

Abstract
Interferon-α (IFN-α) inhibits the replication of hepatitis B virus (HBV) in vivo and in vitro, but the molecular mechanism of this inhibition has been elusive. We found that while HBV replication in transfected human hepatoma Huh-7 cell was severely inhibited by IFN-α treatment as reported previously, this inhibition was markedly impaired in the cell in which the expression of IFN-inducible, double- stranded RNA-dependent protein kinase (PKR) was stably and specifically suppressed through RNA-interference. Intracellular level of viral capsids was down-regulated likewise in a PKR-dependent manner, whereas that of HBV transcripts including the viral RNA pregenome was not affected by IFN-α treatment. Ectopic expression of PKR also resulted in the reduction of viral capsids with concomitant increase of phosphorylated eIF2α. These results suggested that PKR functions as a key mediator of IFN-α in opposing HBV replication, most likely through the inhibition of protein synthesis.
Keywords: antiviral mechanism, Hepatitis B virus, IFN-α, PKR
INTRODUCTION
Hepatitis B virus (HBV) causes acute and chronic hepatitis in humans. More than 350 million people worldwide suffer from chronic HBV infection and face the risk of its progression to liver cirrhosis and hepatocellular carcinoma (Lok and McMahon, 2007). HBV contains a 3.2-kb long, partially double-stranded, circular DNA genome, which serves as a template for viral mRNA transcription by the cellular RNA polymerase II after being converted to a covalently closed form. Among the viral transcripts, the 3.4 kb pregenomic RNA (pgRNA) is encapsidated within the viral capsid along with the virus-encoded reverse transcriptase and converted through reverse transcription into the genomic DNA of progeny viruses (Seeger et al., 2007).
IFN-α plays an important role in controlling HBV infection and in the treatment of chronically infected patients (Hoofnagle and di Bisceglie, 1997). However, mechanisms by which IFN-α inhibits HBV replication have not been clearly defined. IFN-α restricts viral replication by transcriptionally activating many antiviral proteins (Katze et al., 2002). One of the key cellular antiviral proteins induced by IFN-α is the double-stranded RNA (dsRNA)-dependent protein kinase (PKR), a serine/threonine kinase that is activated through dsRNA-dependent autophosphorylation (Proud, 1995). Activated PKR catalyzes the phosphorylation of translation initiation factor eIF-2α, which leads to the inhibition of translation (Samuel, 1993). Phosphorylation of eIF2-α by PKR during virus infection has been generally considered to cause the inhibition of viral protein synthesis and a block in viral replication. The fact that numerous viruses have functions that counteract PKR attests to the importance of the enzyme’s antiviral function (Langland et al., 2006).
While PKR has been shown to work against a range of viruses, the potential role of PKR in mediating the antiviral effect of IFN-α against HBV has not been fully addressed. Guidotti et al. (2002) showed a modest elevation of HBV replication in a transgenic mouse that had been crossed with a PKR-knockout mouse. However, viral replication in the animal was still sensitive to intravenous injection of poly(I)-poly(C), a potent inducer of IFN-α.
In this study, we investigated the effect of RNAi-mediated PKR knockdown on the efficacy of IFN-α in inhibiting HBV replication in transfected human hepatoma Huh-7 cells. Our results show that PKR is the primary mediator of the anti-HBV activity of IFN-α and suggest that PKR functions through the reduction of replication-competent viral capsids.
MATERIALS AND METHODS
Cell culture and other reagents
Human hepatoma Huh-7 cells were cultured in a Dulbecco’s modified Eagle Medium (DMEM) supplemented with 25 mM HEPES, 100 units/ml of penicillin, 100 μg/ml of streptomycin (Invitrogen), and 5% fetal bovine serum (Sigma) at 37℃ in 5% CO2. Huh-7 cells stably expressing either PKR siRNA (PKR-si cells) or siRNA of random sequence (Control-si cells) were previously described (Kang et al., 2009) and maintained in media with 0.5 μg/ml of puromycin (Sigma). When indicated, cells were treated with 100-1,000 U/ml of IFN-α (R&D System Inc.). Lipofectamine PLUS reagent (Invitrogen) was used for transfection of cells according to the manufacturer’s instructions.
Plasmids
The replication-competent pHBV1.2 plasmid contains one fulllength HBV DNA genome (ayw subtype, 3,182 nt) and 20% of the genome (nt 1,419-2,003) in addition in pGEM4Z+ vector (Cha et al., 2004). Plasmids expressing the HA-tagged wildtype or kinase-defective (K296R) PKR, both of which were engineered to be siRNA-insensitive were previously described (Kang et al., 2009). The CAT gene reporter pEnhI-CAT was constructed to contain the HBV enhancer I and X gene promoter sequences (nt 1,069-1,373) inserted into the pCAT3- basic vector (Promega).
HBV DNA and capsid analyses
Four days after transfection of pHBV1.2, Huh-7 cells grown in a 60-mm plate were washed with PBS and resuspended in 1 ml of lysis buffer [50 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1% NP-40, and 100 mM NaCl]. The lysate was incubated on ice for 20 min and clarified by centrifugation at 15,000 × g. To measure HBV DNA in the viral capsid, the lysate was treated with 100 μg/ml of DNase I and 100 μg/ml of RNase A (Promega) in 10 mM MgCl2 solution for 2 h at 37℃. Polyethylene glycol was added at 7% (w/v), and after incubation overnight at 4℃, viral capsids were precipitated by centrifugation for 5 min at 13,000 rpm. Viral DNA was extracted by treating the pellet with 1 mg/ml of proteinase K for 1 h at 50℃ in 10 mM Tris (pH 8), 100 mM NaCl, and 1 mM EDTA, followed by phenol extraction. Viral DNA was resolved by electrophoresis in a 1.0% agarose gel in TAE buffer, denatured and transferred to a Nytran Super- Charge nylon membrane (Schleicher & Schuell), and hybridized overnight at 65℃ with 32P-labeled HBV DNA. Autoradiography was obtained and band intensity was quantitated with TINA image analysis software.
To examine the capsids an aliquot of cell lysate (~2% of the total volume) was directly applied to a 1.2% agarose gel and resolved by electrophoresis in TAE buffer. The capsid proteins were transferred to a Protran nitrocellulose membrane (Schleicher & Schuell) by capillary transfer in 20× SSC and immunoblotted with an anti-HBc antibody (Dako). Protein bands were visualized with the West Pico ECL detection reagents (Thermo Scientific). Replication-competent viral capsid was measured in the endogenous polymerase assay (EPA) as described (Sohn et al., 2006). Briefly, viral capsids were isolated from cell lysate with protein A-Sepharose CL-4B beads (Pharmacia) that had been pre-incubated with anti-HBc antibody. The beads were precipitated by centrifugation, washed with PBS, and incubated at 37℃ overnight in 50 μl of buffer [50 mM Tris-HCl (pH 7.5), 75 mM NH4Cl, 1 mM EDTA, 20 mM MgCl2, 0.1% β-mercaptoethanol, 0.5% NP-40, 0.4 mM dATP, 0.4 mM dGTP, 0.4 mM dTTP, and 10 μCi of (α-32P) dCTP (3,000 Ci/ mmol, Perkin-Elmer)]. Contaminating DNA in the sample was removed by treatment with 0.25 mg/ml of DNase I for 30 min at 37℃. Capsids were disrupted by incubation for 30 min at 37℃ in 50 μl of 1% SDS, 10 mM Tris-HCl (pH 7.5), 10 mM EDTA, 0.6 mg/ml of proteinase K, and 0.8 mg/ml of yeast tRNA. Viral DNA was extracted with phenol, precipitated and electrophoresed through 1% agarose gel in TAE buffer. Gel was dried by vacuum and autoradiography was obtained.
Northern blotting
Total RNA was isolated from transfected cells by Trizol extraction (Gibco-BRL) according to the manufacturer’s protocol. RNA (~10 μg) was separated through 1% agarose gel containing 2.2 M formaldehyde and 20 mM MOPS (pH 7.0), transferred to a Nytran nylon membrane (Schleicher & Schuell) by capillary transfer in 20× SSC buffer, and hybridized with a 32Plabeled HBV DNA.
Immunoblotting
Protein in the cell lysate was resolved in 10 or 12% polyacrylamide gels under denaturing conditions and transferred to a Protran nitrocellulose membrane (Schleicher & Schuell). The membrane was blocked for 2 h in 5% skim milk in TBS-T [10 mM Tris (pH 8.0), 150 mM NaCl, and 0.05% Tween-20] and incubated with antibodies against PKR-HA, Stat-1, phospho- Stat-1, actin, Hsp70, eIF2α (all from Santa Cruz), phospho- PKR or phospho-eIF2α (from Cell Signaling). Protein bands were visualized on X-ray film with the West Pico ECL detection reagents (Thermo Scientific).
RESULTS
PKR is a key mediator of the anti-HBV activity of IFN-α
IFN-α has been shown to inhibit HBV replication in cultured human hepatoma cells (Hayashi and Koike, 1989; Rang et al., 1999). We confirmed the early findings that IFN-α inhibits viral replication in Huh-7 cells transfected with the replicationcompetent HBV1.2 DNA. Southern blot analysis indicated that in four days of IFN-α (500 U/ml) treatment the encapsidated viral DNA decreased by ~72% (Fig. 1A). We found a similar result in transfected HepG2 cells, another human hepatoma cell (data not shown).
Fig. 1. Inhibition of HBV replication by IFN-α. Huh-7 cells transfected with 2 μg of pHBV1.2 were treated with 500 U/ml of IFN-α for four days. (A) Intracellular viral capsids were precipitated from cell lysate and the encapsidated viral DNA was analyzed by Southern blotting. The relaxed circular (RC), double-stranded linear (DL), and singlestranded (SS) forms of viral DNA are indicated on the left. Signal intensity of four similar experiments was quantified using TINA image analysis software and an average value was shown on the right. (B) An aliquot of cell lysate was immunoblotted for PKR. Hsp70 was used as a loading control.
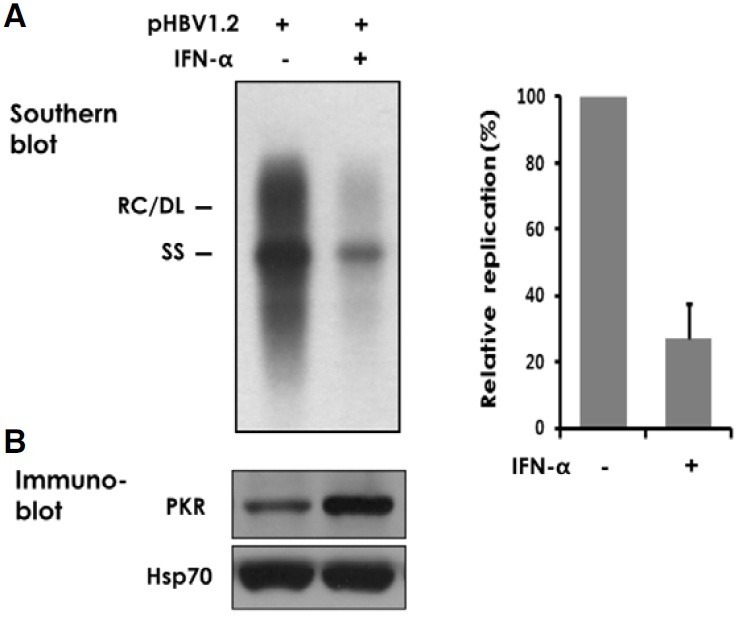
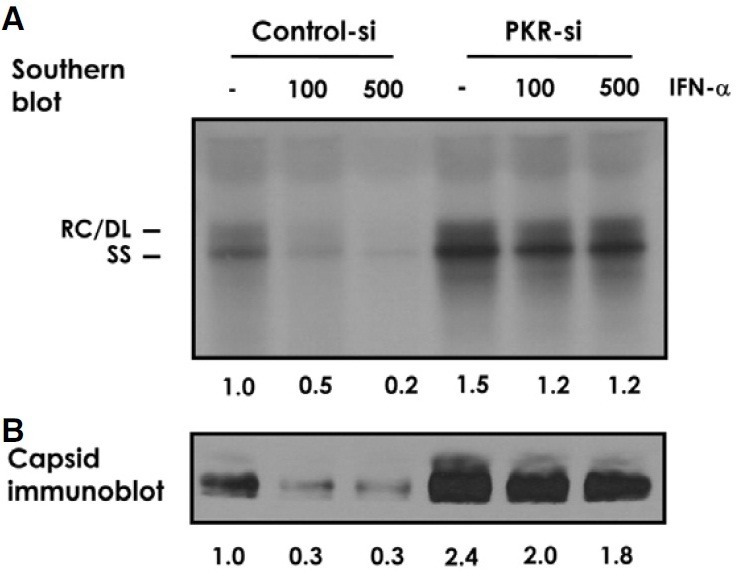
To further define the inhibitory mechanisms of IFN-α, we focused on the PKR, a cellular serine/threonine protein kinase whose expression is induced by IFN-α (Fig. 1B). While PKR has been implicated against a range of viruses (Garcia et al., 2007), its role in mediating the antiviral effect of IFN-α against HBV has not been sufficiently addressed. It was reported that HBV replication was modestly elevated in a transgenic mouse that had been crossed with a PKR-knockout mouse. However, viral replication in the animal was still sensitive to intravenous injection of poly(I)-poly(C) and the extent of inhibition was not profoundly different from that of a corresponding control animal (Guidotti et al., 2002). To address whether and to what degree PKR contributes to the inhibitory effect of IFN-α on HBV replication in human cells, we examined viral replication in a HBV1.2 transfected Huh-7 cell in comparison to another Huh-7 cell (named PKR-si) in which PKR expression is specifically and stably suppressed through RNA-interference. A 50-80% inhibition of viral DNA replication was observed in control cells treated with 100 or 500 U/ml IFN-α. In contrast, PKR-si cells showed ~15% inhibition at the respective IFN-α concentrations (Fig. 2A). In addition, PKR-si cells supported ~50% more viral replication than control cells receiving no exogeneous IFN-α treatment. We found that the level of intracellular viral capsids was also regulated by IFN-α, in a PKR-dependent fashion (Fig. 2B). The profoundly attenuated response of viral DNA and capsids to IFN-α in the PKR knockdown cells strongly suggested that PKR plays a primary role in mediating the anti-HBV activity of IFN-α.
Fig. 2. PKR-dependent anti-HBV activity of IFN-α. Control and stable PKR knock-down (PKR-si) Huh-7 cells were transfected with pHBV1.2 and treated for four days with 100 or 500 U/ml of IFN-α. (A) Viral DNA in the capsid was analyzed by Southern blotting. (B) Aliquots of viral capsid were separated by native agarose-gel electrophoresis and immunoblotted with anti-core antibody. Similar result was obtained from more than three repeated experiments. Representative blots were shown with quantified signal intensity indicated below the image.
PKR-mediated inhibition occurs at posttranscriptional steps
To address which steps of HBV replication were affected, we measured viral transcripts by Northern blotting. In both control and PKR-si cells IFN-α treatment had no apparent effect on the steady-state level of all four HBV transcripts, including the 3.4 kb pgRNA (Fig. 3A). We also found in both control and PKR-si cells that the transcript of a transfected CAT gene construct controlled by the HBV enhancer I and the HBX promoter was not affected by IFN-α treatment. Enhancer I overlaps the HBx promoter, but it up-regulates transcription of all HBV promoters (Su and Yee, 1992). These results suggested that the inhibition of HBV by IFN-α occurs at posttranscriptional steps.
Fig. 3. Post-transcriptional down-regulation of viral capsids. (A) Control and PKR-si Huh-7 cells transfected with pHBV1.2 were treated with 500 U/ml of IFN-α for two days. Total cellular RNA was extracted and viral transcripts were measured by Northern blotting. Expected sizes of the four viral transcripts are indicated on the right. Analyzed in parallel was mRNA of a co-transfected CAT gene construct controlled by the HBV enhancer I and the HBX promoter. The latter construct also served a control for transfection efficiency. (B) Control and PKR-si Huh-7 cells transfected with pHBV1.2 were treated with 500 U/ml of IFN-α for four days. Viral capsid immunoprecipitated with anti-core antibody was subjected to the endogenous polymerase assay. Radiolabelled DNA products were extracted and electrophoresed on a 1% agarose gel. An autoradiograph of three similar experiments was shown with quantified signal intensity. (C) Control and PKR-si Huh-7 cells were treated for two days with 1,000 U/ml of IFN-α, and cellular proteins were immunoblotted with the antibodies against PKR, phospho-PKR, STAT-1, phospho-STAT-1, or actin (as a loading control).
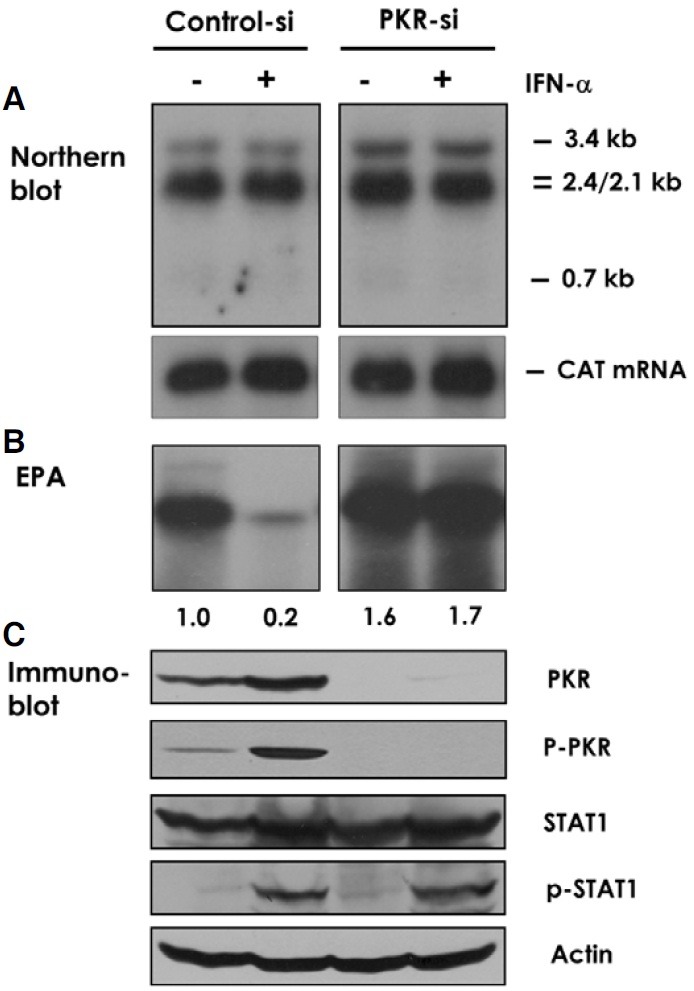
Because IFN-α treatment did not down-regulate HBV transcripts, we measured DNA synthesis in the isolated viral capsids in the endogenous polymerase assay (EPA). This assay measures DNA synthesizing activity associated with the viral capsids, which contain the RNA pregenome and the viral polymerase associated within the capsid particles. IFN-α inhibited EPA activity by ~80%. In contrast, this inhibition was remarkably and almost completely reversed in PKR-si cells (Fig. 3B). Together, these results suggested that IFN-α treatment decreases the level of total and replication-competent viral capsids by a PKR-dependent mechanism.
To address whether the different response to IFN-α observed in the PKR-si cells and control cells is due specifically to the PKR suppression, we examined PKR expression and IFN-α signaling in these cells. IFN-α binds type I IFN receptor and transduces signals through activating the cytoplasmic tyrosine kinases Jak1 and Tyk2, and the latent transcription factor STAT. Phosphorylated STAT-1 binds STAT-2 and p48 to form the ISGF3 complex, which translocates to the nucleus. Here, the complex binds the IFN-stimulated response element (ISRE) and activates the transcription of IFN-stimulated genes (Darnell, 1997). Immunoblot showed that in the control cells, PKR expression increased about two-fold by IFN-α treatment (Fig. 3C). In contrast, PKR expression was blocked in the PKR-si cells, regardless of whether the cells were also treated with IFN-α. Accordingly, we noted phosphorylation of PKR in the control cells but not in the PKR-si cells. To the contrary, both the control and PKR-si cells showed the same pattern of STAT-1 expression and phosphorylation in response to IFN-α treatment. Accordingly, we observed the induction of another IFN-α-inducible protein MxA by IFN-α in both the control and the PKR-si cells (data not shown). This result indicated that PKR siRNA does not interfere with other components of IFN-α pathway.
Antiviral activity of PKR requires its kinase function
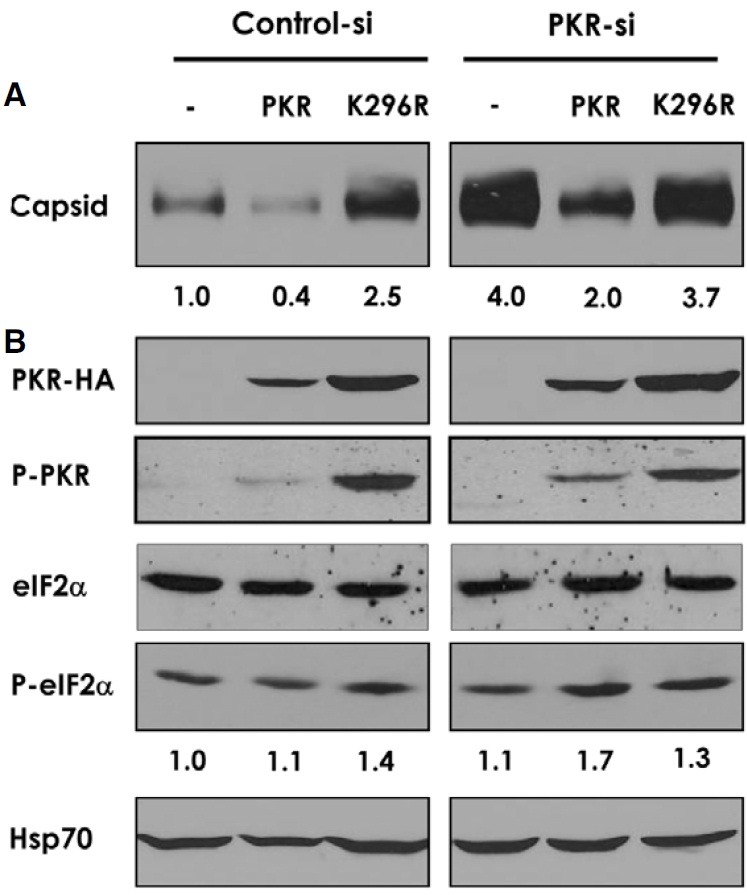
The antiviral function of PKR was addressed more directly through co-transfection of PKR with HBV1.2 constructs. Ectopic expression of PKR down-regulated the viral capsid level by 50-60% in the control and PKR-si cells (Fig. 4A). The inhibitory effect of ectopically expressed PKR on viral capsid was correlated with the concomitant increase in the phosphorylated eIF2α, which was more evident in PKR-si cells while total amount of eIF2α remained unaffected (Fig. 4B). In contrast to the wildtype PKR, expression of a kinase-defective PKR (K296R) rather increased (in control cells) the capsid level or had no inhibitory effect (in PKR-si cells). The capsid-increasing effect of K296R (in control cells) was considered due to the dominantnegative inhibition of endogenous PKR by the mutant. These results, together, suggested that the anti-HBV activity of PKR depends on its kinase function, and most likely through inhibition of viral protein synthesis. We noted that K296R was expressed more than wild-type PKR, probably due to the loss of its autoregulatory function. We also noted phosphorylation of K296R, probably due to trans-phosphorylation by the endogeneous PKR.
Fig. 4. Kinase-dependent antiviral activity of PKR. Control and PKR-si cells were co-transfected with pHBV1.2 and siRNA-insensitive PKR constructs either expressing the wild-type or the kinase-defective (K296R) enzymes. Cells were harvested in 48 h. (A) Viral capsid was separated by native agarose-gel electrophoresis and immunoblotted with anti-core antibody. A representative blot was shown with signal intensity indicated below the image. (B) Cellular proteins were immunoblotted with the antibodies against PKR (HA-tag), phospho-PKR, eIF2α, phospho-eIF2α (intensity indicated below), or Hsp70 (for a loading control).
DISCUSSION
The mechanisms by which IFN-α inhibits HBV replication have been the focus of many studies, but these remain elusive to date. Ohata et al. (2003) have shown that IFN-α blocks NF-κB activation which is triggered by transfection of Huh-7 cells with either a full HBV genome or HBX gene. We found in our transfection model that a HBV1.2 construct that does not produce HBX protein was also inhibited by IFN-α treatment (data not shown).
In a transgenic mouse model (Wieland et al., 2000) and cultured murine cells (Pasquetto et al., 2002; Wieland et al., 2005), it was shown that the IFN response does not affect the steadystate levels of HBV mRNA, but it does prevent the accumulation of pgRNA-containing capsids. These results are consistent with ours. However, in another study when IFN response was induced in model animals by acute MCMV infection or by multiple injection of poly(I)-poly(C), HBV gene expression was inhibited at the transcriptional level (Uprichard et al., 2003). In addition to the difference in experimental system, the extent and the way IFN response was induced might have contributed to the differences between these and our results. IFN response was induced by the injection of poly(I)-poly(C) in the aforementioned studies on HBV-transgenic mice (Guidotti et al., 2002; Wieland et al., 2000).
As diverse cellular proteins are induced by IFN-α, its antiviral activity is likely mediated by more than a single protein, through a specific mechanism by each mediator. In the current study, we showed that the PKR-mediated inhibition constitutes a major antiviral measure of IFN-α against HBV. We provided evidences indicating that PKR reduces the level of replicationcompetent viral capsids in the cell. We observed a similar effect of PKR in down-regulating viral envelope proteins (not shown). Together, these results suggest that PKR functions most likely through the inhibition of viral protein synthesis.
In addition to the inhibition of viral protein synthesis, an accelerated decay of viral proteins or destabilization of capsids might also contribute to the antiviral activity of IFN-α against HBV. It was shown in cultured murine cells that IFN-induced anti-HBV activity could be blocked with proteasome inhibitors while the exp ression of IFN-inducible genes was not affected (Robek et al., 2002). Further, Xu et al. (2010) reported recently that IFN-α and IFN-γ can trigger in a proteasome-dependent manner the decay of replication-competent HBV nucleocapsids but not that of empty capsids. Although currently no data bears on the activation of proteasome by PKR, given the profound effect of PKR on the level of viral capsid as seen in this work, it might be worth checking if PKR is involved also in the destabilization of capsids.
IFN treatment rarely leads to the complete clearance of chronic HBV infection, while it induces strong systemic inflammatory symptoms. The direct induction or activation of PKR might be an alternative strategy for treating chronically infected patients.
Acknowledgments
We thank W.S. Ryu for kindly providing us with pHBV1.2. This work was supported by the Health Technology Research Grant (#A080599) from the Ministry of Health, Welfare and Family Affairs of the Republic of Korea.
References
- 1.Cha M.Y., Kim C.M., Park Y.M., Ryu W.S. Hepatitis B virus X protein is essential for the activation of Wnt/betacatenin signaling in hepatoma cells. Hepatology. (2004);39:1683–1693. doi: 10.1002/hep.20245. [DOI] [PubMed] [Google Scholar]
- 2.Darnell J.E. Jr. STATs and gene regulation. Science. (1997);277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 3.Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: virus and cell control. Biochimie. (2007);89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Guidotti L.G., Morris A., Mendez H., Koch R., Silverman R.H., Williams B.R., Chisari F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. (2002);76:2617–2621. doi: 10.1128/JVI.76.6.2617-2621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayashi Y., Koike K. Interferon inhibits hepatitis B virus replication in a stable expression system of transfected viral DNA. J. Virol. (1989);63:2936–2940. doi: 10.1128/jvi.63.7.2936-2940.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoofnagle J.H., di Bisceglie A.M. The treatment of chronic viral hepatitis. N. Engl. J. Med. (1997);336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- 7.Kang J.I., Kwon S.N., Park S.H., Kim Y.K., Choi S.Y., Kim J.P., Ahn B.Y. PKR protein kinase is activated by hepatitis C virus and inhibits viral replication through translational control. Virus Res. (2009);142:51–56. doi: 10.1016/j.virusres.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Katze M.G., He Y., Gale M., Jr. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. (2002);2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 9.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. (2006);119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Lok A.S., McMahon B.J. Chronic hepatitis B. Hepatology. (2007);45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 11.Ohata K., Ichikawa T., Nakao K., Shigeno M., Nishimura D., Ishikawa H., Hamasaki K., Eguchi K. Interferon alpha inhibits the nuclear factor kappa B activation triggered by X gene product of hepatitis B virus in human hepatoma cells. FEBS Lett. (2003);553:304–308. doi: 10.1016/s0014-5793(03)01034-2. [DOI] [PubMed] [Google Scholar]
- 12.Pasquetto V., Wieland S.F., Uprichard S.L., Tripodi M., Chisari F.V. Cytokine-sensitive replication of hepatitis B virus in immortalized mouse hepatocyte cultures. J. Virol. (2002);76:5646–5653. doi: 10.1128/JVI.76.11.5646-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proud C.G. PKR: a new name and new roles. Trends Biochem. Sci. (1995);20:241–246. doi: 10.1016/s0968-0004(00)89025-8. [DOI] [PubMed] [Google Scholar]
- 14.Rang A., Gunther S., Will H. Effect of interferon alpha on hepatitis B virus replication and gene expression in transiently transfected human hepatoma cells. J. Hepatol. (1999);31:791–799. doi: 10.1016/s0168-8278(99)80279-7. [DOI] [PubMed] [Google Scholar]
- 15.Robek M.D., Wieland S.F., Chisari F.V. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J. Virol. (2002);76:3570–3574. doi: 10.1128/JVI.76.7.3570-3574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samuel C.E. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. (1993);268:7603–7606. [PubMed] [Google Scholar]
- 17.Seeger C., Zoulim F., Mason W.S. Hepadnaviruses. Field’s virology, D.M., Knipe, and P.M. Howley, eds. Lippincott Williams & Wilkins; Philadelphia, USA: (2007). pp. 2977–3029. [Google Scholar]
- 18.Sohn S.Y., Kim S.B., Kim J., Ahn B.Y. Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J. Gen. Virol. (2006);87:1883–1891. doi: 10.1099/vir.0.81684-0. [DOI] [PubMed] [Google Scholar]
- 19.Su H., Yee J.K. Regulation of hepatitis B virus gene expression by its two enhancers. Proc. Natl. Acad. Sci. USA. (1992);89:2708–2712. doi: 10.1073/pnas.89.7.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uprichard S.L., Wieland S.F., Althage A., Chisari F.V. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc. Natl. Acad. Sci. USA. (2003);100:1310–1315. doi: 10.1073/pnas.252773599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wieland S.F., Guidotti L.G., Chisari F.V. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J. Virol. (2000);74:4165–4173. doi: 10.1128/jvi.74.9.4165-4173.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wieland S.F., Eustaquio A., Whitten-Bauer C., Boyd B., Chisari F.V. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc. Natl. Acad. Sci. USA. (2005);102:9913–9917. doi: 10.1073/pnas.0504273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu C., Guo H., Pan X.B., Mao R., Yu W., Xu X., Wei L., Chang J., Block T.M., Guo J.T. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J. Virol. (2010);84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]