

Abstract
Glucagon-like peptide-1 (GLP-1) stimulates insulin secretion from pancreatic β-cells in a glucose-dependent manner. However, factors other than glucose that regulate the β-cell response to GLP-1 remain poorly understood. In this study, we examined the possible involvement of insulin and receptor tyrosine kinase signaling in regulation of the GLP-1 responsiveness of β-cells. Pretreatment of β-cells with HNMPA, an insulin receptor inhibitor, and AG1478, an epidermal growth factor receptor inhibitor, further increased the cAMP level and Erk phosphorylation in the presence of exendin-4 (exe-4), a GLP-1 agonist. When β-cells were exposed to a high concentration of glucose (25 mM), which stimulates insulin secretion, exe-4-induced cAMP formation declined gradually as exposure time was increased. This decreased cAMP formation was not observed in the presence of HNMPA. HNMPA was able to further increase the exe-4-induced insulin secretion when β-cells were exposed to high glucose for 18 h. Treatment of β-cells with insulin significantly decreased exe-4- induced cAMP formation in a dose-dependent manner. Lowering the phospho-Akt level by HNMPA or LY294002, a PI3K inhibitor, further augmented exe-4-induced cAMP formation and Erk phosphorylation. These results suggest that insulin contributes to fine-tuning of the β-cell response to GLP-1.
Keywords: β-cells, cAMP, Erk, GLP-1, insulin, RTK
INTRODUCTION
Glucagon-like peptide-1 (GLP-1) is an important incretin hormone released from intestinal L-cells. GLP-1 regulates glucose homeostasis at multiple levels, including stimulation of glucosedependent insulin secretion and inhibition of glucagon secretion, appetite, and gastric emptying. GLP-1 also enhances β-cell mass through regulation of β-cell proliferation and inhibition of β-cell apoptosis (Bregenholt et al., 2005; Hansotia and Drucker, 2005; Nauck et al., 1993). GLP-1 exerts its multiple biological effects through binding of its specific G-protein-coupled receptor, GLP1R. GLP1R is mainly expressed by pancreatic β-cells, and activation of this receptor in response to ligand stimulation increases the intracellular cAMP level, leading to stimulation of insulin secretion by two different pathways, PKA-dependent and PKA-independent exchange protein directly activated by cAMP (EPAC) pathways (Drucker et al., 1987; Fehmann et al., 1995). Subsequently, PKA and EPAC increase protein phosphorylation and intracellular Ca2+ concentration (Kieffer et al., 1999), causing increased synthesis and secretion of insulin by β-cells. Activated GLP1R is phosphorylated by GPCR kinase (GRK) and is internalized to the cytosol by binding to β-arrestin (Jorgensen et al., 2007). Receptor-bound β-arrestin induces Erk phosphorylation. Activation of Erk also affects insulin secretion and proliferation of β-cells (Sonoda et al., 2008). Activation of the phosphatidylinositol 3 kinase (PI3K) pathway by GLP-1 has also been reported either through direct activation by the β/γ-subunits of Gs (Kerchner et al., 2004) or through an indirect pathway involving c-src-mediated transactivation of the epidermal growth factor receptor (EGFR) (Buteau et al., 2003).
Molecular crosstalk between GLP1R and other signaling molecules is a matter of concern regarding the fine-tuning of insulin secretion and GLP-1 responsiveness of β-cells. GPCRs and receptor tyrosine kinases (RTKs) are transmembrane receptors that initiate intracellular signaling cascades in response to their ligands. Recent studies have shown that signal transduction initiated by GPCRs and RTKs is organized in mutually related signaling cassettes, leading to crosstalk between the RTK and GPCR signaling pathways (Natarajan et al., 2006). In pancreatic β-cells, in addition to GLP-1 and glucose-dependent insulinotropic polypeptide, insulin, insulin-like growth factor (IGF), and other growth factors are known to regulate insulin secretion as well as proliferation and apoptosis of β-cells (Creutzfeldt and Ebert, 1985; Loreti et al., 1974; Shepherd, 2004). Insulin secretion and proliferation by β-cells are inhibited in model systems in which the insulin receptor (IR), IGF receptor (IGFR), or IRS (IR substrate, an IR and IGFR downstream molecule) are knocked out/down (Da Silva Xavier et al., 2004). However, in β- cells exposed to insulin for an extended period, insulin secretion was inhibited, suggesting a possible autoregulation mechanism underlying insulin secretion by insulin signaling pathways (Loreti et al., 1974), although this possibility is still under debate (Kulkarni et al., 1999; Withers et al., 1998). Additionally, GLP-1-regulated insulin secretion with regard to the insulin autoregulatory process is of interest in exploring the fine-tuning of glucose homeostasis. However, the regulatory signaling process, including crosstalk between RTKs and GPCRs controlling the GLP-1 responsiveness of β-cells, is poorly understood. This information may provide critical clues for understanding signaling mechanisms for GLP-1-regulated glucose homeostasis. This study examined the possible involvement of RTKs, particularly insulin signaling, in regulation of the GLP-1 responsiveness of β-cells.
MATERIALS AND METHODS
Cell transfection and luciferase assays
INS-1 cells, rat pancreatic β-cells, were grown in RPMI 1640 medium containing 10% fetal bovine serum (FBS) at 37℃. MIN6 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) in the presence of 15% FBS. HEK293T cells were cultured in DMEM in the presence of 10% FBS. For luciferase assays, 1 day before transfection, cells were plated in 48-well plates and transfected with Effectene reagent (Qiagen, USA) according to the manufacturer’s instructions. Approximately 48 h after transfection, cells were treated with exendin-4 (exe-4), a GLP-1 agonist, for 6 h. Cells were then harvested, and luciferase activity in cell extracts was determined using a luciferase assay system according to the standard methods for a Wallac 1420 VICTOR3 multilabel counter (Perkin-Elmer, USA).
cAMP accumulation assay
Twenty-four hours before transfection, INS-1 cells were seeded into 12-well plates. Forty-eight hours later, cells were labeled with 2 μCi/ml [3H]adenine (NEN Life Science Products, USA) for 24 h. Cells were washed twice with phosphate-buffered saline (PBS) and then incubated at 37℃ for 20 min in serum-free RPMI containing 1 mM 1-methyl-3-isobutylxanthine and stimulated by exe-4 with or without signaling inhibitors for 15 min. The reactions were terminated by replacing medium with ice-cold 5% trichloroacetic acid containing 1 mM ATP and 1 mM cAMP. [3H]cAMP and [3H]ATP were separated on an AG50W-X4 resin (Bio-Rad, USA) and alumina columns as previously described (Oh et al., 2005). The cAMP accumulation was expressed as [3H]cAMP/([3H]ATP + [3H]cAMP) × 1000.
Western blotting
INS-1 cells were maintained in 6-well plates to reach 90% confluency. Before protein preparation for western blotting, cells were incubated for at least 16 h in serum-free medium. Prior to agonist stimulation, cells were washed and incubated with KRB buffer [118.5 mM NaCl, 4.8 mM KCl, 2.7 mM CaCl2, 1.2 mM KH2PO4, 1.1 mM MgSO4·7H2O, 25 mM NaHCO3, and 4% bovine serum albumin (BSA; pH 7.4)] and pretreated with various signaling inhibitors for 2 h. After agonist stimulation, cells were lysed with RIPA buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA (pH 8.0), 0.1% sodium dodecyl sulfate, and 0.5% sodium deoxycholate] containing a protease inhibitor cocktail (Roche, Germany). Protein concentrations were measured using the Bradford dye (Bio-Rad). Equal amounts of cellular extract were separated on 10% polyacrylamide gels and then transferred to nitrocellulose membranes for immunoblotting. EGFR and IR were detected by immunoblotting using mouse monoclonal antibodies (Santa Cruz Biotechnology, USA). Phosphorylated Erk1/2 and total Erk1/2 were detected by immunoblotting using mouse monoclonal anti-phospho-p44/42 MAPK (Santa Cruz Biotechnology) and anti-p44/42 MAPK (Cell Signaling Technology, USA), respectively. Phosphorylated Akt (Ser-473) and total Akt were detected by immunoblotting using rabbit polyclonal anti-phospho-Akt (Cell Signaling Technology) and anti-Akt (Cell Signaling Technology), respectively. Chemiluminescence detection was performed using the ECL reagent.
Insulin secretion
INS-1 cells were cultured in 24-well plates to reach 90% confluency. Cells were stimulated with a high concentration of glucose (25 mM). Prior to the assay, cells were washed and incubated with KRB buffer and treated with 25 mM glucose for 2 h at 37℃. Insulin secretion in KRB buffer was then determined using an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (Millipore, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA from INS-1 cells was extracted using TRI Reagent (Sigma-Aldrich, USA) according to the manufacturer’s instructions. Total RNA was then reverse transcribed using random hexamers and MMLV reverse transcriptase (Promega, USA). The cDNAs served as templates for subsequent PCR amplification of partial clones of EGFR and IR. PCR conditions were as follows: denaturation at 95℃ for 5 min, followed by different cycles at 95℃ for 30 s, 57℃ for 30 s, and 72℃ for 30 s.
Data analysis
All data are presented as means ± standard error of the mean (SEM) of at least three independent experiments. Group means were compared using Student’s t-test or one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test. P < 0.05 was accepted to indicate statistical significance.
RESULTS
Inhibition of IR and EGFR signaling up-regulates GLP1R-mediated signaling in pancreatic β-cells
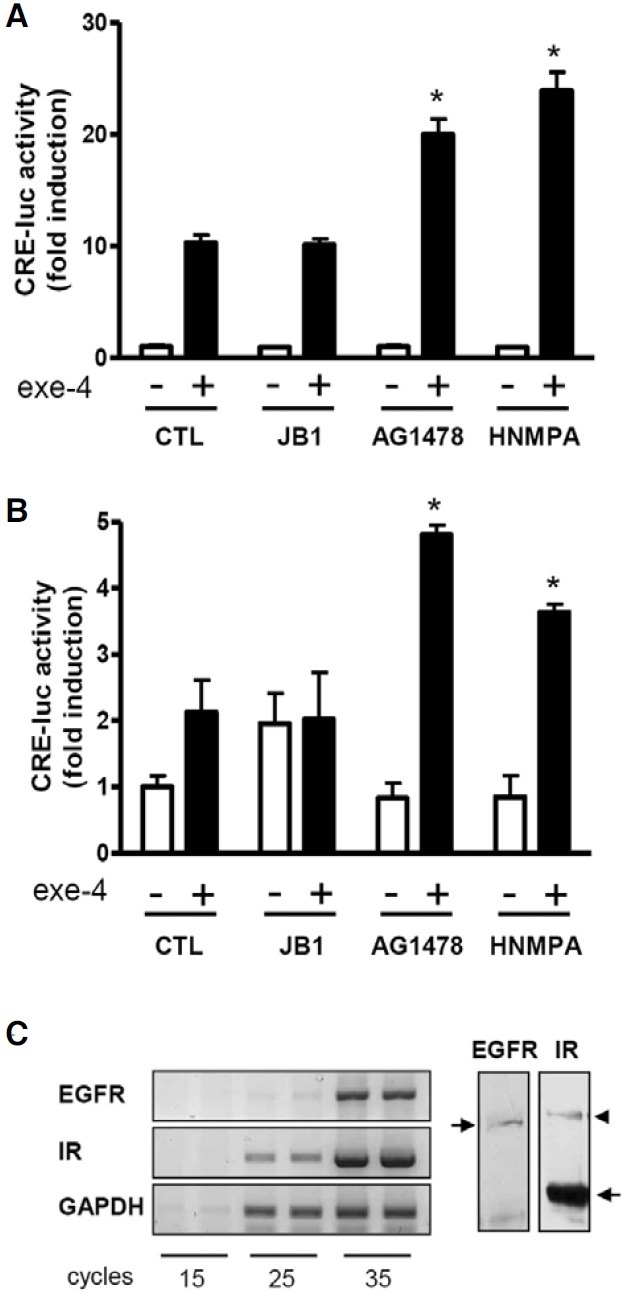
Pancreatic β-cells are known to express many types of RTKs such as IR, IGF1R, and EGFR (Harbeck et al., 1996; Huotari et al., 1998; Van Schravendijk et al., 1987). To investigate involvement of RTK signaling in the control of the GLP1R-mediated signaling cascade, INS-1 cells, rat pancreatic β-cells, were pretreated with the IGFR inhibitor JBI, the EGFR inhibitor AG1478, and the IR inhibitor HNMPA for 2 h prior to treatment with exe-4, a GLP-1 agonist. GLP1R activity was determined using the CRE-luc assay system. Exe-4 significantly increased CRE-luc activity by ~10-fold. Pretreatment of the cells with AG1478 and HNMPA, but not JB1, further increased the exe-4- induced CRE-luc activity (Fig. 1A). Similar results were obtained using MIN6 cells (Fig. 1B). The presence of EGFR and IR transcripts and proteins in INS-1 cells were examined using RT-PCR and Western blotting, respectively (Fig. 1C). Thus, there is a possibility that the IR and EGFR in β-cells under cultured condition can be continuously activated by insulin secreted from β-cells and serum components of the media. Although β-cells are known to express IGFR (Van Schravendijk et al., 1987), the contribution of IGFR-mediated signaling to GLP-1 response is likely marginal under cultured condition.
Fig. 1. Effect of RTK inhibitors on exe-4-induced CRE-luc activity in pancreatic β-cells. (A, B) AG1478 and HNMPA increase exe-4- induced CRE-luc activity. INS-1 cells (A) and MIN6 cells (B) harboring the CRE-luc reporter gene were pretreated with RTK inhibitors (JB1, a IGF1R inhibitor; 10 μM; AG1478, a EGFR inhibitor; 10 μM; HNMPA, an IR inhibitor; 2.5 mM) for 2 h before treatment with exe- 4 (0.1 μM). GLP1R activity by exe-4 was measured using the luciferase assay system. (C) EGFR and IR expression in INS-1 cells. Expression levels of EGFR and IR were determined by RT-PCR and western blotting in INS-1 cells. Arrows in western blot indicate EGFR and IR. Arrowhead shows IR precursor (* P < 0.05).
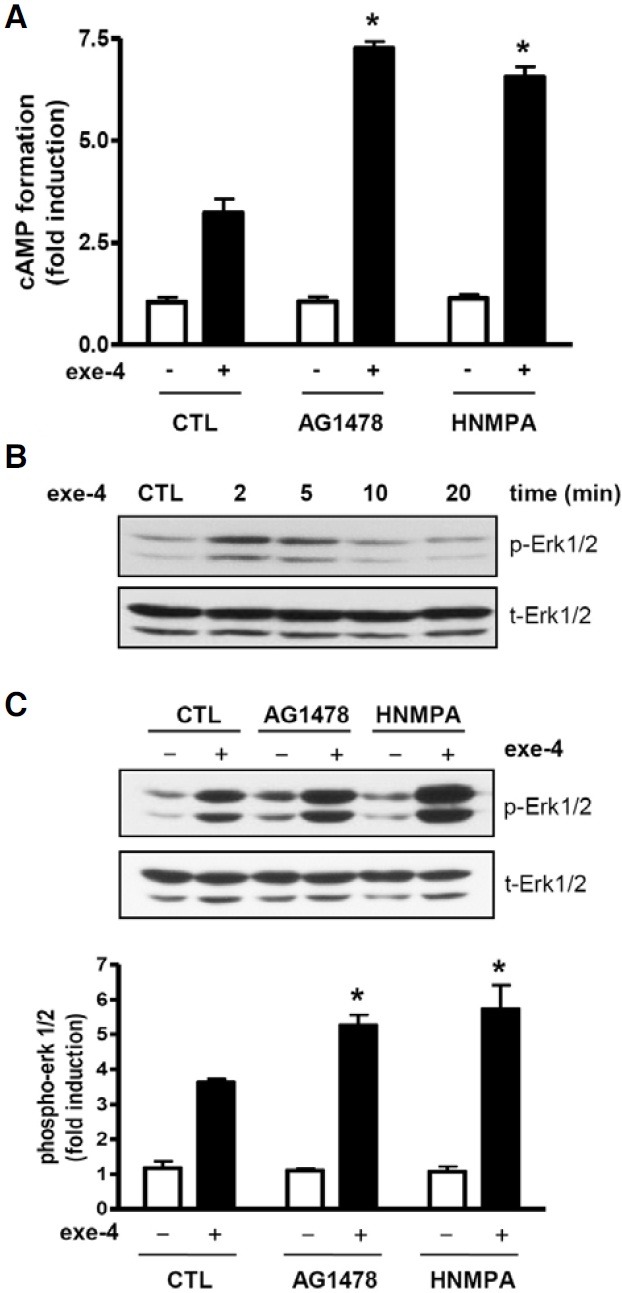
To further verify the effect of AG1478 and HNMPA on GLP1R-mediated signaling, cAMP accumulation and Erk phosphorylation in response to exe-4 in the presence or absence of AG1478 and HNMPA were determined in INS-1 cells. Exe-4 stimulated cAMP formation, which was significantly increased in AG1478- and HNMPA-pretreated INS-1 cells (Fig. 2A). This result agrees well with the finding obtained using the CRE-lucassay system (Fig. 1). We then examined the effect of AG1478 and HNMPA on exe-4-induced Erk phosphorylation. Exe-4 increased Erk phosphorylation in a time-dependent manner with a peak at ~2 to 5 min after exe-4 treatment and a return to basal levels 20 min after treatment (Fig. 2B). AG1478 and HNMPA significantly increased exe-4-induced Erk phosphorylation compared with control groups (Fig. 2C). These results suggest that EGFR and IR are likely involved in control of the β-cell response to GLP-1.
Fig. 2. AG1478 and HNMPA increase exe-4-induced cAMP formation and Erk phosphorylation. (A) AG1478 and HNMPA increase exe-4-induced cAMP accumulation. INS-1 cells were pretreated with AG1478 and HNMPA for 2 h before treatment with exe-4. cAMP accumulation was then measured. (B) Exe-4 induces Erk1/2 phosphorylation. INS-1 cells were exposed to exe-4 for indicated times. Exe-4-induced phosphorylation of Erk1/2 was determined using a specific phospho-Erk1/2 antibody. (C) AG1478 and HNMPA increase exe-4-induced Erk1/2 phosphorylation. INS-1 cells were pretreated with AG1478 and HNMPA for 2 h following treatment of exe-4 for 2 min. Immunoblotting for phosphorylated Erk1/2 was determined (* P < 0.05).
Effect of insulin on exe-4-induced cAMP formation and insulin release
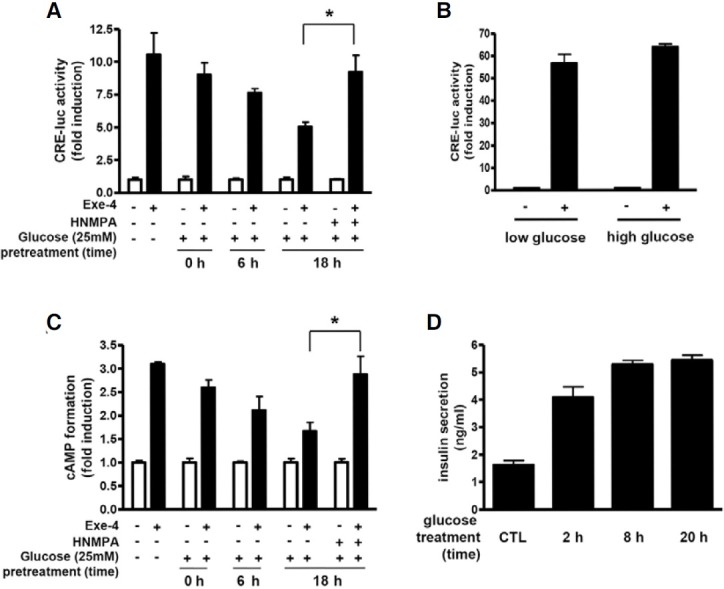
GLP-1 controls glucose homeostasis through stimulation of insulin secretion from pancreatic β-cells. However, the effect of insulin signaling on the β-cell response to GLP-1 has been poorly understood. Thus, we focused more on the effect of insulin signaling on GLP1R-mediated cellular responses. We examined GLP1R-mediated signaling under a high glucose condition (25 mM), which led to increased insulin secretion from β-cells. In INS-1 cells exposed to a high glucose, exe-4-induced CRE-luc activity was lowered continuously as the exposure time was extended (Fig. 3A). This decreased CRE-luc activity was rescued by pretreatment with HNMPA. Exe-4-induced CRE-luc activity in HEK293T cells was not affected by the high glucose concentration, indicating that this glucose effect is specific for INS-1 cells (Fig. 3B). We further confirmed the high glucose effect on GLP1R-mediated signaling by mea-suring cAMP accumulation by exe-4. Exe-4-induced cAMP formation was significantly decreased when INS-1 cells are exposed to a high glucose condition in a time-dependent manner. This decreased cAMP formation was significantly restored by pretreatment with HNMPA (Fig. 3C). The high glucose condition indeed led to increased insulin secretion from INS-1 cells (Fig. 3D).
Fig. 3. Time-dependent effect of high concentration of glucose on exe-4-induced cAMP formation in INS-1 cells. (A) Effect of glucose concentration on exe-4-mediated CRE-luc activity. INS-1 cells expressing the CRE-luc reporter gene were pretreated with 25 mM glucose for indicated times and then exposed with exe-4 for 6 h. HNMPA was pretreated for 2 h before exe-4 treatment. Luciferase activity by exe-4 was measured. (B) Effect of glucose concentration on exe-4-mediated CRE-luc activity in HEK293T cells. HEK293T cells harboring the CRE-luc reporter gene were pretreated with 25 mM glucose for 2, 8 or 20 h before exe-4-induced CRE-luc activity was measured. (C) Effect of glucose concentration on exe-4-mediated cAMP accumulation. INS-1 cells were pretreated with 25 mM glucose at different times with or without HNMPA. Exe-4-induced cAMP formation was measured. (D) Temporal changes in insulin secretion in a high glucose condition. INS-1 cells were stimulated with a high concentration of glucose (25 mM) for 2, 8, or 20 h. Secreted insulin in the medium was detected by ELISA (* P < 0.05).
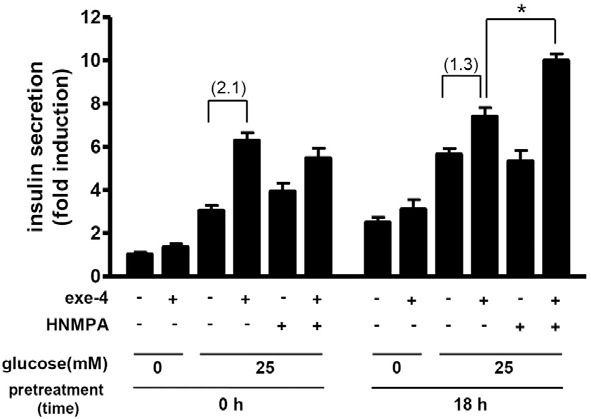
We further determined insulin release in response to exe-4 in this high glucose condition. Exe-4 significantly increased insulin release from INS-1 cells in the presence of glucose in both high glucose-pretreated and non-pretreated group but HNMPA alone failed to do so. It is of interest to note that the exe-4- induced insulin secretion in the high glucose-pretreated group was just 1.3-fold higher than the control (glucose only), while that in the non-pretreated group was 2.1-fold higher than the control (Fig. 4). HNMPA significantly increased exe-4-induced insulin secretion in the high glucose-pretreated group, while this HNMPA effect was not observed in the non-pretreated group (Fig. 4).
Fig. 4. Effect of high glucose on exe-4-induced insulin secretion. INS-1 cells were plated in 24-well plates and grown in RPMI 1640 medium with 10% FBS in the absence or presence of high glucose (25 mM) for 18 h. Insulin secretion in response to exe-4 and/or HNMPA was determined during another 2-h incubation of cells with KRB buffer in the absence of glucose or presence of high glucose. Secreted insulin in the medium was determined using ELISA. Fold-increases are in the parentheses (* P < 0.05).
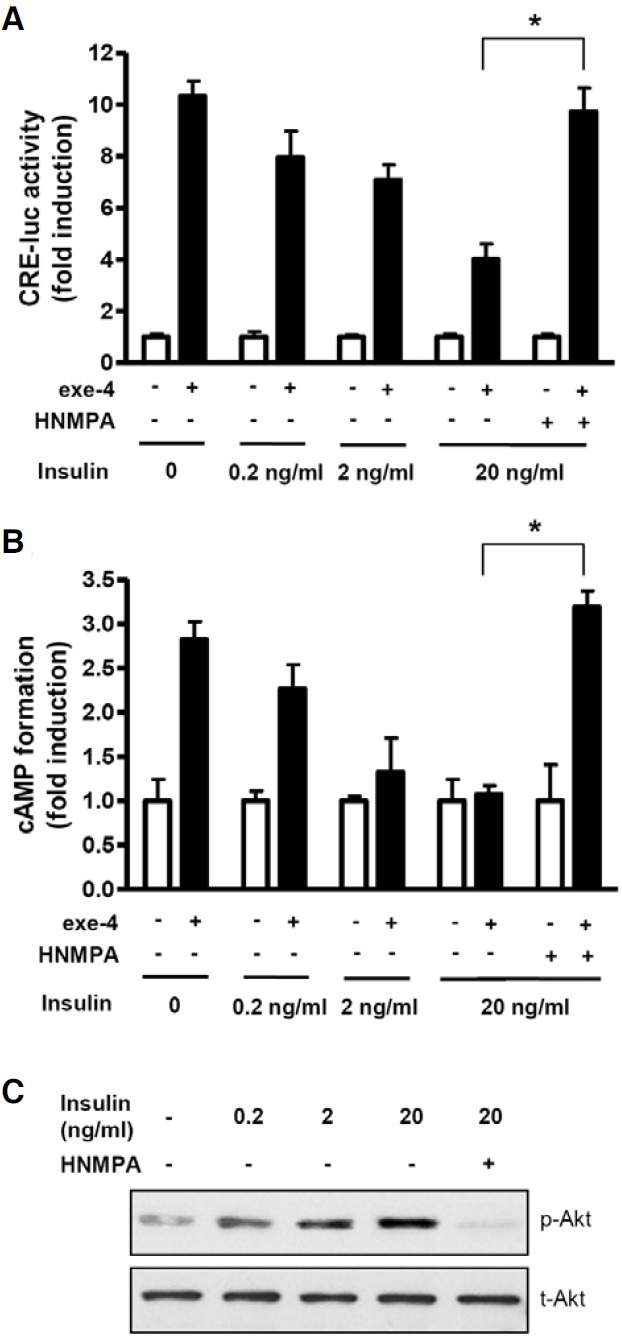
To confirm whether this inhibitory effect of high glucose concentration is due to secreted insulin, we directly treated INS-1 cells with increasing concentrations of insulin for 2 h prior to exe-4 treatment. Insulin pretreatment decreased exe-4-induced CRE-luc activity (Fig. 5A) and cAMP formation (Fig. 5B). These insulin-induced decreases in CRE-luc activity and cAMP formation were rescued by pretreatment with HNMPA (Figs. 5A and 5B). In β-cells, the PI3K/Akt pathway is a well-known insulin downstream signaling pathway (Leibiger et al., 1998; 2010). To further confirm whether this signaling cascade functions in INS- 1 cells, phospho-Akt levels were determined after treatment with increasing concentrations of insulin in the absence or presence of HNMPA. Insulin increased phospho-Akt levels in a dose-dependent manner, whereas pretreatment with HNMPA decreased phospho-Akt levels (Fig. 5C).
Fig. 5. Insulin inhibits exe-4-induced cAMP formation. (A) Effect of insulin on CRE-luc activity. INS-1 cells expressing the CRE-luc reporter gene were pretreated with 0.2, 2, or 20 ng/ml insulin in the absence or presence of HNMPA. CRE-luc activity by exe-4 was measured using the luciferase assay system. (B) Effect of insulin on cAMP accumulation. INS-1 cells were pretreated with an increasing concentration of insulin with or without HNMPA. Exe-4-mediated cAMP accumulation was measured. (C) Dose-dependent activity of insulin on Akt phosphorylation. INS-1 cells were pretreated with HNMPA for 2 h before treatment with insulin. Phosphorylation of Akt by different concentrations of insulin was determined by western blotting (* P < 0.05).
PI3K/Akt signaling is involved in the insulin-induced decrease in the β-cell response to GLP-1
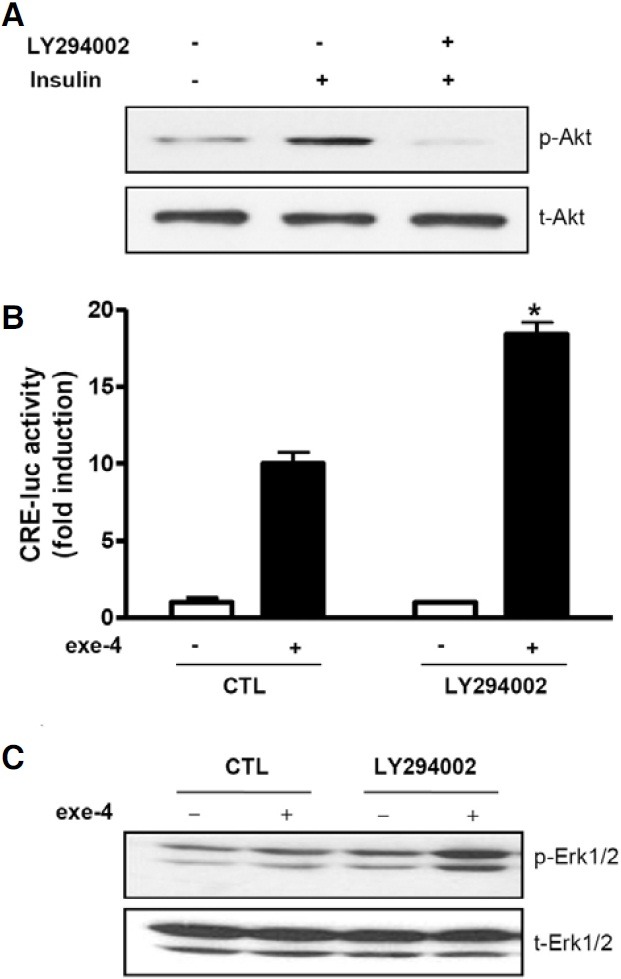
To determine whether the PI3K/Akt signaling cascade is involved in the insulin-induced decrease in the β-cell response to GLP-1, we treated INS-1 cells with LY294002, a PI3K inhibitor. LY294002 decreased phospho-Akt induced by insulin in INS-1 cells (Fig. 6A). This decreased phospho-Akt level was accompanied by an increased response of β-cells to GLP-1. LY294002 treatment significantly increased exe-4-induced cAMP formation (Fig. 6B) and Erk phosphorylation (Fig. 6C), indicating that insulin-mediated PI3K and Akt signaling are involved in the β- cell response to GLP-1.
Fig. 6. LY294002 augments exe-4-induced cAMP formation and Erk phosphorylation. (A) LY294002 inhibits insulin-mediated Akt phosphorylation. INS-1 cells were pretreated with LY294002 (10 μM) for 2 h. Phosphorylation of Akt by insulin (20 ng/ml) was determined by western blotting. (B) LY294002 increases exe-4-induced CRE-luc activity. INS-1 cells expressing the CRE-luc reporter gene were pretreated with LY294002 for 2 h before exe-4 treatment followed by detection of luciferase activity. (C) LY294002 increases exe-4-mediated Erk1/2 phosphorylation. INS-1 cells were pretreated with LY294002 for 2 h following treatment of exe-4 for 2 min. Immunoblotting for phosphorylated Erk1/2 was determined (* P < 0.05).
DISCUSSION
GLP-1 has attracted much attention for treatment of diabetes because of its multimodal actions to control glucose homeostasis. In pancreatic β-cells, GLP-1 stimulates insulin secretion by increasing cAMP levels. However, this insulin secretory action of GLP-1 is highly dependent on the glucose level. GLP-1 and forskolin, an activator of adenylyl cyclase, fail to enhance insulin secretion in the absence of glucose (Yajima et al., 1999). This glucose dependency is rather beneficial because it avoids hypoglycemia caused by inappropriate insulin secretion. This incretin effect of GLP-1 is greatly reduced in patients with type 2 diabetes mellitus, although similar GLP-1 secretion levels in response to oral glucose in both patients with type 2 diabetes and normal subjects have been observed (Kjems et al., 2003; Nauck et al., 1986). Thus, GLP-1 must be infused or administered at supraphysiologic levels in patients with type 2 diabetes; physiologic levels of GLP-1 generated by intravenous infusions have demonstrated little antiglycemic effect in these patients (Nauck, 2011). However, regulatory mechanisms to control the responsiveness of β-cells to GLP-1 have been poorly understood.
In this study, we demonstrated that insulin signaling lowers the β-cell response to GLP-1. HNMPA further augmented exe-4-induced cAMP levels and Erk phosphorylation in INS-1 cells. INS-1 cells express a substantial amount of IR as demonstrated by others (Harbeck et al., 1996; Huotari et al., 1998; Kulkarin et al., 1999) and in our present study. Insulin secreted from INS-1 cells likely contributes to this lowered response to GLP-1 through IR. Exposure of INS-1 cells to a high glucose condition increases insulin secretion in INS-1 cells. Under this condition, exe-4-induced cAMP levels were decreased as the exposure time was extended. HNMPA again attenuated the decrease in exe-4-induced cAMP levels caused by prolonged exposure to a high glucose condition. Direct treatment of INS-1 cells with increasing concentrations of insulin also caused a reduced GLP-1 response of the cells. As β-cells under a high glucose condition are exposed to insulin concentrations that are higher than those in the periphery (Zawalich et al., 1975), it is likely that glucose exerts a stimulatory effect on the β-cell response to GLP-1 in the initial phase but high concentrations of secreted insulin from β-cells decrease the β-cell response to GLP-1 in the late phase.
The concept that insulin inhibits its own secretion has been suggested for a long time (Best and Haist, 1941; Loreti et al., 1974). However, this autocrine effect of secreted insulin on β-cell function has been a matter of debate (Leibiger, 2002). Chronic administration of insulin or transplantation of insulinomas reduced pancreatic insulin content (Best and Haist, 1941; Koranyi et al., 1992). In contrast to these results, disruption of insulin signaling in the IRS-2 knockout mouse or in the β-cellrestricted IR knockout mouse led to reduction in the pancreatic insulin content in the adult state (Kulkarni et al., 1999; Withers et al., 1998) and selective loss of glucose-stimulated acute insulin secretion (Kulkarni et al., 1999). These debates, however, largely focused on the insulin effects on insulin secretion, biosynthesis, insulin content, and β-cell mass. No experiment regarding the insulin effect on the β-cell response to GLP-1 has been yet reported.
This inhibitory effect of insulin on the GLP-1 response of β-cells is likely mediated by the PI3K/Akt signaling pathway because lowering the phospho-Akt level by HNMPA and the PI3K inhibitor increased exe-4-induced cAMP levels and Erk phosphorylation. A major component of the IR-mediated signaling pathway in β-cells is activation of PI3K followed by Akt phosphorylation. Indeed, this signaling pathway is known to be involved in glucose/insulin-dependent up-regulation of insulin gene transcription in β-cells (Leibiger et al., 1998; 2010). Thus, the PI3K/Akt pathway likely plays a dual role in insulin-mediated β-cell control, stimulation of insulin gene expression and attenuation of the β-cell response to GLP-1. Regarding how Akt modulates the GLP-1 response, Akt may activate phosphodiesterase 3B (PDE3B), which then hydrolyzes cAMP to AMP. Increased activity of PDE3B may cause rapid breakdown of cAMPs that are produced by GLP-1 activation (Härndahl et al., 2002). PDE3B is highly expressed in β-cells and can be activated by interaction with Akt (Ahmad et al., 2007; Kitamura et al., 1999). Indeed, our preliminary experiment showed that pretreatment of INS-1 cells with cilostamide, a specific inhibitor of PDE3B, significantly increased exe-4-induced cAMP accumulation and Erk phosphorylation (data not shown). However, the detailed mechanism underlying the insulin-regulated β-cell response to GLP-1 needs to be further investigated.
In conclusion, understanding the regulatory mechanism to control the GLP-1 responsiveness of β-cells is important both physiologically and clinically. Our study suggests that insulin signaling through the PI3K/Akt pathway participates in regulation of the GLP-1 responsiveness of β-cells, which may contribute to the fine-tuning of insulin secretion in β-cells under physiological conditions.
Acknowledgments
This work was supported by grants from the Korea Research Foundation Grant (313-2008-2-E00262).
References
- 1.Ahmad F., Lindh R., Tang Y., Weston M., Degerman E., Manganiello V.C. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem. J. (2005);404:257–268. doi: 10.1042/BJ20060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Best C.H., Haist R.E. The effect of insulin administration on the insulin content of the pancreas. J. Physiol. (1941);100:142–146. doi: 10.1113/jphysiol.1941.sp003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bregenholt S., Moldrup A., Blume N., Karlsen A.E., Nissen Friedrichsen B., Tornhave D., Knudsen L.B., Petersen J.S. The long-acting glucagon-like peptide-1 analogue, liraglutide, inhibits beta-cell apoptosis in vitro. Biochem. Biophys. Res. Commun. (2005);330:577–584. doi: 10.1016/j.bbrc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Buteau J., Foisy S., Joly E., Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. (2003);52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- 5.Creutzfeldt W., Ebert R. New developments in the incretin concept. Diabetologia. (1985);28:565–573. doi: 10.1007/BF00281990. [DOI] [PubMed] [Google Scholar]
- 6.Da Silva Xavier G., Rutter J., Rutter G.A. Involvement of Per-Arnt-Sim (PAS) kinase in the stimulation of preproinsulin and pancreatic duodenum homeobox 1 gene expression by glucose. Proc. Natl. Acad. Sci. USA. (2004);101:8319–8324. doi: 10.1073/pnas.0307737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drucker D.J., Philippe J., Mojsov S., Chick W.L., Habener J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA. (1987);84:3434–3438. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fehmann H.C., Goke R., Goke B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose- dependent insulin releasing polypeptide. Endocr. Rev. (1995);16:390–410. doi: 10.1210/edrv-16-3-390. [DOI] [PubMed] [Google Scholar]
- 9.Hansotia T., Drucker D.J. GIP and GLP-1 as incretin hormones: lessons from single and double incretin receptor knockout mice. Regul. Pept. (2005);128:125–134. doi: 10.1016/j.regpep.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Härndahl L., Jing X.J., Ivarsson R., Degerman E., Ahrén B., Manganiello V.C., Renström E., Holst L.S. Important role of phosphodiesterase 3B for the stimulatory action of cAMP on pancreatic beta-cell exocytosis and release of insulin. J. Biol. Chem. (2002);277:37446–3755. doi: 10.1074/jbc.M205401200. [DOI] [PubMed] [Google Scholar]
- 11.Harbeck M.C., Louie D.C., Howland J., Wolf B.A., Rothenberg P.L. Expression of insulin receptor mRNA and insulin receptor substrate 1 in pancreatic islet beta-cells. Diabetes. (1996);45:711–717. doi: 10.2337/diab.45.6.711. [DOI] [PubMed] [Google Scholar]
- 12.Huotari M.A., Palgi J., Otonkoski T. Growth factormediated proliferation and differentiation of insulin-producing INS-1 and RINm5F cells: identification of betacellulin as a novel beta-cell mitogen. Endocrinology. (1998);139:1494–1499. doi: 10.1210/endo.139.4.5882. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen R., Kubale V., Vrecl M., Schwartz T.W., Elling C.E. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor beta-arrestin recruitment and signaling through Galpha(s). J. Pharmacol. Exp. Ther. (2007);322:148–154. doi: 10.1124/jpet.107.120006. [DOI] [PubMed] [Google Scholar]
- 14.Kerchner K.R., Clay R.L., Mccleery G., Watson N., Mcintire W.E., Myung C.S., Garrison J.C. Differential sensitivity of phosphatidylinositol 3-kinase p110gamma to isoforms of G protein betagamma dimers. J. Biol. Chem. (2004);279:44554–44562. doi: 10.1074/jbc.M406071200. [DOI] [PubMed] [Google Scholar]
- 15.Kieffer T.J., Habener J.F. The glucagon-like peptides. Endocr. Rev. (1999);20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura T., Kitamura Y., Kuroda S., Hino Y., Ando M., Kotani K., Konishi H., Matsuzaki H., Kikkawa U., Ogawa W., et al. Insulin-induced phosphorylation and activa-tion of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol. Cell. Biol. (1999);19:6286–6296. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kjems L.L., Holst J.J., Volund A., Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. (2003);52:380–386. doi: 10.2337/diabetes.52.2.380. [DOI] [PubMed] [Google Scholar]
- 18.Koranyi L., James D.E., Kraegen E.W., abd Permutt M.A. Feedback inhibition of insulin gene expression by insulin. J. Clin. Invest. (1992);89:432–436. doi: 10.1172/JCI115602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni R.N., Brüning J.C., Winnay J.N., Postic C., Magnuson M.A., Kahn C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. (1999);96:329–339. doi: 10.1016/s0092-8674(00)80546-2. [DOI] [PubMed] [Google Scholar]
- 20.Leibiger I.B., Leibiger B., Moede T., Berggren P.O. Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 s6 kinase and CaM kinase pathways. Mol. Cell. (1998);1:933–938. doi: 10.1016/s1097-2765(00)80093-3. [DOI] [PubMed] [Google Scholar]
- 21.Leibiger I.B., Leibiger B., Berggren P.O. Insulin feedback action on pancreatic beta-cell function. FEBS Lett. (2002);532:1–6. doi: 10.1016/s0014-5793(02)03627-x. [DOI] [PubMed] [Google Scholar]
- 22.Leibiger B., Moede T., Uhles S., Barker C.J., Creveaux M., Domin J., Berggren P.O., Leibiger I.B. Insulin-feedback via PI3K-C2alpha activated PKBalpha/Akt1 is required for glucosestimulated insulin secretion. FASEB J. (2010);24:1824–1837. doi: 10.1096/fj.09-148072. [DOI] [PubMed] [Google Scholar]
- 23.Loreti L., Dunbar J.C., Chen S., Foa P.P. The autoregulation of insulin secretion in the isolated pancreatic islets of lean (obOb) and obese-hyperglycemic (obob) mice. Diabetologia. (1974);10:309–315. doi: 10.1007/BF02627732. [DOI] [PubMed] [Google Scholar]
- 24.Natarajan K., Berk B.C. Crosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinases. Methods Mol. Biol. (2006);332:51–77. doi: 10.1385/1-59745-048-0:51. [DOI] [PubMed] [Google Scholar]
- 25.Nauck M.A. Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am. J. Med. (2011);124:S3–18. doi: 10.1016/j.amjmed.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Nauck M., Stockmann F., Ebert R., Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. (1986);29:46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 27.Nauck M.A., Heimesaat M.M., Orskov C., Holst J.J., Ebert R., Creutzfeldt W. Preserved incretin activity of glucagon- like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Invest. (1993);91:301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh D.Y., Song J.A., Moon J.S., Moon M.J., Kim J.I., Kim K., Kwon H.B., Seong J.Y. Membrane-proximal region of the carboxyl terminus of the gonadotropin-releasing hormone receptor (GnRHR) confers differential signal transduction between mammalian and nonmammalian GnRHRs. Mol. Endocrinol. (2005);19:722–731. doi: 10.1210/me.2004-0220. [DOI] [PubMed] [Google Scholar]
- 29.Shepherd P.R. Secrets of insulin and IGF-1 regulation of insulin secretion revealed. Biochem. J. (2004);377:e1–2. doi: 10.1042/BJ20031747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonoda N., Imamura T., Yoshizaki T., Babendure J.L., Lu J.C., Olefsky J.M. Beta-Arrestin-1 mediates glucagonlike peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc. Natl. Acad. Sci. USA. (2008);105:6614–6619. doi: 10.1073/pnas.0710402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Schravendijk C.F., Foriers A., Van Den Brande J.L., Pipeleers D.G. Evidence for the presence of type I insulin- like growth factor receptors on rat pancreatic A and B cells. Endocrinology. (1987);121:1784–1788. doi: 10.1210/endo-121-5-1784. [DOI] [PubMed] [Google Scholar]
- 32.Withers D.J., Gutierrez J.S., Towery H., Burks D.J., Ren J.M., Previs S., Zhang Y., Bernal D., Pons S., Shulman G.I., et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. (1998);391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 33.Yajima H., Komatsu M., Schermerhorn T., Aizawa T., Kaneko T., Nagai M., Sharp G.W., Hashizume K. cAMP enhances insulin secretion by an action on the ATP-sensitive K channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes. (1999);48:1006–1012. doi: 10.2337/diabetes.48.5.1006. [DOI] [PubMed] [Google Scholar]
- 34.Zawalich W.S., Karl R.C., Ferrendelli J.A., Matschinsky F.M. Factors governing glucose induced elevation of cyclic 3′5′ AMP levels in pancreatic islets. Diabetologia. (1975);11:231–235. doi: 10.1007/BF00422327. [DOI] [PubMed] [Google Scholar]