects of vitamin E. J. Wound Care. (2000);9:36–40. doi: 10.12968/jowc.2000.9.1.25939. [DOI] [PubMed] [Google Scholar]
- 15.Huang C.Y., Fujimura M., Noshita N., Chang Y.Y., Chan P.H. SOD1 down-regulates NF-kappaB and c-Myc expression in mice after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. (2001);21:163–173. doi: 10.1097/00004647-200102000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Ito Y., Katagiri H., Ishii K., Kakita A., Hayashi I., Majima M. Effects of selective cyclooxygenase inhibitors on ischemia/reperfusion-induced hepatic microcirculatory dysfunction in mice. Eur. Surg. Res. (2003);35:408–416. doi: 10.1159/000072174. [DOI] [PubMed] [Google Scholar]
- 17.Jung J.Y., Kim W.J. Involvement of mitochondrial- and Fas-mediated dual mechanism in CoCl2-induced apoptosis of rat PC12 cells. Neurosci. Lett. (2004);371:85–90. doi: 10.1016/j.neulet.2004.06.069. [DOI] [PubMed] [Google Scholar]
- 18.Jung J.Y., Mo H.C., Yang K.H., Jeong Y.J., Yoo H.G., Choi N.K., Oh W.M., Oh H.K., Kim S.H., Lee J.H., et al. Inhibition by epigallocatechin gallate of CoCl2-induced apoptosis in rat PC12 cells. Life Sci. (2007);80:1355–1363. doi: 10.1016/j.lfs.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 19.Kang Y.J., Wingerd B.A., Arakawa T., Smith W.L. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. (2006);177:8111–8122. doi: 10.4049/jimmunol.177.11.8111. [DOI] [PubMed] [Google Scholar]
- 20.Kim K.S., Rajagopal V., Gonsalves C., Johnson C., Kalra V.K. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. (2006);177:7211–7224. doi: 10.4049/jimmunol.177.10.7211. [DOI] [PubMed] [Google Scholar]
- 21.Kotake-Nara E., Saida K. Characterization of CoCl2- induced reactive oxygen species (ROS): inductions of neurite outgrowth and endothelin-2/vasoactive intestinal contractor in PC12 cells by CoCl2 are ROS dependent, but those by MnCl2 are not. Neurosci. Lett. (2007);422:223–227. doi: 10.1016/j.neulet.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Kotake-Nara E., Takizawa S., Quan J., Wang H., Saida K. Cobalt chloride induces neurite outgrowth in rat pheochromocytoma PC12 cells through regulation of endothelin-2/vasoactive intestinal contractor. J. Neurosci. Res. (2005);81:563–571. doi: 10.1002/jnr.20568. [DOI] [PubMed] [Google Scholar]
- 23.Kwak H.J., Park K.M., Choi H.E., Lim H.J., Park J.H., Park H.Y. The cardioprotective effects of zileuton, a 5-lipoxygenase inhibitor, are mediated by COX-2 via activation of PKC delta. Cell. Signal. (2010);22:80–87. doi: 10.1016/j.cellsig.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Lazarides M.K., Giannoukas A.D. The role of hemodynamic measurements in the management of venous and ischemic ulcers. Int. J. Low. Extrem. Wounds. (2007);6:254–261. doi: 10.1177/1534734607306878. [DOI] [PubMed] [Google Scholar]
- 25.Li B., Li Y.M., Li X., Shi B., He M.Y., Zhu X.L., Zhou W.C., Wachtel M.S., Frezza E. COX-2 inhibition improves immune system homeostasis and decreases liver damage in septic rats. J. Surg. Res. (2009);157:43–47. doi: 10.1016/j.jss.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Liu W., Reinmuth N., Stoeltzing O., Parikh A.A., Tellez C., Williams S., Jung Y.D., Fan F., Takeda A., Akagi M., et al. Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. Cancer Res. (2003);63:3632–3636. [PubMed] [Google Scholar]
- 27.Montopoli M., Froldi G., Comelli M.C., Prosdocimi M., Caparrotta L. Aescin protection of human vascular endothelial cells exposed to cobalt chloride mimicked hypoxia and inflammatory stimuli. Planta Med. (2007);73:285–288. doi: 10.1055/s-2007-967118. [DOI] [PubMed] [Google Scholar]
- 28.Mustoe T.A., O’Shaughnessy K., Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast. Reconstr. Surg. (2006);117:35S–41S. doi: 10.1097/01.prs.0000225431.63010.1b. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama M., Uchimura K., Zhu R.L., Nagayama T., Rose M.E., Stetler R.A., Isakson P.C., Chen J., Graham S.H. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc. Natl. Acad. Sci. USA. (1998);95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh Y.T., Lee J.Y., Lee J., Lee J.H., Kim J.E., Ha J., Kang I. Oleamide suppresses lipopolysaccharide-induced expression of iNOS and COX-2 through inhibition of NF-kappaB activation in BV2 murine microglial cells. Neurosci. Lett. (2010);474:148–153. doi: 10.1016/j.neulet.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Peirce S.M., Skalak T.C., Rieger J.M., Macdonald T.L., Linden J. Selective A(2A) adenosine receptor activation reduces skin pressure ulcer formation and inflammation. Am. J. Physiol. Heart Circ. Physiol. (2001);281:H67–74. doi: 10.1152/ajpheart.2001.281.1.H67. [DOI] [PubMed] [Google Scholar]
- 32.Pieper G.M., Olds C., Hilton G., Lindholm P.F., Adams M.B., Roza A.M. Antioxidant treatment inhibits activation of myocardial nuclear factor kappa B and inhibits nitrosylation of myocardial heme protein in cardiac transplant rejection. Antioxid. Redox Signal. (2001);3:81–88. doi: 10.1089/152308601750100542. [DOI] [PubMed] [Google Scholar]
- 33.Schmalz G., Schweikl H., Hiller K.A. Release of prostaglandin E2, IL-6 and IL-8 from human oral epithelial culture models after exposure to compounds of dental materials. Eur. J. Oral Sci. (2000);108:442–448. doi: 10.1034/j.1600-0722.2000.108005442.x. [DOI] [PubMed] [Google Scholar]
- 34.Shibata A., Nakagawa K., Yamanoi H., Tsuduki T., Sookwong P., Higuchi O., Kimura F., Miyazawa T. Sulforaphane suppresses ultraviolet B-induced inflammation in HaCaT keratinocytes and HR-1 hairless mice. J. Nutr. Biochem. (2010);21:702–709. doi: 10.1016/j.jnutbio.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Shrivastava A., Aggarwal B.B. Antioxidants differentially regulate activation of nuclear factor-kappa B, activator protein-1, c-jun amino-terminal kinases, and apoptosis induced by tumor necrosis factor: evidence that JNK and NF-kappa B activation are not linked to apoptosis. Antioxid. Redox Signal. (1999);1:181–191. doi: 10.1089/ars.1999.1.2-181. [DOI] [PubMed] [Google Scholar]
- 36.Talalay P., Fahey J.W., Healy Z.R., Wehage S.L., Benedict A.L., Min C., Dinkova-Kostova A.T. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc. Natl. Acad. Sci. USA. (2007);104:17500–17505. doi: 10.1073/pnas.0708710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsutakawa S., Kobayashi D., Kusama M., Moriya T., Nakahata N. Nicotine enhances skin necrosis and expression of inflammatory mediators in a rat pressure ulcer model. Br. J. Dermatol. (2009);161:1020–1027. doi: 10.1111/j.1365-2133.2009.09349.x. [DOI] [PubMed] [Google Scholar]
- 38.Tu X.K., Yang W.Z., Shi S.S., Wang C.H., Chen C.M. Neuroprotective effect of baicalin in a rat model of permanent focal cerebral ischemia. Neurochem. Res. (2009);34:1626–1634. doi: 10.1007/s11064-009-9953-4. [DOI] [PubMed] [Google Scholar]
- 39.Wagner M., Klein C.L., Kleinert H., Euchenhofer C., Forstermann U., Kirkpatrick C.J. Heavy metal ion induction of adhesion molecules and cytokines in human endothelial cells: the role of NF-kappaB, I kappaB-alpha and AP-1. Pathobiology. (1997);65:241–252. doi: 10.1159/000164135. [DOI] [PubMed] [Google Scholar]
- 40.Wagner M., Klein C.L., van Kooten T.G., Kirkpatrick C.J. Mechanisms of cell activation by heavy metal ions. J. Biomed. Mater. Res. (1998);42:443–452. doi: 10.1002/(sici)1097-4636(19981205)42:3<443::aid-jbm14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 41.Yun Y., Duan W.G., Chen P., Wu H.X., Shen Z.Q., Qian Z.Y., Wang D.H. Down-regulation of cyclooxygenase-2 is involved in ischemic postconditioning protection against renal ischemia reperfusion injury in rats. Transplant. Proc. (2009);41:3585–3589. doi: 10.1016/j.transproceed.2009.06.209. [DOI] [PubMed] [Google Scholar]
- 42.Zou W., Yan M., Xu W., Huo H., Sun L., Zheng Z., Liu X. Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. J. Neurosci. Res. (2001);64:646–653. doi: 10.1002/jnr.1118. [DOI] [PubMed] [Google Scholar]