

Abstract
We have previously shown that Ras mediates NO-induced BNIP3 expression via the MEK-ERK-HIF-1 pathway in mouse macrophages, and that NO-induced death results at least in part from the induction of BNIP3. In the present study, we describe another aspect of Ras regulation of BNIP3 expression in pancreatic cancer cells. Human BNIP3 promoter-driven luciferase activity was efficiently induced by activated Ras in AsPC-1, Miapaca-2, PK-1 and PANC-1 cells. However, expression of endogenous BNIP3 was not induced, and BNIP3 up-regulation by hypoxia was also inhibited. Treatment of the cells with the DNMT inhibitor, 5- aza-2-deoxycytidine, restored BNIP3 induction, indicating that DNA methylation of the BNIP3 promoter was responsible for the inhibition of BNIP3 induction. Furthermore, inhibition of the MEK pathway with U0126 reduced DNMT1 expression, but not that of DNMT3a and 3b, and restored the hypoxia-inducibility of BNIP3, suggesting that the DNA methylation of the BNIP3 promoter was mediated by DNMT1 via the MEK pathway.
Keywords: BNIP3, DNMT1, MEK, methylation, Ras
INTRODUCTION
Pancreatic cancers are highly resistant to chemical and radiation therapy and have an extremely poor prognosis (Ghaneh et al., 2007). The 80-95% of pancreatic cancers have functional mutations of p16INK4A; more than 50% have alteration in the p53 locus and 75-90% have dominant mutations in K-ras. Several intracellular signaling pathways are activated in pancreatic cancers, including the Jak/STAT, PI3K, EGFR and Ras-MEK pathways, and it has been a challenge to develop anti-cancer agents targeting these pathways.
Bcl-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3) was discovered in a yeast two-hybrid screen for proteins that interact with adenovirus E1B 19 kDa, a homologue of Bcl-2 (Boyd, 1994; Boyd et al., 1994; Lee and Paik, 2006). BNIP3 is a pro-apoptotic protein that contains a single Bcl-2 homology 3 (BH3) domain and thus belongs to the BH3-only subfamily (Chen et al., 1997; Yasuda et al., 1998). It is present in the outer mitochondrial membrane with its N-terminus in the cytoplasm and its C-terminus inside the mitochondrion. Overexpression of BNIP3 leads to opening of the mitochondrial permeability transition pore (PTP) thereby abolishing the proton electrochemical gradient, and this is followed by chromatin condensation and DNA fragmentation (Kim et al., 2002; Vande Velde et al., 2000). It has been proposed that BNIP3 induces a novel necrosis-like form of cell death. This is independent of caspases and nuclear translocation of AIF, a mitochondrial flavoprotein. It is not a ubiquitous apoptotic protein. Under physiological conditions, BNIP3 protein is present at a low level in skeletal muscle and brain, and its expression can be induced in both normal and cancer tissues that experience hypoxia or hypoxia-like conditions. Expression is markedly up-regulated in breast and non-small cell lung cancers and down-regulated in almost all cases of pancreatic cancer and in a significant proportion of hematopoietic, colorectal, and gastric cancers (Giatromanolaki et al., 2004; Murai et al., 2005a; 2005b; Okami et al., 2004; Sowter et al., 2003).
The promoter of BNIP3 is located within a CpG island and is methylated in most pancreatic cancer cell lines (Abe et al., 2005; Akada et al., 2005; Erkan et al., 2005). Restoration of BNIP3 expression by the methyltransferase inhibitor, 5-aza-2- deoxycytidine, leads to the death of pancreatic cancer cells in response to hypoxia. Methylation is often considered a process blocking the expression of tumor suppressor genes. Therefore silencing of BNIP3 by methylation has been suggested to be essential for the survival of pancreatic cancer cells under hypoxic condition. Hypoxia-induced expression of BNIP3 is also impaired in other cancer cell lines, although there are differences in the extent of methylation and silencing (de Angelis et al., 2004; Kennedy et al., 2000). Murai and colleagues detected methylation of BNIP3 in 66% of primary colorectal and 49% of gastric cancers, 15% of acute lymphocytic and 17% of acute myelogenous leukaemias, and about 21% of multiple myelomas, but not in adjacent normal tissues (Murai et al., 2005a; 2005b). Therefore, inactivation of BNIP3 appears to play an important role in the progression of various types of cancers. It is also thought to be one of the factors affecting the aggressiveness of cancers and their resistance to treatment.
We previously showed that nitric oxide treatment led to activation of the BNIP3 promoter in RAW264.7 murine macrophage cells under normoxic conditions (Yook et al., 2004) and that BNIP3 induction by nitric oxide was mediated by Ras, followed by MEK, ERK and HIF-1 (An et al., 2006). Ras activation also induced cell death in these macrophages, and antisense silencing of BNIP3 expression greatly reduced death, suggesting that it resulted from up-regulation of BNIP3. Here we report that human BNIP3 promoter activity was efficiently induced by Ras in pancreatic cancers, but not the expression of endogenous BNIP3. Treatment of the cells with the DNMT inhibitor, 5- aza-2 deoxycytidine, restored BNIP3 induction, suggesting that methylation of the BNIP3 promoter was responsible for the loss of BNIP3 induction. Moreover, inhibition of the MEK pathway with U0126 reduced DNMT1 expression and restored the hypoxia- inducibility of BNIP3, indicating that DNA methylation of the BNIP3 promoter was due to DNMT1.
MATERIALS AND METHODS
Cell culture and reagents
Pancreatic cancer cell lines AsPC-1, Miapaca-2, PK-1, PANC-1, Hs766T and CFPAC-1 were cultured in Dulbecco’s modified Eagle’s medium (Welgene) supplemented with 10% FBS (JRS) and antibiotics/antimycotics (Invitrogen). Cells were grown at 37℃ in a humidified atmosphere with 5% CO2. For hypoxia treatment, they were exposed to 1% oxygen (5% CO2 and 94% N2) in a hypoxia chamber (Billups-Rothenberg Inc., USA). 5- aza-2-deoxycytidine (A3656) was purchased from Sigma (USA), and Go6976 (365250) and U0126 (662005) were from Calbiochem (USA).
Construction of expression plasmids
A putative human Bnip3 promoter fragment (-753 to -1 bp; +1 indicates the translation start site) was amplified with forward primer 5′-AGATCTCCCGGCGGGGCGGGCAAAGA(Bgl2)-3′ and reverse primer 5′-CCATGGCGCCAGAGGGCAACTGCG (Nco1)-3′, using human genomic DNA as template, and the product was cloned upstream of luc+ in pGL3-Basic vector (Promega, USA). Putative BNIP3 promoter sequences for transcription factor binding sites were identified with the MatInspector program of Genomatix software (http://www.genomatix.de). Sequencing of the human BNIP3 promoter revealed two HRE (CACGT) sites at -249 bp and -613 bp, respectively, relative to the translation start site. The pcDNA3.1(+)-Ras-Q61L, pcDNA3.1 (+)-Ras-S17N vectors were constructed by cloning the corresponding inserts into the BamHI site of pcDNA3.1(+) vector (An et al., 2006).
Transient transfection and dual-luciferase assays
Transfections were performed with Transfectin™ lipid reagent (Bio-Rad). The 2 μg of reporter plasmids and 0.3 μg of pRL-TK (an internal control plasmid expressing the Renilla luciferase gene; Promega) were used in the transfections. Firefly and Renilla luciferase activities in cell lysates were measured in succession using the Dual-Luciferase reporter assay system (Promega) with a VICTOR3 multilabel reader (Perkin Elmer Life Sciences).
Immunoblotting
Cultured cells were lysed in lysis buffer (1 mM Tris, 5 mM NaCl, 0.5 mM EDTA, 10% NP-40, 100 mM PMSF). Lysates were incubated on ice for 15 min and cleared by centrifugation. Aliquots of protein were resolved on SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore, USA) in a Mighty Small Transphor unit (Amersham Biosciences). Anti- DNMT1 (sc-10221), anti-DNMT3a (sc-20703) and anti-DNMT3b (sc-20704) antibodies were purchased from Santa Cruz Biotechnology, (USA). Anti-BNIP3 (ab10433) was from Abcam plc. (UK) and anti-β-actin monoclonal antibody (A5060) was from Sigma-Aldrich (USA).
RT-PCR analysis
Total RNA was reverse transcribed with M-MLV Reverse Transcriptase (Promega, USA), and semi-quantitative PCR was performed with the following primer pairs: human BNIP3 forward 5′-CCCGGGATGCAGGAGGAGA-3′, reverse 5′-CGTGC GCTTCGGGTGTTTA-3′; β-actin forward 5′-GGAGTCCTGT GGCATCCACG-3′, reverse 5′-CTAGAAGCATTTGCGGTGGA- 3′. PCR products were resolved by electrophoresis on 1% agarose gels followed by ethidium bromide staining. All reactions were performed in duplicate.
Ras activity
Ras activity was measured with a Ras Activation Assay Kit (Upstate) that detects Ras bound to the Ras-binding domain of Raf-1 (Raf-1 RBD), following manufacturers’ instruction.
RESULTS
Ras induces Bnip3 promoter activity but not endogenous mRNA in pancreatic cancer cells
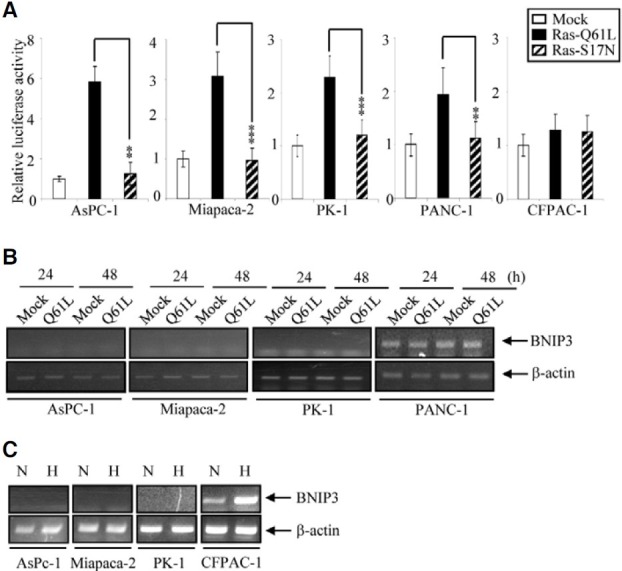
In a previous report we showed that Ras induces promoter activity and expression of endogenous BNIP3 via the MEKERK- HIF-1 pathway in RAW264.7 mouse macrophages (An et al., 2006). Here, we investigated the same pathway in cancer cell lines, because it has been reported that the promoter of BNIP3 is methylated in most such lines, so that induction of its expression is inhibited. We used the pancreatic cancer cell lines, AsPC-1, Miapaca-2, PK-1, PANC-1, CFPAC-1 and Hs766T, First we analyzed activation of the human BNIP3 promoter by Ras using plasmids bearing constitutively active Ras (Q61L mutant) and dominant-negative Ras (S17N mutant) mutations. The reporter plasmid harbors a putative human Bnip3 promoter fragment (-753 to -1 bp; +1 indicates the translation start site) that contains HRE (hypoxia response element, CACGT) sites, at -249 bp and -613 bp. Ras-Q61L activated the BNIP3 promoter 2-6 fold in AsPC-1, Miapaca-2, PK-1 and PANC-1 cells, while the ras-S17N mutant was completely ineffective (Fig. 1A). We next examined the production of endogenous BNIP3 mRNA in cells harboring Ras-Q61L. The endogenous level of BNIP3 was low in AsPC-1, Miapaca-2 and PK-1 cells, and was not increased by Ras-Q61L expression (Fig. 1B). It was high in PANC-1 cells, and was not changed by Ras-Q61l expression. Hypoxia-induced BNIP3 up-regulation was also inhibited in AsPC-1, Miapaca-2 and PK-1 cells (Fig. 1C). On the other hand BNIP3 was rapidly induced by hypoxia treatment in CFPAC-1 cells, implying that their level of promoter methylation differed from that in AsPC-1, Miapaca-2 and PK-1 cells. Together, these results show that Ras induces BNIP3 promoter activity in AsPC-1, Miapaca-2, PK-1 and PANC-1 cells but cannot induce expression of BNIP3 mRNA in these cells.
Fig. 1. BNIP3 promoter activity and mRNA expression in response to Ras in pancreatic cancer cells. (A) Cells were co-transfected with constitutively active Ras (Ras-Q61L) or a dominant negative Ras (Ras-S17N) plus a BNIP3 promoter reporter construct. Luciferase activities were measured after 24 h of transfection and were plotted against the activity of mock transfected cells. *P < 0.05, **P < 0.01 and ***P < 0.001. (B) At the indicated times after transfection with Ras-Q61L, total RNA was prepared and BNIP3 mRNA was measured by semiquantitative RT-PCR. A 585 bp fragment of human BNIP3 cDNA was amplified. The β-actin was used as a control for RNA quality and loading. (C) After 48 h of hypoxia, total RNA was prepared and BNIP3 mRNA was measured by semi-quantitative RTPCR. N, normoxia, H, hypoxia.
Inhibition of DNA methylation restores BNIP3 mRNA synthesis
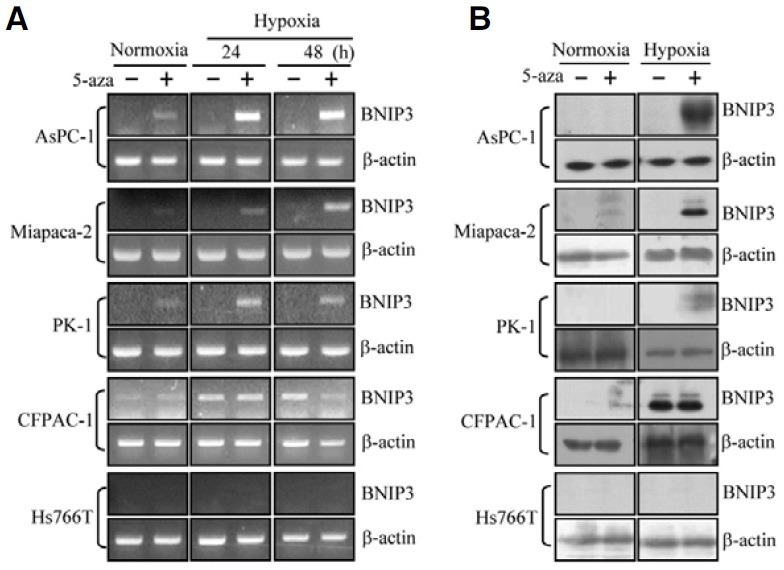
To see whether the inhibition of BNIP3 induction was due to methylation of the BNIP3 promoter, cells were treated with the DNMT inhibiter, 5-aza-2-deoxycytidine, and hypoxia-dependent expression of BNIP3 was examined. After 5-aza-2-deoxycytidine treatment BNIP3 mRNA increased in response to hypoxia in AsPC-1, Miapaca-2 and PK-1 cells (Fig. 2A). BNIP3 mRNA was even enhanced by 5-aza-2-deoxycytidine treatment without exposure to hypoxia. In agreement with the results in Fig. 1, substantial BNIP3 expression was induced by hypoxia in CFPAC-1 cells, and expression was not affected by 5-aza-2- deoxycytidine. BNIP3 protein expression paralleled mRNA expression and could also be induced by treatment with 5-aza- 2-deoxycytidine in the AsPC-1, Miapaca-2 and PK-1 cells (Fig. 2B). In Hs766T cells hypoxia plus 5-aza-2-deoxycytidine failed to induce any BNIP3 mRNA and protein expression. These results show that DNA methylation of the BNIP3 promoter occurs in the majority of pancreatic cancer cell lines but not all.
Fig. 2. Restoration of BNIP3 expression in pancreatic cancer cell lines following treatment with the DNMT inhibitor, 5-aza-2-deoxycytidine. Cells were treated with 1 μM 5-aza-2-deoxycytidine (5-aza) for 6 days and exposed to hypoxia for 24-48 h. (A) Total RNA was prepared at the indicated times and expression of BNIP3 was measured by RTPCR. β-actin was used as a control for RNA quality and loading. (B) BNIP3 protein was examined by immunoblotting using a monoclonal anti-BNIP3 antibody.
Inhibition of the MEK pathway reduces DNMT1 expression and restores the hypoxia-inducibility of BNIP3
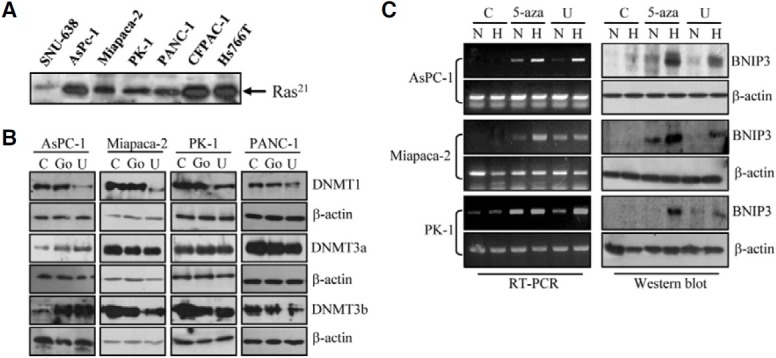
Next, we examined the relationship between BNIP3 induction and the signaling pathway downstream of Ras. It is known that dominant active mutations of K-ras are present in 75-90% of pancreatic cancer cells (Ghaneh et al., 2007). Therefore we examined Ras activity in the pancreatic cancer cell lines using pull-down assays with the Ras binding domain of Raf-1 followed by immunoblotting with anti-Ras. As expected, most of the pancreatic cancer lines had higher levels of activated Ras than SNU-638, a representative gastric cancer cell line (Fig. 3A). When we treated cells with U0126, a MEK inhibitor, to inhibit Ras downstream signaling, DNMT1 expression was strongly inhibited in AsPc-1, Miapaca-2, PK-1 and PANC-1 cells, whereas DNMT3a and DNMT3b levels were unaffected, and even increased in AsPC-1 cells (Fig. 3B). As a control, cells were treated with Go6976, a PKC inhibitor, and there was no consistent change of DNMT expression. Finally we tested whether BNIP3 expression could be restored by U0126 as it is by 5-aza-2-deoxycytidine. In AsPC-1, Miapaca-2 and PK-1 cells, BNIP3 mRNA expression was clearly up-regulated by U0126 treatment alone or together with hypoxia (Fig. 3C). BNIP3 protein expression was consistent with the mRNA expression. This outcome was similar to that observed with 5-aza-2-deoxycytidine treatment, suggesting that DNA methylation of the BNIP3 promoter is mediated by DNMT1 via the MEK pathway.
Fig. 3. Effect of U0126 treatment on the expression of DNMT and BNIP3. (A) The 30 μg of total cell extract of each cell line was incubated with a Raf-1-RBD GST fusion protein in glutathione agarose. The captured protein was eluted, and analyzed by Western blotting using anti-Ras monoclonal antibody. (B) The indicated pancreatic cancer cell lines were treated with vehicle only (C), 10 μM U0126 (U) or 5 μM Go6976 (Go) for 6 days. Cell lysates were analyzed for expression of DNMTs by immunoblotting with antibodies against DNMT1, DNMT3a, and DNMT3b. Data are representative of three independent experiments. β-actin was used as an internal control. (C) Cells were treated with vehicle only (C), 1 μM 5-aza-2-deoxycytidine (5-aza) or 10 μM U0126 (U) for 6 days before exposure to hypoxic condition for a further 48 h. BNIP3 mRNA (left panel) and protein (right panel) levels were examined by RT-PCR and immunoblotting, respectively. The β-actin was used as a control. N, normoxia, H, hypoxia.
DISCUSSION
In the present study we have shown that silencing of BNIP3 in several pancreatic cancer cell lines is the result of promoter methylation by DNMT1 induced via the MEK pathway. This conclusion is based on the following evidence: a) Ras induced Bnip3 promoter activity but not endogenous mRNA expression in the pancreatic cancer cells (Fig. 1); b) BNIP3 expression could be restored by treatment with a DNA methylation inhibitor (Fig. 2); c) inhibition of the MEK pathway reduced DNMT1 expression and restored the hypoxia-inducibility of BNIP3 (Fig. 3).
We used six different pancreatic cancer cell lines and showed that BNIP3 up-regulation was inhibited in AsPC-1, Miapaca-2, PK-1 and PANC-1 cells, and that BNIP3 expression could be restored by treatment with a DNA methylation inhibitor. Our results are consistent with previous findings. Okami et al. detected hypermethylation of the BNIP3 promoter in most of the BNIP3-silenced pancreatic cancer cell lines examined, including AsPC-1, Miapaca-2 and PANC-1 and in eight of ten primary pancreatic cancer tissues (Okami et al., 2004). Abe et al. 2005) showed that BNIP3 promoter methylation was strong in the Miapaca-2, PK-8, PK-9 and PK-59 cell lines but weak in PANC- 1 and CFPAC-1. For our study, we selected AsPC-1, Miapaca- 2, PK-1 and PANC-1 for further investigation of BNIP3 promoter methylation and regulation of expression.
In a previous report we showed that Ras activated the BNIP3 promoter and induced expression of BNIP3 protein in murine RAW264.7 macrophages and this was mediated by MEK, ERK and HIF-1 (An et al., 2006). Ras activation also induces cell death in macrophages. In the present study, the promoter activity of human BNIP3 was efficiently induced in the pancreatic cancer cells, in agreement with the previous data. The promoter region in the reporter plasmid contains two HRE sites and also a CpG island (AT/CG ratio; 20.6%), as in genomic DNA. Obviously the plasmid cannot be hypermethylated since it is transiently transfected into the cells. The reporter was clearly induced by dominant active Ras (Q61L mutant), although induction was relatively weak since pancreatic cancer cells often have elevated basal levels of activated Ras (Fig. 3A). On the other hand, induction of the endogenous BNIP3 by activated Ras or hypoxia was blocked but could be restored by inhibition of DNA methyltransferase. Thus, Ras may have two opposite effects on BNIP3 expression: induction of BNIP3 expression by activating HIF-1 and inhibition of BNIP3 expression by inducing DNMT1. The latter seems to be the major downstream signaling pathway activated by Ras in pancreatic cancer cells. These findings are consistent with other reports. Chang et al. (2006) showed that Ras activation inhibited expression of the metastasis suppressor RECK via histone deacetylation and promoter methylation, and that DNMT3b played a role in the DNA methylation. Lu et al. (2007) reported that inhibition of the ERK-MAPK pathway using PD98059, rottlerin or MEK siRNA, attenuated DNMT1 expression and led to demethylation of the promoters of p16INK4A and p21WAF1. DNMT1 and DNMT3b are often expressed at high levels and catalyze genomic DNA methylation in cancer cells (Rhee et al., 2002).
Epigenetic modification of tumor suppressors, cell growthand apoptosis-related molecules and intracellular signaling molecules, has recently been the subject of intense investigation in relation to carcinogenesis (Na et al., 2010; Shin et al., 2011). At the same time methylation inhibitors have been developed as potential therapies for various kinds of human cancer. Therefore, further investigation of the detailed mechanisms and signaling pathways involved in DNA methylation is needed in order to clarify the molecular basis of this process and to aid in the development of effective treatments.
Acknowledgments
This study was financially supported by the research fund of Chungnam National University in 2009.
References
- 1.Abe T., Toyota M., Suzuki H., Murai M., Akino K., Ueno M., Nojima M., Yawata A., Miyakawa H., Suga T., et al. Upregulation of BNIP3 by 5-aza-2′-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J. Gastroenterol. (2005);40:504–510. doi: 10.1007/s00535-005-1576-1. [DOI] [PubMed] [Google Scholar]
- 2.Akada M., Crnogorac-Jurcevic T., Lattimore S., Mahon P., Lopes R., Sunamura M., Matsuno S., Lemoine N.R. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin. Cancer Res. (2005);11:3094–3101. doi: 10.1158/1078-0432.CCR-04-1785. [DOI] [PubMed] [Google Scholar]
- 3.An H.J., Maeng O., Kang K.H., Lee J.O., Kim Y.S., Paik S.G., Lee H. Activation of Ras up-regulates pro-apoptotic BNIP3 in nitric oxide-induced cell death. J. Biol. Chem. (2006);281:33939–33948. doi: 10.1074/jbc.M605819200. [DOI] [PubMed] [Google Scholar]
- 4.Boyd Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. (1994);79:1121. [PubMed] [Google Scholar]
- 5.Boyd J.M., Malstrom S., Subramanian T., Venkatesh L.K., Schaeper U., Elangovan B., D’Sa-Eipper C., Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. (1994);79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 6.Chang H.C., Cho C.Y., Hung W.C. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. (2006);66:8413–8420. doi: 10.1158/0008-5472.CAN-06-0685. [DOI] [PubMed] [Google Scholar]
- 7.Chen G., Ray R., Dubik D., Shi L., Cizeau J., Bleackley R.C., Saxena S., Gietz R.D., Greenberg A.H. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. (1997);186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Angelis P.M., Fjell B., Kravik K.L., Haug T., Tunheim S.H., Reichelt W., Beigi M., Clausen O.P., Galteland E., Stokke T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int. J. Oncol. (2004);24:1279–1288. [PubMed] [Google Scholar]
- 9.Erkan M., Kleeff J., Esposito I., Giese T., Ketterer K., Buchler M.W., Giese N.A., Friess H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene. (2005);24:4421–4432. doi: 10.1038/sj.onc.1208642. [DOI] [PubMed] [Google Scholar]
- 10.Ghaneh P., Costello E., Neoptolemos J.P. Biology and management of pancreatic cancer. Gut. (2007);56:1134–1152. doi: 10.1136/gut.2006.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giatromanolaki A., Koukourakis M.I., Sowter H.M., Sivridis E., Gibson S., Gatter K.C., Harris A.L. BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin. Cancer Res. (2004);10:5566–5571. doi: 10.1158/1078-0432.CCR-04-0076. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy A.S., Harrison G.H., Mansfield C.M., Zhou X.J., Xu J.F., Balcer-Kubiczek E.K. Survival of colorectal cancer cell lines treated with paclitaxel, radiation, and 5-FU: effect of TP53 or hMLH1 deficiency. Int. J. Cancer. (2000);90:175–185. [PubMed] [Google Scholar]
- 13.Kim J.Y., Cho J.J., Ha J., Park J.H. The carboxy terminal C-tail of BNip3 is crucial in induction of mitochondrial permeability transition in isolated mitochondria. Arch. Biochem. Biophys. (2002);398:147–152. doi: 10.1006/abbi.2001.2673. [DOI] [PubMed] [Google Scholar]
- 14.Lee H., Paik S.G. Regulation of BNIP3 in normal and cancer cells. Mol. Cells. (2006);21:1–6. [PubMed] [Google Scholar]
- 15.Lu R., Wang X., Chen Z.F., Sun D.F., Tian X.Q., Fang J.Y. Inhibition of the extracellular signal-regulated kinase/mitogen- activated protein kinase pathway decreases DNA methylation in colon cancer cells. J. Biol. Chem. (2007);282:12249–12259. doi: 10.1074/jbc.M608525200. [DOI] [PubMed] [Google Scholar]
- 16.Murai M., Toyota M., Satoh A., Suzuki H., Akino K., Mita H., Sasaki Y., Ishida T., Shen L., Garcia-Manero G., et al. Aberrant DNA methylation associated with silencing BNIP3 gene expression in haematopoietic tumours. Br. J. Cancer. (2005a);92:1165–1172. doi: 10.1038/sj.bjc.6602422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai M., Toyota M., Suzuki H., Satoh A., Sasaki Y., Akino K., Ueno M., Takahashi F., Kusano M., Mita H., et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. (2005b);11:1021–1027. [PubMed] [Google Scholar]
- 18.Na Y.K., Lee S.M., Hong H.S., Kim J.B., Park J.Y., Kim D.S. Hypermethylation of growth arrest DNA-damage-inducible gene 45 in non-small cell lung cancer and its relationship with clinicopathologic features. Mol. Cells. (2010);30:89–92. doi: 10.1007/s10059-010-0092-1. [DOI] [PubMed] [Google Scholar]
- 19.Okami J., Simeone D.M., Logsdon C.D. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. (2004);64:5338–5346. doi: 10.1158/0008-5472.CAN-04-0089. [DOI] [PubMed] [Google Scholar]
- 20.Rhee I., Bachman K.E., Park B.H., Jair K.W., Yen R.W., Schuebel K.E., Cui H., Feinberg A.P., Lengauer C., Kinzler K.W., et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. (2002);416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 21.Shin J.E., Park S.H., Jang Y.K. Epigenetic up-regulation of leukemia inhibitory factor (LIF) gene during the progression to breast cancer. Mol. Cells. (2011);31:181–189. doi: 10.1007/s10059-011-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sowter H.M., Ferguson M., Pym C., Watson P., Fox S.B., Han C., Harris A.L. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J. Pathol. (2003);201:573–580. doi: 10.1002/path.1486. [DOI] [PubMed] [Google Scholar]
- 23.Vande Velde C., Cizeau J., Dubik D., Alimonti J., Brown T., Israels S., Hakem R., Greenberg A.H. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. (2000);20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yasuda M., Theodorakis P., Subramanian T., Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. (1998);273:12415–12421. doi: 10.1074/jbc.273.20.12415. [DOI] [PubMed] [Google Scholar]
- 25.Yook Y.H., Kang K.H., Maeng O., Kim T.R., Lee J.O., Kang K.I., Kim Y.S., Paik S.G., Lee H. Nitric oxide induces BNIP3 expression that causes cell death in macrophages. Biochem. Biophys. Res. Commun. (2004);321:298–305. doi: 10.1016/j.bbrc.2004.06.144. [DOI] [PubMed] [Google Scholar]