

Abstract
Members of polo-like kinases (collectively, Plks) have been identified in various eukaryotic organisms and play pivotal roles in cell proliferation. They are characterized by the presence of a distinct region of homology in the C-terminal noncatalytic domain, called polo-box domain (PBD). Among them, Plk1 and its functional homologs in other organisms have been best characterized because of its strong association with tumorigenesis. Plk1 is overexpressed in a wide spectrum of cancers in humans, and is thought to be an attractive anti-cancer drug target. Plk1 offers, within one molecule, two functionally different drug targets with distinct properties-the N-terminal catalytic domain and the C-terminal PBD essential for targeting the catalytic activity of Plk1 to specific subcellular locations. In this review, we focused on discussing the recent development of small-molecule and phosphopeptide inhibitors for their potency and specificity against Plk1. Our effort in understanding the binding mode of various inhibitors to Plk1 PBD are also presented.
Keywords: cancer therapy, inhibitors, Plk1, polo-box domain (PBD), polo-like kinase
INTRODUCTION
Polo-like kinase 1 (Plk1) is a conserved Ser/Thr protein kinase required for proper M-phase progression. A growing body of evidence suggests that it regulates various mitotic events, such as mitotic entry, spindle formation, chromosome segregation, and cytokinesis (Archambault and Glover, 2009; Barr et al., 2004; Petronczki et al., 2008; van de Weerdt et al., 2006; Takaki et al., 2008). In mammalian cells, three closely related members (Plk1-3) and one distantly-related member (Plk4) exist and they appear to exhibit differential functions and tissue distributions. Comparative analyses of the primary sequences of these proteins reveal conserved kinase domains (KDs) (29% identity among all four Plks and 48% identity among Plk1-3 at the amino acid level) that closely resemble those from Aurora kinases and calcium/calmodulin-dependent kinases. Alignment of the C-terminal noncatalytic polo-box domain (PBD) sequences from these four Plks (for simplicity, we will call them PBD1 to PBD4, respectively, hereafter) show that PBD1-3 exhibit a high level of homology in two distinct polo-box motifs-39-43% in PB1 (aa 411-aa 489 in Plk1) and 36-39% in PB2 (aa 511-aa 592 in Plk1). PBD4 has a highly divergent sequence and the structure of PBD4 is distinct from that of PBD1-3 (Elia et al., 2003a; Leung et al., 2002).
As with the importance of Plk1 in cell proliferation, Plk1 is overexpressed in various human cancers. Furthermore, its overexpression appears to be sufficient to override cellular checkpoints and induce genetic instability, thus promoting tumorigenesis (Eckerdt et al., 2005; Strebhardt et al., 2006; Takai et al., 2005). Consistent with these observations, the level of Plk1 transcript positively correlates with aggressiveness of tumor progression, and that patients with high Plk1 expression in their tumors exhibit a significantly poorer rate of survival than those with low Plk1 expression (Knecht et al., 1999; 2000; Kneisel et al., 2002; Strebhardt et al., 2000). Interference with Plk1 function effectively induces apoptotic cell death in most tumor cells but not in normal cells (Liu et al., 2006; Luo et al., 2009; Sur et al., 2009), and prompts tumor regression in mouse xenograft models (Elez et al., 2003; Steegmaier et al., 2007). Notably, genome-wide RNAi screens have distinguished Plk1 from the rest of the members of the kinase family as the only kinase selectively required for the viability of activated Ras or inactivated p53 mutation-bearing cancer cells, but less for the respective normal cells (Luo et al., 2009; Sur et al., 2009). Based on these and other related studies, Plk1 is thought to be one of the most attractive anti-cancer drug targets.
Plk1 offers two functionally crucial target sites within one molecule-the N-terminal KD and the C-terminal PBD. Recently, several articles reviewed the role of Plk1 during the cell cycle and cancer development and the recent advances in the development of small-molecule Plk1 inhibitors (Archambault et al., 2009; Petronczki et al., 2008; Strebhardt et al., 2010). In this communication, we will therefore focus on discussing the molecular and structural basis of various small-molecule and peptide- derived inhibitors that target either the kinase domain or the PBD.
Targeting the kinase domain of Plk1
For more than a decade, numerous inhibitors against the KD of various protein kinases have been developed and their potential as therapeutic agents in anti-cancer therapy has been assessed. Despite a substantial progress in protein kinase drug discovery, design of potent inhibitors with high degrees of selectivity during lead optimization remains a major challenge. Many kinase inhibitors have failed in preclinical or clinical development, due to the lack of selectivity that induces intolerable side effects. This is largely because the catalytic cleft of various kinases is highly conserved not only in sequence but also in conformation (Liao, 2007).
As of today, several crystal structures of Plk1 KD have been determined by multiple groups. These include the structure of Plk1 KD in complex with a designed ankyrin-repeat protein (DARPin) (Bandeiras et al., 2008), an ATP pocket-binding ligand such as a nonhydrolysable ATP analogue adenylylimidodiphosphate (Kothe et al., 2007a), a pyrrolo-pyrazole inhibitor (PHA-680626) (Kothe et al., 2007a), a purine mimetic inhibitor (Elling et al., 2008b), a quinazoline mimetic inhibitor (Beria et al., 2010), a pyrazolopyridine inhibitor (Hanan et al., 2008), wortmannin (Elling et al., 2008a), BI 2536 (Kothe et al., 2007b), or BI 6727 (Rudolph et al., 2009). All of these structures display the typical kinase fold in which the ATP binding site is located in a cleft formed between the N-terminal lobe composed predominantly of anti-parallel β sheet and the primarily α-helical C-terminal lobe. The two lobes are connected by a hinge region that allows specific interactions between the backbone and each of the bound ligands. Targeting a Phe183 residue at the bottom of the ATP pocket together with a Cys67 in the roof of the Leu132-generated binding pocket is thought to be important to achieve selectivity against various Plks. Based on the above reported crystal structures, the interactions between the Plk1 KD and the small molecule inhibitors are summarized in Figs. 1 and 2. All the Plk1 kinase domain inhibitors that are in clinical trials and preclinical development are summarized in Tables 1 and 2, respectively.
Fig. 1. Crystal structure of the ATP binding site of zPlk1 KD in complex with adenosine diphosphate (PDB ID: 3D5W) was depicted in ribbon structure with key residues. Ribbon structure depicts various binding pockets such as adenine (blue), ribose (pink), phosphate (red), hydrophobic (green and gold), adaptive pocket (black) and hinge region (violet) along with the corresponding interacting residues with stick models. Outer box shows all the reported Plk1 kinase inhibitors and the engaged atoms with the hydrogen bonding, π-π stacking etc are highlighted with corresponding color in the pockets.
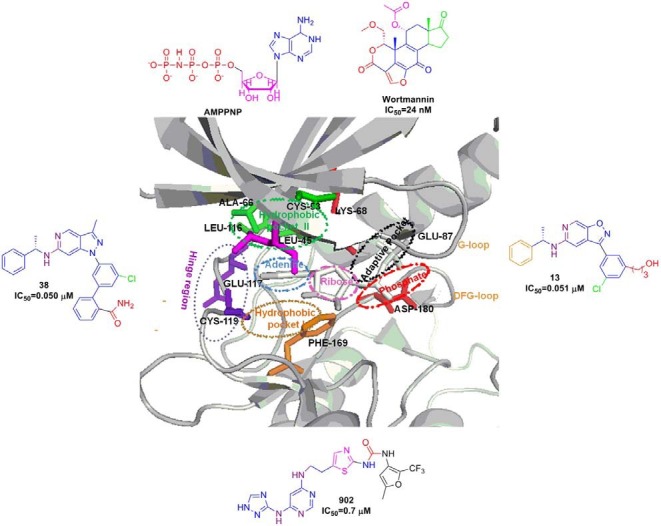
Fig. 2. Crystal structure of ATP binding site of hPlk1 KD complexed with BI 2536 inhibitor (PDB ID: 2RKU) was depicted in ribbon structure with key residues. Ribbon structure depicts various binding pockets like adenine (blue), ribose (pink), phosphate (red), hydrophobic (green and gold), adaptive pocket (black) and hinge region (violet) along with the corresponding interacting residues with stick models. Outer box shows all the reported Plk1 kinase inhibitors and the engaged atoms with the hydrogen bonding, π-π stacking etc are highlighted with corresponding color in the pockets.
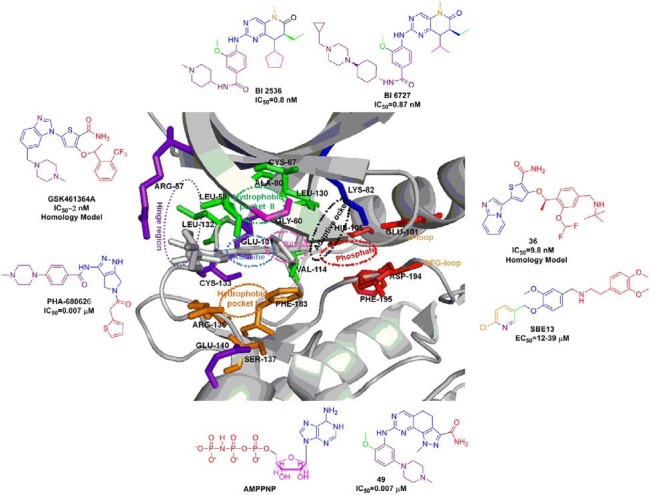
Table 1.
Plk1 kinase Inhibitors in clinical phase I/II/III development
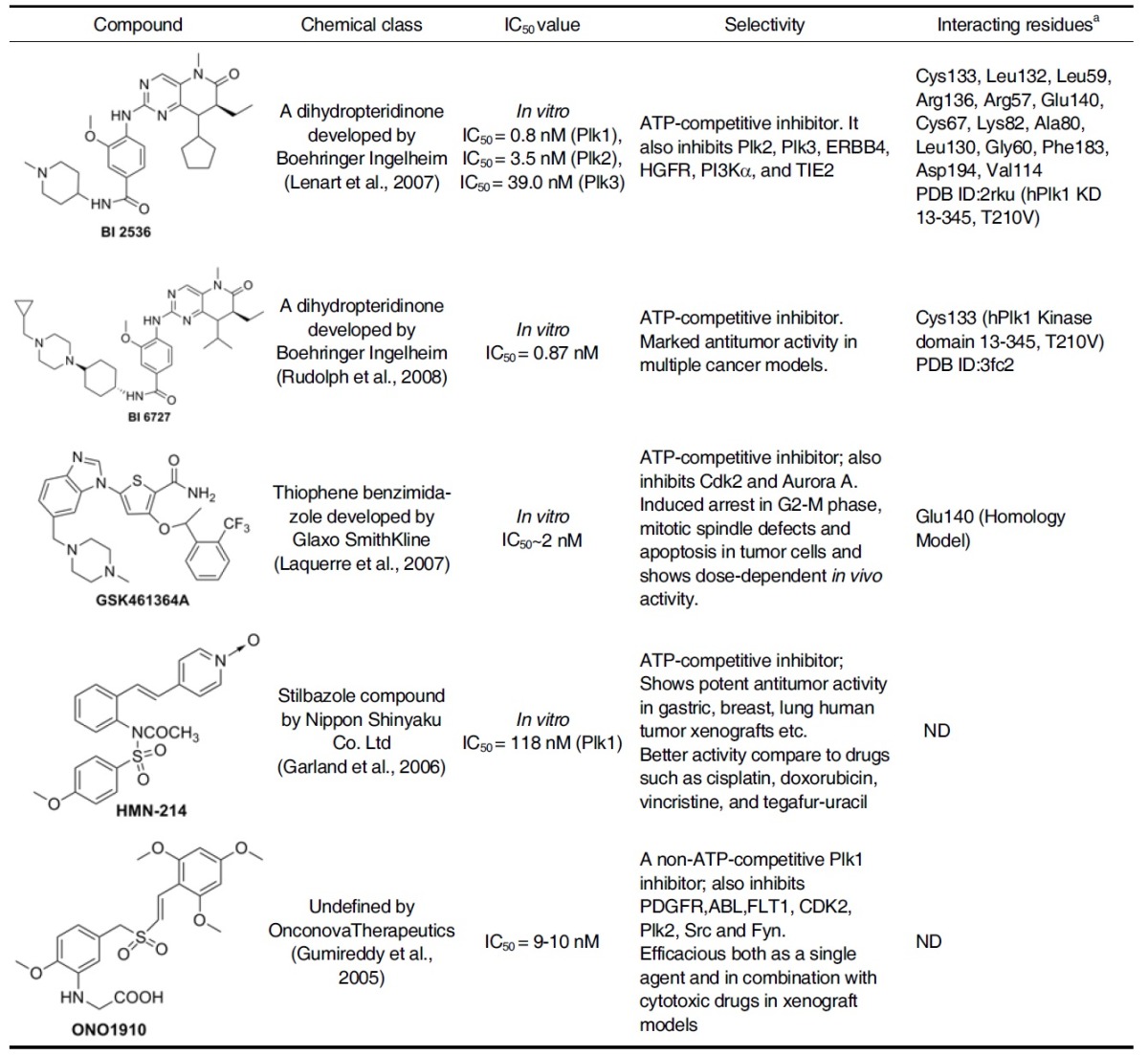
a Interacting residues by hydrogen bonding etc. are evaluated based on crystal structure or homology models.
Table 2.
Plk1 kinase Inhibitors in preclinical development
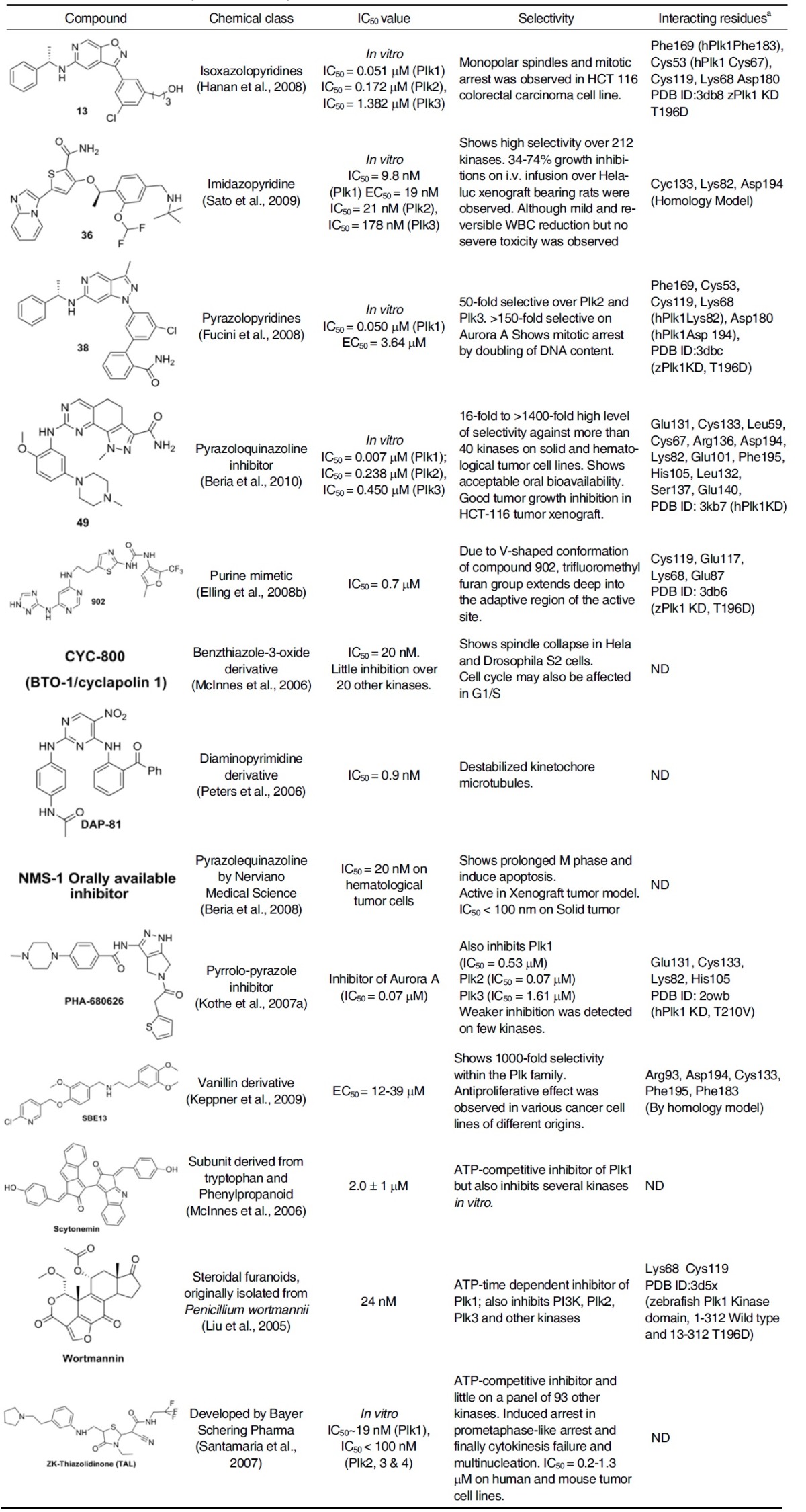
a Interacting residues by hydrogen bonding etc. are evaluated based on crystal structure or homology models.
Scytonemin
The natural product scytonemin was one of the first Plk1 inhibitors to be characterized. Scytonemin has been shown to inhibit the ability of Plk1 to phosphorylate Cdc25C in a concentrationdependent manner with an in vitro IC50 of 2 ± 0.1 μM (McInnes et al., 2006; Stevenson et al., 2002). However, it was later turned out to be a nonselective molecule that also inhibits myelin transcription factor 1 (MYT1), cyclin-dependent kinase 1 (CDK1), checkpoint kinase 1 (CHK1), and protein kinase C (PKC) with similar potencies (McInnes et al., 2005).
ON01910
This compound is a water-soluble benzyl styryl sulfone analogue that was initially reported to inhibit Plk1 in a substratedependent and an ATP-independent manner (Gumireddy et al., 2005). ON01910 has been shown to inhibit the cell proliferation in >100 cancer cell lines including several drug-resistant cell lines with IC50 values ranging from 50 to 250 nM. However, later studies have shown that ON01910 exhibits little activity against Plk1 in vitro. Cell-based phenotypes do not correlate with Plk1 inhibition but are consistent with affecting microtubule dynamics (Peters et al., 2006). Synergism was observed when it was combined with paclitaxel, oxaliplatin, doxorubicin, irinotecan, or vincristine in tumor cell lines in culture, or in xenograft models. Clinical trials in cancer patients are currently underway to test the effects of ON01910 both as a monotherapy and in combination with conventional chemotherapy.
Wortmannin
This was originally characterized as a phosphatidylinositol 3- kinase (PI3K) inhibitor, was later found to inhibit Plk1 as well with an IC50 value of 24 nM (Liu et al., 2005). The mode of the interaction between wortmannin and Plk1 KD was recently evaluated by the crystallographic study, which is in good agreement with the already reported homology model (Elling et al., 2008a).
Crystal structure of the zebrafish Plk1 (zPlk1) KD Thr196Asp mutant in complex with wortmannin shows that both the activation segment and the alpha C helix became ordered, even though wortmannin is positioned >20 Å away from the Cα atom of Asp196. It also confirms that the compound is indeed covalently linked to the catalytic Lys68 and reveals a single direct hydrogen-bonding interaction between the O atom (five-membered keto group) of wortmannin and the amide N atom of Cys119. Water mediated hydrogen bonds between the carbonyl O atoms on the exocyclic ester group of wortmannin and the surrounding residues are not immediately obvious owing to the limited resolution of the structure (Fig. 1). Unlike BI 2536 or PHA-680626 bound to Plk1, which will be discussed below, wortmannin also appears to hydrogen bond to Lys68 in addition to forming a covalent bond with the same residue (Elling et al., 2008a). Furthermore, Plk1 was found to be the only family member that is covalently modified by time-dependent inhibition of wortmannin. Though both Plk2 and Plk3 were inhibited with Ki values comparable to that for Plk1, time-dependent inhibition was not observed (Johnson et al., 2007).
BI 2536
A dihydropteridinone developed by Boehringer Ingelheim, Vienna, Austria, exhibits a potent inhibitory activity against Plk1 in vitro with an IC50~1 nM. It also inhibits Plk2 and Plk3 at 4 nM and 9 nM, respectively, but shows little activity against a panel of 63 other kinases tested (Lenart et al., 2007). Cell-based phenotypes observed with BI 2536 treatment is consistent with those with Plk1 inhibition, suggesting that it may serve as a good tool for probing Plk1 function (Lenart et al., 2007). It has been demonstrated that the intravenous (i.v.) administration of BI 2536 at well-tolerated dose inhibits tumor growth and induces tumor regression in several human carcinoma xenograft mouse models (Steegmaier et al., 2007).
In a Phase I dose-escalation study, BI 2536 has been well tolerated when administered to patients with relapsed or refractory non-Hodgkin lymphoma (NHL) or advanced solid tumors (Mross et al., 2008; Vose et al., 2008). Three phase II studies in patients with small or non-small cell lung cancer, hormone-refractory prostate cancer (HRPC), and advanced pancreatic cancer have also been completed with BI 2536 (Gandhi et al., 2009; Pandha et al., 2008; Von Pawel et al., 2008). Recently, BI 2536 was found to inhibit proliferation of imatinib-sensitive and imatinib-resistant chronic myeloid leukemia (CML) cells (Gleixner et al., 2010).
Crystallization studies of this inhibitor with Plk1 KD has revealed several features that might be important for its high selectivity (Kothe et al., 2007b). Binding of aminopyrimidine portion of BI 2536 with the hinge region of Cys133 places the pteridinone moiety and the cyclopentyl group in the adenine and ribose portion of the ATP pocket, respectively, with the N-methyl- piperidine group pointing into the solvent. The phenyl ring of BI 2536 is sandwiched between the hydrophobic pockets (I & II shown in Fig. 2) created by the side chain of Leu59 from the top N-lobe and the side chain of Arg136 from the bottom C-lobe. The methoxy group is accommodated in a pocket created by the Leu132 residue from the hinge region. Presence of bulky tyrosine or phenylalanine in place of Leu132 in other kinases is predicted to clash with methoxy group of BI 2536, thus providing the specificity for Plk1. The amide linker of the inhibitor mediates the NH and carbonyl forming hydrogen bonds with the main chain carbonyl of Leu59 of the glycine-rich loop and the side chain of Arg57, respectively. The side chain of Arg136, stabilized by water-mediated hydrogen bonds with the side chain of Glu140 and the main chain carbonyl of Leu59, interacts with the amide linker as well as the piperidine through van der Waals contacts, thereby providing additional binding affinity.
At the roof of the binding site, ethyl group is accommodated in a small pocket formed by Cys67, Lys82, Ala80 and Leu130 residues, and the cyclopentyl is positioned near Leu59, Gly60, and Cys67 residues. Binding affinity was further enhanced by the π-π stacking interaction between the pteridinone moiety and the Phe183 at the bottom of the binding site. Two watermediated hydrogen bondings were also observed from the carbonyl group of pteridinone to the side chain of Lys82 and the backbone NH of Asp194. Finally, the methyl group is buried in a pocket created near the Val114, Phe183, and the gatekeeper residue Leu130 (Kothe et al., 2007b).
BI 6727
This is a potent and selective inhibitor of Plk1 developed by Boehringer Ingelheim as a second-in class dihydropteridinone derivative. It binds to the ATP-binding pocket of the kinase and induces the formation of monopolar spindles. BI 6727 induces a distinct prometaphase “polo-arrest” phenotype and subsequently apoptosis. BI 6727 is active against a broad range of tumor cells in vitro. BI 6727 strongly inhibits Plk1, Plk2, and Plk3 at IC50 values of 0.87, 5, and 56 nM, respectively (Rudolph et al., 2008). BI 6727 is highly efficacious not only in standard nude mouse xenograft models of human cancers such as colon, pancreas, bread cancer, and NSCLC tumor models but also in the taxane-resistant CXB1 (human colon carcinoma tumor) xenograft model of colorectal cancer. It was well tolerated in the phase I dose escalation study with advanced or metastatic solid tumors (Schoffski et al., 2008).
Crystallization studies with the human Plk1 Thr210Val mutant kinase domain reveal the binding mode of BI 6727 in the ATP-binding pocket. The inhibitor binds to the hinge region via two hydrogen bonds from aminopyrimidine portion of the core to the backbone NH and carbonyl of Cys133. This placed the pteridinone moiety in the adenine pocket, along with the piperidine group pointing into the solvent (Rudolph et al., 2009).
GSK461364A
GSK461364A, developed by Glaxo Smith Kline, is a selective i.v. thiophene amide inhibitor that displays at least 1000-fold greater potency for Plk1 than for Plk2 and Plk3 (Gilmartin et al., 2009). GSK461364A is in Phase I clinical trials; this molecule has been derived via chemical optimization from a benzimidazolyl- thiophene precursor molecule called compound 1 (Lansing et al., 2007). In an external kinase panel screened against 260 protein kinases, GSK461364A demonstrated an IC50 value of < 1 μM against only 10 other kinases. Interestingly, the docking studies with an analogue of GSK461364A reveal that the basic nitrogens pointing to cavity may interact with Glu140. Although it is not understood whether this interaction is critical for delivering kinase selectivity, both Plk2 and Plk3 possess a His residue at the position analogous to that of Glu140 in Plk1, and therefore fail to generate favorable salt-bridge interaction (Emmitte et al., 2009).
While the observed selectivity is yet to be further validated, this GSK461364A is broadly active against several hundred tumor cell lines with proliferation IC50 values of < 100 nM in 91% of all cell lines examined. The predominant phenotype for cells treated with GSK461364A is a pro-metaphase arrest with characteristically collapsed polar polo spindle (Gilmartin et al., 2009). GSK461364A is an ATP-competitive inhibitor that rapidly forms a reversible complex with Plk1. GSK461364A also showed dosedependent in vivo activity (ranging from complete tumor growth inhibition to growth delay) on various established human tumor xenografts (Erskine et al., 2007; Laquerre et al., 2007). In the phase I studies in patients with advanced solid tumors, GSK461364A has been found to be well tolerated (Beria et al., 2010; Olmos et al., 2009). In a recent study, GSK461364A has shown both in vitro and in vivo efficacy in preventing brain metastases of breast cancer, suggesting that Plk1 is a potential target for the prevention of metastases (Palmieri et al., 2009).
HMN-214
HMN-214 is a prodrug of a novel stilbazole compound, HMN-176, developed by Nippon Shinyaku Co. Ltd. (Japan). HMN-176 has potent cytotoxicity with an IC50 value of 118 nM. in vitro studies suggest that HMN-176 downregulates Plk1 function by interfering with its subcellular localization at centrosomes and along the cytoskeletal structures (Yuan et al., 1997). in vivo, HMN-176 exhibits an antitumor activity against a broad spectrum of human tumors in a dosage-dependent manner. HMN- 176 potently inhibits the proliferation activity of gastric, breast, lung, pancreas, prostate, and colorectal human tumor xenografts, and this effect appears to be equal to or better than some of the previously characterized clinically available drugs such as cisplatin, doxorubicin, vincristine, and tegafur-uracil (Takagi et al., 2003; Tanaka et al., 2003). Further preclinical and clinical studies are needed to evaluate the efficacy and safety on patients with solid tumors, lymphomas, or hematological malignancies.
Targeting the PBD of Plk1
A growing body of evidence suggests that PBD represents an attractive alternative target for the development of anti-Plk1 inhibitors (Park et al., 2010). Because PBD is essential for the subcellular localization and mitotic functions of Plk1, it is ideally suited for the development of anti-Plk1 inhibitors that interfere with PBD-dependent protein-protein interactions, rather than with ATP binding. Given a high level of specificity of proteinprotein interactions, the PBD may prove to be a target that allows one to achieve Plk-isoform selectivity (McInnes et al., 2005; Strebhardt et al., 2006). To date, at least seven X-ray crystal structures of the human Plk1 PBD have been solved and reported in the Protein Data Bank. The crystal structures of the PBD of human Plk1 revealed that each of the individual Polo-box repeats (PB1 and PB2) in Plk1 consisted of a six-stranded anti-parallel β-sheet and an α-helix. Moreover, a short α-helix called Polo-cap is found upstream of the PB1 that is thought to hold both Polo-boxes in the correct orientation. Close examination of co-crystal structures of the PBD in complex with phosphopeptides reveals that ligand-binding results from a zipper-like contribution of residues alternatively arising from each PB, and highlights the importance of Trp414, His538, and Lys540 residues in the interaction (Elia et al., 2003a; 2003b). As of today, several small molecule inhibitors and peptide mimetics that bind to the Plk1 PBD have been isolated. Figure 3 provides an overview of the polo-box domain binding interaction with the small molecule or peptide-derived inhibitors based on crystal structures and homology models. Table 3 summarized all the Plk1 PBD inhibitors that have been reported so far.
Fig. 3. Crystal structure of the phosphopeptide binding site of hPlk1 PBD in complex with PLHSpT peptide (PDB ID: 3VFH) was depic-ted with key regions indicated by different color. Outer box shows all the reported Plk1 PBD binding ligands based on their crystal structure or homology model.
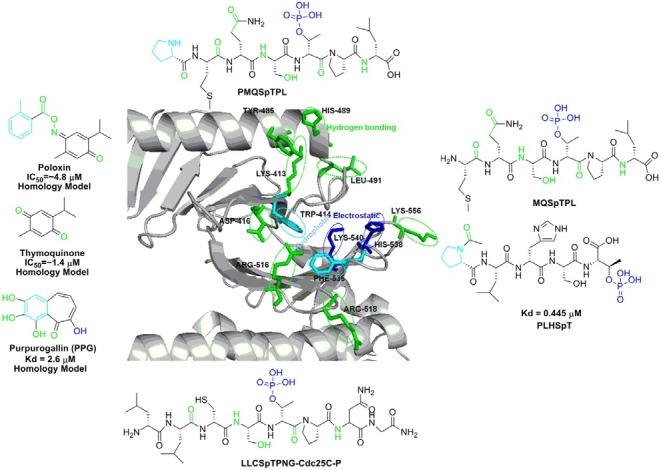
Table 3.
Plk1 Polo-box domain inhibitors
a Interacting residues by hydrogen bonding etc. are evaluated based on crystal structure or homology models.
SMALL MOLECULE PBD INHIBITORS
Thymoquinone and its derivative, Poloxin
A fluorescence polarization-based screening led to the identification of an anti-neoplastic thymoquinone and its derivative named Poloxin. Subsequent studies showed that Poloxin inhibits Plk1 PBD-dependent interaction with an apparent IC50 of ~1.4 and 4.8 μM. Although thymoquinone displays less desirable specificity profile, Poloxin exhibits a low level of Plk1 PBD specificity with 4-10 times higher IC50 values for Plk2 and Plk3. Poloxin does not appear to significantly inhibit other types of phosphopeptide-binding domains such as FHA, WW, and SH2 domains. Remarkably, inhibition of PBD with these natural inhibitors induces similar phenotypic changes (i.e., chromosome congression defects, mitotic arrest, and apoptosis) as observed by the treatment of ATP-competitive Plk1 inhibitors (Reindl et al., 2008).
It should be noted that Thymoquinone and Poloxin do not bear a negatively charged group functionally equivalent to the phosphate anchor of PBD-binding phosphopeptides, and Poloxin fails to significantly disrupt the interaction between Plk1 PBD and one of its interacting phosphopeptides derived from a kinetochore component, PBIP1 (Liao et al., 2010). This finding suggests that the mode of Poloxin binding to Plk1 PBD could be rather atypical.
Purpurogallin (PPG)
This compound is a benzotropolone-containing natural compound derived from nutgalls. It is isolated as a PBD inhibitor through the high throughput screening of 2,500 compounds stored in RIKEN Natural Product Depository, Saitama, Japan. PPG inhibits the PBD-dependent binding at IC50 of 0.3 μM and exhibits a similar in vitro specificity profile as Poloxin. PPG has also been shown to inhibit several other targets, such as tyrosin- specific protein kinases, human immunodeficiency virus 1 integrase, DNA synthesis of tumor cells, prolyl endopeptidases, and the Bcl-XL-BH3 peptide interaction. PPG not only delayed the onset of mitosis but also prolonged the progression of mitosis in HeLa cells. Interestingly, however, PPG-like compounds lacking the 4-hydroxyl group failed to exhibit the inhibitory activity, indicating that the 4-hydroxyl group of PPG is essential for the observed activity (Watanabe et al., 2009).
Schrodinger’s Induced Fitting Docking Protocol was used to explore the binding modes of PPG. The results suggest that the negatively charged enolized hydroxyl group of the bound PPG forms an electrostatic interaction with the positively charged His538. The other three phenolic hydroxyl groups and the carbonyl group form four hydrogen bonds with the positively charged Lys540, the backbone NH of Trp414, the indole ring of Trp414, and the backbone carbonyl of Leu491. Moreover, the phenyl ring and one double bond in the tropolone ring of PPG form π-π stacking interactions with the pyrrole ring and the phenyl ring (both embedded in the indole ring) of Trp414, respectively. The analogue of PPG lacking the 4-hydroxyl group was unable to form reasonable docking poses (Liao et al., 2010), underlying the importance of the 4-hydroxyl group for PBD inhibition.
Poloxipan
Poloxipan, a pan-specific inhibitor of the PBDs of Plk1-3, was recently isolated through the screening of chemical libraries against the PBD of Plk1. Poloxipan inhibited the function of all three PBDs with the IC50 value of 3.2 μM for Plk1 PBD, 1.7 μM for Plk2 PBD, and 3.0 μM for Plk3 PBD. The use of higher concentrations of poloxipan was not feasible due to its limited solubility in aqueous buffers. Inhibition of the Plk1 PBD by poloxipan did not significantly increase over time. Despite the presence of a Michael acceptor system in its core structure (Reindl et al., 2009), this observation argues against the formation of a covalent bond between the proteins and the inhibitor.
PEPTIDE-DERIVED PBD INHIBITORS
MQSpTPL
The design of the inhibitor peptides for the PBD of Plk1 was based on an elegant peptide library screening approach carried out by Yaffe and his co-workers (Elia et al., 2003a; 2003b). The results demonstrate that PBDs from yeast, Xenopus, and human Plks recognize similar phosphoserine/threonine (pSer/ pThr)-containing motifs. With PMQSpTPL and MQSpTPL as the preferred binding motifs of the Plk1 PBD originating from the MAXXXXSpTXXXXAKK framework (X, any amino acid residue), PoloBoxtide designed as MAGPMQSpTPLNGAKK (see below) was successfully used in various biochemical assays (Elia et al., 2003a; 2003b).
Crystal structure of the PBD of human Plk1 with bound phosphopeptide, MQSpTPL, shows that the phosphopeptide sits in a pocket that is formed by PB1 and PB2. Lys540 and His538 residues are the only residues that make direct contact with the phosphate group in a form of ‘pincer grip’, which is stabilized by a network of van der Waals interactions and hydrogen bonds between water molecules and other conserved residues within the PBD (Cheng et al., 2003; Elia et al., 2003a). The pThr- or pSer-containing phosphopeptide interacts in an extended conformation with one end of a planar gap that is constituted between the two PBs. The explanation for the Ser preference at the -1 position seems to be that the side-chain of Ser forms a hydrogen bond with Trp414-a highly conserved residue in all PBDs. Significantly, this finding is consistent with an earlier functional study that had implicated Trp414 in strategic ligand binding by showing that a Trp414Phe mutation disrupts Plk1 localization to spindle poles and abrogates its function (Lee et al., 1998).
MAGPMQSpTPLNGAKK
The phosphopeptides named PoloBoxtides were experimentally determined to be the optimal sequence for PBD-dependent binding (Elia et al., 2003a). PoloBoxtide effectively interferes with Plk1 PBD-dependent binding in vitro in a concentration-dependent manner with an in vitro IC50 of 5 μM and Kd of 280 nM (Elia et al., 2003a; 2003b; Watanabe et al., 2009). Minimal PoloBoxtides, PMQSpTPL and MQSpTPL, described above have been used for many experiments.
LLCSpTPNG
The crystal structures of the PBD in complex with Cdc25C phospho-target peptide, LLCSpTPNG, revealed that the Trp414 of PB1 is fundamental in their recognition regardless of its phosphorylation status. Binding measurements demonstrated that a Trp414Phe mutation abolishes molecular recognition and diminishes centrosomal localization. Therefore, in addition to the His538 and Lys540 residues involved in phosphorylated target binding (Elia et al., 2003a), Plk1 centrosomal localization may also be promoted by the Trp414-dependent interaction (Garcia-Alvarez et al., 2007).
PLHSpT
Although the above phosphopeptides optimized for Plk1 PBD revealed the key interacting residues, the molecular basis of achieving the Plk1 PBD-binding specificity remains elusive. Among various Plk1 PBD-binding proteins, PBIP1 appears to exhibit a high level of binding affinity and specificity via its p-T78 motif (Kang et al., 2006).
Systematic analyses of the Plk1 PBD-PBIP1 p-T78 motif interaction led to the identification of a minimal PBD-binding sequence, PLHSpT. Structural analyses of the PBD in complex with the minimal p-T78 peptide revealed that the N-terminal Pro residue plays an important role in conferring the specificity by docking its side chain into a hydrophobic core surrounded by the Trp414, Phe535, and Arg516 residues, while concomitantly participating in a hydrogen bonding interaction between its carbonyl oxygen and the guanidinium moiety of Arg516 of the PBD. The PBDs of Plk2 and Plk3 possess Lys and Tyr residues at positions corresponding to the Arg516 and Phe535 residues in Plk1 PBD, respectively, thus failing to interact with the N-terminal Pro residue (Fig. 3).
PLHSpT exhibits a dose-dependent Plk1 PBD inhibition with a Kd of 0.445 μM (Yun et al., 2009). However, the hydrolytic lability of phosphoryl esters to phosphatases limits the use of phosphopeptides in cellular contexts. Development of hydrolytically- stable mimetics, in which the labile phosphoryl ester oxygen is replaced with non-hydrolysable methylene or difluoromethylene groups, offers one approach to circumvent this limitation (Yun et al., 2009). The phosphatase-resistant pThr mimetic (2S, 3R)-2-amino-3-methyl-4-phosphonobutyric acid (Pmab)- containing peptide, PLHS-Pmab, binds to Plk1 PBD with an undiminished affinity and specificity, and efficiently induces mitotic arrest, when microinjected into HeLa cells (Yun et al., 2009). This finding provide the proof of principle that specific inhibition of Plk1 PBD can be achievable by small PBD-binding mimetic peptides.
CONCLUSION
As expected from the importance of protein kinases in cell growth and signaling pathway, protein kinases represent the largest class of drug targets for pharmaceutical industry and academia alike. As of today, more than 10 kinase inhibitors have already reached the market and more than 200 kinase inhibitors are either in phase I or in phase II studies.
First-generation inhibitors that are designed based on the active conformation of kinase domain as shown in Tables 1 and 2 except SBE13, have been shown to be effective at reducing tumor cell growth both in vitro and in vivo. Although substantial progress has been made, this approach has confronted with limited specificity largely because of the conserved structures of various kinases. For the past decade, efforts have been made to improve the potency and specificity of kinase inhibitors and to overcome the off-target effects by using less-conserved inactive conformation of kinase domain (Liu and Gray, 2006). In an inactive conformation, an additional hydrophobic pocket with less conserved surrounding residues is frequently created. As an example of this second approach, virtual screening using a homology model of inactive human Plk1 led to the identification of 13 candidate compounds that exhibit anti-proliferative effects in HeLa cells. Among them, SBE13 shows EC50 values of 12- 39 μM in various cancer cells of different origin (Keppner et al., 2009), brightening the future of the development of second generation inhibitors.
Notably, however, although development of anti-kinase domain inhibitors have been widely sought by a large number of pharmaceutical companies, one of the common problems associated with this class of inhibitors is a high level of crossreactivity, as demonstrated by a recent study that closely examined previously characterized 38 kinase inhibitors (Karaman et al., 2008). Mainly due to the highly conserved ATP binding site within the catalytic domain of all the protein kinases, most of the currently developed kinase domain inhibitors exhibit less than acceptable selectivity profile. In addition, many of these inhibitors also display a broad spectrum of inhibition with a high level of cross-reactivity, because they have certain combinations of active site residues in the ATP binding site prone to developing ligand promiscuity.
Accumulating evidence suggests that targeting an essential non-catalytic domain could be an alternative strategy to overcome the cross-reactivity and drug resistance commonly associated with kinase domain inhibitors. One attractive direction that has recently gained momentum is to develop small molecule protein-protein interaction inhibitors that target pivotal biological processes required for cancer cell viability. It has been well documented that peptide-derived inhibitors, such as peptide mimetics or peptoids, are extremely potent and highly specific with few toxicological problems, thus avoiding many of their disadvantages that small molecule inhibitors do have. Also, they do not accumulate in organs or suffer from drug-drug interactions as many small molecules do. In this regard, targeting the polo-box domain of Plk1 may serve as an attractive approach to overcome many of the problems associated with inhibitors targeting the kinase domain of Plk1. Because PBDs are unique to the family of Plks, inhibition of Plk1 PBD was proposed to obtain potentially more specific inhibitors. In fact, two of the small-molecule inhibitors of the Plk1 PBD reported subsequently were shown to inhibit mitotic progression of cancer cells with promising specificity (Table 3). However, peptidebased inhibitors have their own drawbacks such as poor stability and transcellular permeability. Cyclic peptides are of considerable interest as drug candidates as their conformational rigidity and membrane permeability through the elimination of the charged termini may enhance their binding affinity for target protein while reducing the entropy loss.
On the basis of advanced technologies that allow efficient structure-based drug design with powerful screening platforms for drug activity and target selectivity, it is expected that novel compounds with increased potency, kinase specificity, and drug-like properties will soon be available for clinical evaluation. Furthermore, ongoing studies would provide new template molecules for designing inhibitors for docking sites on ATP and PBD-binding region. In summary, Plk1 represents an appealing target for anti-cancer therapy. Further evaluation is necessary to discern the utility of Plk1 inhibitors in a clinical setting and to determine the performance of these inhibitors in comparison to other currently available anti-mitotic agents.
Acknowledgments
The authors gratefully acknowledge financial support from the Korea Basic Science Institute’s International Joint Research Program grant F30601 (J.K.B).
References
- 1.Archambault V., Glover D.M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. (2009);10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 2.Bandeiras T.M., Hillig R.C., Matias P.M., Eberspaecher U., Fanghanel J., Thomaz M., Miranda S., Crusius K., Putter V., Amstutz P., et al. Structure of wild-type Plk1 kinase domain in complex with a selective DARPin. Acta Cryst. (2008);D64:339–353. doi: 10.1107/S0907444907068217. [DOI] [PubMed] [Google Scholar]
- 3.Barr F., Sillje H., Nigg E. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. (2004);5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 4.Beria I., Valsasina B., Brasca M.G., Caruso M., Ferguson R.R., Lansen J., Moll J., Pesenti E., Posteri H., Rocchetti M. Antitumoral activity of pyrazoloquinazoline derivatives as potent oral Plk1 specific inhibitors. Eur. J Cancer. (2008);6:136. [Google Scholar]
- 5.Beria I., Ballinari D., Bertrand J.A., Borghi D., Bossi R.T., Brasca M.G., Ferguson R.D., Lansen J., Rocchetti M., Storici M., et al. Identification of 4,5-Dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as a new class of orally and selective Polo-like kinase 1inhibitors. J. Med. Chem. (2010);53:3532–3551. doi: 10.1021/jm901713n. [DOI] [PubMed] [Google Scholar]
- 6.Cheng K.Y., Lowe E.D., Sinclair J., Nigg E.A., Johnson L.N. The crystal structure of the human polo-like kinase 1 polo box domain and its phosphor-peptide complex. EMBO J. (2003);22:5757–5768. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckerdt F., Yuan J., Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. (2005);24:267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 8.Elez R., Piiper A., Kronenberger B., Kock M., Brendel M., Hermann E., Pliquett U., Neumann. E., Zeuzem S. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene. (2003);22:69–80. doi: 10.1038/sj.onc.1206038. [DOI] [PubMed] [Google Scholar]
- 9.Elia A.E., Rellos P., Haire L.F., Chao J.W., Ivins F.J., Hoepker K., Mohammad D., Cantley L.C., Smerdon S.J., Yaffe M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. (2003a);115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 10.Elia A.E., Cantley L.C., Yaffe M.B. Proteomic screen finds pSer/pThr binding domain localizing Plk1 to mitotic substrates. Science. (2003b);299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 11.Elling R.A., Fucini R.V., Romanowski M.J. Structures of the wild-type and activated catalytic domains of Brachydanio rerio Polo-like kinase 1 (Plk1): changes in the active-site conformation and interactions with ligands. Acta Cryst. (2008a);D64:909–918. doi: 10.1107/S0907444908019513. [DOI] [PubMed] [Google Scholar]
- 12.Elling R.A., Fucini R.V., Hanan E.J., Barr K.J., Zhu J., Paulvannan K., Yang W., Romanowski M.J. Structure of the Brachydanio rerio Polo-like kinase 1 (Plk1) catalytic domain in complex with an extended inhibitor targeting the adaptive pocket of the enzyme. Acta Cryst. (2008b);F64:686–691. doi: 10.1107/S1744309108019623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmitte K.A., Adjebang G.M., Andrews C.W., Badiang J.G., Bambal R., Chamberlain S.D., Davis-Ward R.G., Dickson H.D., Hassler D.F., Hornberger K.R., et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg. Med. Chem. Lett. (2009);19:1694–1697. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- 14.Erskine S., Madden L., Hassler D., Smith G., Copeland R., Gontarek R. Biochemical characterization of GSK461364A: A novel, potent, and selective inhibitor of Polo-like kinase 1 (Plk1).; Presented at the 98th American Association for Cancer Research Annual Meeting; April 14-18; Los Angeles. (2007). [Google Scholar]
- 15.Fucini R.V., Hanan E.J., Romanowski M.J., Elling R.A., Lew W., Barr K.J., Zhu J., Yoburn J.C., Liu Y., Fahr B.T., et al. Design and synthesis of 2-amino-pyrazolopyridines as Polo-like kinase 1 inhibitors. Bioorg. Med. Chem. Lett. (2008);18:5648–5652. doi: 10.1016/j.bmcl.2008.08.095. [DOI] [PubMed] [Google Scholar]
- 16.Gandhi L., Chu Q.S., Stephenson J., Johnson B.E., Govindan R., Bonomi P., Eaton K., Fritsch H., Munzert G., Socinski M. An open label phase II trial of the Plk1 inhibitor BI 2536, in patients with sensitive relapsed small cell lung cancer (SCLC). J. Clin. Oncol. (2009);27:8108. [Google Scholar]
- 17.Garcia-Alvarez B., de Carcer G., Ibanez S., Bragado-Nilsson E., Montoya G. Molecular and structural basis of pololike kinase 1 substrate recognition: Implications in centrosomal localization. Proc. Natl. Acad. Sci. USA. (2007);104:3107–3112. doi: 10.1073/pnas.0609131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garland L.L., Taylor C., Pilkington D.L., Cohen J.L., Von Hoff D.D. A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase 1 interacting properties, in patients with advanced solid tumors. Clin. Cancer Res. (2006);12:5182–5189. doi: 10.1158/1078-0432.CCR-06-0214. [DOI] [PubMed] [Google Scholar]
- 19.Gilmartin A.G., Bleam M.R., Richter M.C., Erskine S.G., Kruger R.G., Madden L., Hassler D.F., Smith G.K., Gontarek R.R., Courtney M.P., et al. Distinct concentration-dependent effects of the polo-like kinase 1 specific inhibitor GSK461364A, including differential effect on apoptosis. Cancer Res. (2009);69:6969–6977. doi: 10.1158/0008-5472.CAN-09-0945. [DOI] [PubMed] [Google Scholar]
- 20.Gleixner K., Ferenc V., Peter B., Gruze A., Meyer R., Hadzijusufovic, Cerny-Reiterer S., Mayerhofer M., Pickl W.F., Valent P., et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. (2010);15:1513. doi: 10.1158/0008-5472.CAN-09-2181. [DOI] [PubMed] [Google Scholar]
- 21.Gumireddy K., Reddy M.V., Cosenza S.C., Boominathan R., Baker S.J., Papathi N., Jiang J., Holland J., Reddy E.P. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. (2005);7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Hanan E.J., Fucini R.V., Romanowski M.J., Elling R.A., Lew W., Purkey H.E., Vanderporten E.C., Yang W. Design and synthesis of 2-amino-isoxazolopyridines as Polo-like kinase inhibitors. Bioorg. Med. Chem. Lett. (2008);18:5186–5189. doi: 10.1016/j.bmcl.2008.08.091. [DOI] [PubMed] [Google Scholar]
- 23.Johnson E.F., Stewart K.D., Woods K.W., Giranda V.L., Luo Y. Pharmacological and functional comparison of the Polo-like kinase family: insight into inhibitor and substrate specificity. Biochemistry. (2007);46:9551–9563. doi: 10.1021/bi7008745. [DOI] [PubMed] [Google Scholar]
- 24.Kang Y.H., Park J.E., Yu L.R., Soung N.K., Yun S.M., Bang J.K., Seong Y.S., Yu H., Garfield S., Veenstra T.D., et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1- PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell. (2006);24:409–422. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Karaman M.W., Herrgard S., Treiber D.K., Gallant P., Atteridge C.E., Campbell B.T., Chan K.W., Ciceri P., Davis M.I., Edeen P.T., et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. (2008);26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 26.Keppner S., Proschak E., Schneider G., Spankuch B. Identification and validation of a potent type II inhibitor of inactive Polo-like kinase 1. Chem. Med. Chem. (2009);4:1806–1809. doi: 10.1002/cmdc.200900338. [DOI] [PubMed] [Google Scholar]
- 27.Knecht R., Elez R., Oechler M., Solbach C., von Ilberg C., Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. (1999);59:2794–2797. [PubMed] [Google Scholar]
- 28.Knecht R., Oberhauser C., Strebhardt K. PLK (pololike kinase), a new prognostic marker for oropharyngeal carcinomas. Int. J. Cancer. (2000);89:535–536. [PubMed] [Google Scholar]
- 29.Kneisel L., Strebhardt K., Bernd A., Wolter M., Binder A., Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J. Cutan. Pathol. (2002);29:354–358. doi: 10.1034/j.1600-0560.2002.290605.x. [DOI] [PubMed] [Google Scholar]
- 30.Kops G.J., Weaver B.A., Cleveland D.W. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. (2005);5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 31.Kothe M., Kohls D., Low S., Coli R., Cheng A.C., Jacques S.L., Johnson T.L., Lewis C., Loh C., Ding Y-H., et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry. (2007a);46:5960–5971. doi: 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- 32.Kothe M., Kohls D., Low S., Coli R., Rennie G.R., Feru F., Kuhn C., Ding Y-H. Selectivity-determining residues in Plk1. Chem. Biol. Drug Dis. (2007b);70:540–546. doi: 10.1111/j.1747-0285.2007.00594.x. [DOI] [PubMed] [Google Scholar]
- 33.Laquerre S., Sung S.-M., Gilmartin A., Courtney M., Ho M., Salovich J., Cheung M., Kuntz K., Huang P., Jackson J. A potent and selective Polo-like kinase 1 (Plk1) inhibitor (GSK461364A) induces cell cycle arrest and growth inhibition of cancer cell.; Presented at the 98th American Association for Cancer Research Annual Meeting; April 14-18; Los Angeles. (2007). [Google Scholar]
- 34.Lee K.S., Grenfell T.Z., Yarm F.R., Erikson R.L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian Polo like kinase. Proc. Natl. Acad. Sci. USA. (1998);95:9301–9306. doi: 10.1073/pnas.95.16.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenart P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J.J., Hoffmann M., Rettig W.J., Kraut N., Peters J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. (2007);17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 36.Leung G.C., Hudson J.W., Kozarova A., Davidson A., Dennis J.W., Sicheri F. The Sak Polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat. Struct. Biol. (2002);9:719–724. doi: 10.1038/nsb848. [DOI] [PubMed] [Google Scholar]
- 37.Liao J.J.-L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. (2007);50:409–424. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- 38.Liao C., Park J.E., Bang J.K., Nicklaus M.C., Lee K.S. Probing binding modes of small molecule inhibitors to the Polo-box domain of human Polo-like kinase. ACS Med. Chem. Lett. (2010);1:110–114. doi: 10.1021/ml100020e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y., Gray N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. (2006);2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y., Shreder K.R., Gai W., Corral S., Ferris D.K., Rosenblum. J.S. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo like kinase. Chem. Biol. (2005);12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Liu X., Lei M., Erikson R.L. Normal cells, but not cancer cells, survive Plk1 depletion. Mol. Cell. Biol. (2006);26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo J., Emanuele M.J., Li D., Creighton C.J., Schlabach M.R., Westbrook T.F., Wong K-K., Elledge S.J. A Genome- wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. (2009);137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McInnes C., Mezna M., Fishcher P.M. Progress in the discovery of polo-like kinase inhibitors. Curr. Top. Med. Chem. (2005);5:181–197. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 44.McInnes C., Mazumdar A., Mezna M., Meades C., Midgley C., Scaerou F., Carpenter L., Mackenzie M., Taylor P., Glover D., et al. Inhibitors of Polo-like kinase reveal roles in spindlepole maintenance. Nat. Chem. Biol. (2006);2:608–617. doi: 10.1038/nchembio825. [DOI] [PubMed] [Google Scholar]
- 45.Mross K., Frost A., Steinbild S., Hedbom S., Rentschler J., Kaiser R., Rouyrre N., Trommeshauser D., Hoesl C.E., Munzert G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. (2008);26:5511–5517. doi: 10.1200/JCO.2008.16.1547. [DOI] [PubMed] [Google Scholar]
- 46.Olmos D., Allred A., Sharma R., Brunetto A., Smith D., Murray S., Barker D., Taegtmeyer A., de Bono J., Blagden S. Phase I first-in-human study of the polo-like kinase 1 selective inhibitor, GSK461364A, in patients with advanced solid tumors. J. Clin. Oncol. (2009);27:3536. [Google Scholar]
- 47.Palmieri D., Hau E., Qian Y. Preclinical studies investigating the efficacy of GSK461364A, an inhibitor of Polo-like kinase 1, for the prevention of breast cancer brain meta-stases.; Presented at the American Association for Cancer Research; April 18-22; Denver. (2009). [Google Scholar]
- 48.Pandha H.S., Protheroe A., Wylie J., Parker C., Chambers J., Bell S., Munzert G. An open label phase II trial of BI 2536, a novel Plk1 inhibitor, in patients with metastatic hormone refractory prostate cancer (HRPC). J. Clin. Oncol. (2008);26:14547. [Google Scholar]
- 49.Park J.-E., Soung N.-K., Johmura Y., Kang Y.H., Liao C., Lee K.H., Park C.H., Nicklaus M.C., Lee K.S. Polo-box domain: a versatile mediator of polo-like kinase function. Cell. Mol. Life Sci. (2010);67:1957–1970. doi: 10.1007/s00018-010-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peters U., Cherian J., Kim J.H., Kwok B.H., Kapoor T.M. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat. Chem. Biol. (2006);2:618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- 51.Petronczki M., Lenart P., Peters J.-M. Polo on the risefrom mitotic entry to cytokinesis with Plk1. Dev. Cell. (2008);14:646–659. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 52.Reindl W., Yuan J., Kramer A., Strebhardt K., Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domaindependent protein-protein interactions. Chem. Biol. (2008);15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 53.Reindl W., Yuan J., Kramer A., Strebhardt T., Berg T. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem. (2009);10:1145–1148. doi: 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- 54.Rudolph D., Steegmaier M., Grauert M., Baum A., Quant J., Garin-Chesa P., Adolf G.R. Characterization of BI 6727, a novel Polo-like kinase inhibitor with a distinct pharmacokinetic profile and efficacy in a model of taxane-resistant colon cancer.; 20th EORTC-NCI-American Association for Cancer Research Symposium on Molecular Targets and Cancer Therapeutics; Oct. 21-24; (2008). p. 430. [Google Scholar]
- 55.Rudolph D., Steegmaier M., Hoffmann M., Grauert M., Baum A., Quant J., Haslinger C., Garin-Chesa P., Adolf G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. (2009);15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 56.Santamaria A., Neef R., Eberspacher U., Eis K., Husemann M., Mumberg D., Prechtl S., Schulze V., Siemeister G., Wortmann L., et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol. Biol. Cell. (2007);18:4024–4036. doi: 10.1091/mbc.E07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato Y., Onozaki Y., Sugimoto T., Kurihara H., Kamijo K., Kadowaki C., Tsujino T., Watanabe A., Otsuki S., Mitsuya M., et al. Imidazopyridine derivatives as potent and selective Pololike kinase inhibitors. Bioorg. Med. Chem. Lett. (2009);19:4673–4678. doi: 10.1016/j.bmcl.2009.06.084. [DOI] [PubMed] [Google Scholar]
- 58.Schoffski P., Awada A., Dumez H., Gil T., Bartholomeus S., Selleslach J., Taton M., Fritsch H., Peter N., Munzert G. A Phase I single dose escalation study of the novel Polo-like kinase 1 inhibitor BI 6727 in patients with advanced solid tumors. Eur. J. Cancer. (2008);6:14–15. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 59.Steegmaier M., Hoffmann M., Baum A., Lenart P., Petronczki M., Krssak M., Gurtler U., Garin-Chesa P., Lieb S., Kraut N., et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. (2007);17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 60.Stevenson C.S., Capper E.A., Roshak A.K., Marquez B., Eichman C., Jackson J.R., Mattern M., Gerwick W.H., Jacobs R.S., Marshall L.A. The identification and characterization of the marine natural product scytonemin as a novel antiproliferative pharmacophore. J. Pharmacol. Exp. Ther. (2002);303:858–866. doi: 10.1124/jpet.102.036350. [DOI] [PubMed] [Google Scholar]
- 61.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. (2010);9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 62.Strebhardt K., Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer. (2006);6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 63.Sur S., Pagliarini R., Bunz F., Rago C., Diaz L.A. Jr., Kinler K.W., Vogelstein B., Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. USA. (2009);106:3964–3969. doi: 10.1073/pnas.0813333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takagi M., Honmura T., Watanabe S., Yamaguchi R., Nogawa M., Nishimura I., Katoh F., Matsuda M., Hidaka H. In vivo antitumor activity of a novel sulfonamide, HMN-214, against human tumor xenografts in mice and the spectrum of cytotoxicity of its active metabolite, HMN-176. Invest. New Drugs. (2003);21:387–399. doi: 10.1023/a:1026282716250. [DOI] [PubMed] [Google Scholar]
- 65.Takai N., Hamanaka R., Yoshimatsu J., Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. (2005);24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 66.Takakai T., Trenz K., Costanzo V., Petronczki M. Polo-like kinase 1 reaches beyond mitosis-cytokinesis, DNA damage response, and development. Curr. Opin. Cell Biol. (2008);20:650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka H., Ohshima N., Ikenoya M., Komori K., Katoh F., Hidaka H. HMN-176, an active metabolite of the synthetic antitumor agent HMN-214, restores chemosensitivity to multidrug-resistant cells by targeting the transcription factor NF-Y. Cancer Res. (2003);63:6942–6947. [PubMed] [Google Scholar]
- 68.Van de Weerdt B.C., Medema R.H. Polo-like kinases: a team in control of the division. Cell Cycle. (2006);5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 69.Von Pawel J., Reck M., Digel W., Kortsik C., Thomas M., Frickhofen N., Schuler M., Gaschler-Markefski B., Hanft G., Sebastian M. Randomized phase II trial of two dosing schedules of BI 2536, a novel Plk1 inhibitor, in patients with relapsed advanced or metastatic non-small cell lung cancer (NSCLC). J. Clin. Oncol. (2008);26:8030. [Google Scholar]
- 70.Vose J., Young A., Friedberg J.W., Waller E.K., Cheson B.D., Trommeshauser D., Munzert G. Phase I doseescalation trial of BI 2536, a polo-like kinase 1 inhibitor, in relapsed and refractory Non-Hodgkin’s lymphoma. Blood. (2008);112:233. [Google Scholar]
- 71.Watanabe N., Sekine T., Takagi M., Iwasaki J., Imamoto N., Kawasaki H., Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domaindependent recognition. (2009);284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 72.Yuan J., Horlin A., Hock B., Stutte H.J., Rubsamen-Waigmann H., Strebhardt K. Polo-like kinase, a novel marker for cellular proliferation. Am. J. Pathol. (1997);150:1165–1172. [PMC free article] [PubMed] [Google Scholar]
- 73.Yun S.M., Moulaei T., Lim D., Bang J.K., Park J.E., Shenoy S. R., Liu F., Kang Y.H., Liao C., Soung N.K., et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. (2009);16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]