

Abstract
Downregulation of the CD99 antigen on the surface of Hodgkin’s lymphoma (HL) cells via EBV LMP1-mediated NF-κB suppression of Sp1 transcriptional activity is known to be associated with the appearance of pathogenic Reed- Sternberg cells. Here, we show that in addition, EBV LMP1 heterologous NF-κB activators such as CD30 and CD40 repress the CD99 promoter, which contains multiple Sp1- binding sites but no NF-κB binding sites. In addition, NF- κB-inducing kinase (NIK) repressed the CD99 promoter while NIK kinase mutants and JNK inhibitory protein failed to do so. Of the NF-κB subunits, NF-κB2 (p52) alone or in combination with other Rel subunits consistently inhibited the CD99, while NF-κB1 (p50) showed a marginal repressive effect. Furthermore, while transfection of LMP1 repressed the CD99 promoter in wild-type or NF-κB1 deficient MEFs, the same repression was not observed in NF- κB2 (p52)-deficient MEFs, indicating that NF-κB2 (p52) is required for LMP1-mediated repression of the CD99 promoter. Consistently, basal activity of the CD99 promoter was significantly higher in IKKα-/- and IKKβ-/- MEFs, but not in IKKγ-/- MEFs compared to the wild-type control MEFs. Sp1-binding sites were directly used in the repression, because a synthetic Sp1 reporter with 10 Sp1-binding sites from the CD99 promoter was repressed by LMP1 or p52 transfection. These data indicate that LMP1-mediated NF- κB2 exhibits the major inhibitory role in the transcription at the CD99 promoter.
Keywords: CD99, Hodgkin’s lymphoma, LMP1, P52, Sp1
INTRODUCTION
Hodgkin’s lymphoma Reed-Sternberg cells (HLRSCs) have been intensively studied since they were first described (Hodgkin, 1832), and it is believed that HLRSCs are derived from B lymphocytes (Kuppers and Rajewsky, 1998; Marafioti et al., 1999; Staudt, 2000; Watanabe et al., 2000). In lymphoid cells, where rearrangement and somatic mutations in the antigen receptor are the most lineage-specific events, analysis of the immunoglobulin heavy chain locus in HLRSCs clearly implicated germinal center B cells as the HLRSC origin (Braeuninger et al., 1997; 1999; Kuppers et al., 1998; Ohno et al., 1997). The neoplastic Reed-Sternberg cells in Hodgkin’s lymphoma (HL) are characterized by constitutively activated NF-κB (Bours et al., 1992), due to over-expression of potent NF-κB-activating TNF receptor family proteins (CD40, CD30, or EBV LMP1) (Cabannes et al., 1999; Sylla et al., 1998) or mutation in the inhibitor of the NF-κBα gene (IκBα) (Emmerich et al., 1999; Krappmann et al., 1999). However, the pathogenesis of HL remains unclear.
Complete lack of expression of a cellular surface antigen called CD99 was observed in lymph nodes from HL patients. Similarly, enforced downregulation of CD99 resulted in the generation of HLRSCs cells, a hallmark of HL, along with upregulation of CD30 and CD15, downregulation of MHC Class I and CD45RB, and deregulation of cytokine secretion. Moreover, the phenotype was reversible by addition of CD99 (Kim et al., 1998; 2000; Lee et al., 2001). CD99, a 32-kDa glycoprotein also known as a Mic2 antigen, is implicated in lymphocyte differentiation, including T cell activation and cell-cell adhesion (Bernard et al., 1995; Hahn et al., 1997), as well as in apoptosis of T cells and Ewing sarcoma cells when engaged (Bernard et al., 1997; Pettersen et al., 2001; Sohn et al., 1998). It is expressed in most lymphocytes, with particularly high levels of expression in most T cell lineages, including the activated/ memory T and B cells, while it is expressed at very low or undetectable levels in malignant myeloid cells, B-cell leukemia, and Epstein Barr virus (EBV)-transformed B lymphoblastoid cell lines (Gelin et al., 1989; Levy et al., 1979; Park et al., 1999). The EBV is a transforming herpes virus, causing latent infections of human primary B lymphocytes, and is associated with Hodgkin’s lymphoma and other malignancies (Kieff and Rickinson, 2001; Rickinson and Kieff, 2001; Shin et al., 2011).
The EBV is present in up to 80% of HL cases, with an average incidence of 50%; its episomes are monoclonal, and EBVencoded latent membrane protein 1 (LMP1) is exclusively expressed in HLRSCs, suggesting that LMP1 is closely linked to pathogenesis and viral infection that occur before the neoplastic tumor clone expands.
LMP1 is expressed in all EBV-positive HLRSCs. Moreover, LMP1 has transforming activity in cultured rodent fibroblast cells and LMP1 transgenic mice, and is essential for B cell transformation. LMP1 activates NF-κB and Jun kinase, and affects many cellular events including cell adhesion and apoptosis, and regulation of cellular genes such as CD23, CD40, Bcl2, and A20. LMP1 has a TRAF-binding domain, by which it recruits TRAFs and activates NF-κB, and is believed to be a ligandindependent, functional homologue of CD40.
The nuclear factor-kappa B (NF-κB) family consists of homoor heterodimers of NF-κB (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel (Ghosh et al., 1998). The NF-κB complex is constitutively activated in HLRSCs (Bours et al., 1992). The mechanisms underlying high NF-κB activity in HLRSCs include mutation of IκB, modification of NF-κBs, constitutive IKKβ expression, and EBV infection (Cabannes et al., 1999; Emmerich et al., 1999; Jundt et al., 1999; Krappmann et al., 1999; Wood et al., 1998). NF-κB1 (p50) and NF-κB2 (p52) are closely related NF-κB family members. Although p50 is highly ubiquitous and usually quite abundant, p52 is expressed at low levels and primarily in hematopoietic cells. Both proteins are generated from precursors by proteolytic processing: p50 is derived from p105, and p52 from p100. Processing of p105 and p100 is regulated by TPL-2 and NIK, respectively. Both precursors behave similarly to the inhibitory protein IκB, although they themselves are not IκBα proteins, owing to the presence of ankyrin repeats, which are characteristic of IκB family members. During processing, these inhibitory ankyrin repeats are phosphorylated and degraded, and the resulting p50 and p52 form homo- or heterodimers with c-Rel, RelA, or RelB.
Expression of the tumor necrosis factor-receptor family proteins (CD30, CD40 and LMP1) has the potential to activate NF- κB (Cabannes et al., 1999; Sylla et al., 1998). Given that HLRSCs are derived from B lymphocytes and that LMP1 is expressed exclusively in EBV-positive HLRSCs, as well as that CD99 repression by LMP1 is associated with Reed-Sternberg cell morphology, it is important to determine how LMP1 is involved in the pathogenesis of HL (Boiocchi et al., 1992; Herbst et al., 1990; 1992a; 1992b).
Here, we report that (1) in addition to EBV LMP1, heterologous TNFR and CD30- and CD40-propagated NF-κB signals repress the Sp1 site-rich CD99 promoter; (2) LMP1 directly uses Sp1 sites for repression; (3) NF-κB2 (p52) activity plays a central inhibitory role in the repression of the Sp1 site-rich CD99 promoter.
MATERIALS AND METHODS
Plasmids and antibodies
The pcDNA-SP1 construct was provided by T. Collins at the Brigham’s Women’s Hospital. RSVp50, RSVP65, RSVp52, and RSVp100 were provided by Dr. Garry Nabel at the University of Michigan Medical Center through the NIH AIDS Research & Reference Reagent Program. The pCDNA/c-Rel construct was from Dr. T.D. Gilmore at Boston University and the pcDNARelB construct was from Dr. T. Wirth at the University of Ulm. Dr. E. Kieff at Brigham and Women’s Hospital provided the expression vectors pCMV-FLAG-LMP1, pcDNAFLMP1CD30, pcDNAFNIK, and NIKKM. The pCMV-FLAG-LMP1 construct encodes Flagtagged latent membrane protein 1 from the Epstein Barr virus. LMP1 aa 1-231 harbors the Transformation Effector Site (TES1 domain (aa 187-231) but not the TES2 domain (aa 232-386). The LMP1 AA construct contains 2 mutations in the region 204PQQAT208, resulting in the generation of the sequence 204PAAAT208 in the TES1 domain, while the LMP1 ID mutant has an Y385I mutation in the TES2 domain (Luftig et al., 2004). The pcDNAFLMP1CD30 construct encodes the LMP1 transmembrane domain fused to the cytoplasmic CD30 signaling domain. Both pcDNAFNIK and NIKKM encode Flag-tagged proteins, NF-κB-inducing kinase and its mutant at K429A/K430A that has defective kinase function, respectively. Previously described constructs used in this study were a constitutive NF-κB inhibitor (IκBαSS32/36AA mutant), JUN kinase dominant negative mutant (JNKDN), JNK inhibitory protein (JIP), and pcDNAFLMP1CD40 encoding a Flag-tagged transmembrane domain (aa 1-187) of LMP1 fused to the cytoplasmic CD40 domain mimicking ligand-independent CD40 (Hatzivassiliou et al., 1998). CD99 reporters have also been previously described (Lee et al., 2001). The synthetic SP1 reporter harboring 10 Sp1 sites from the CD99 promoter was constructed using the following primer pairs: SP1N1S (5′-gggcggccgc ccccgcccgg gtgggggggc ggggc cccgc cc-3′) and SP1N1AS (5′-gccccgcccc cccacccggg cgggg gcggc cgcccGTAC-3′), SP1CS (5′-ttcccgcccc ttggggcggg gcgg gcgccc cgcccccacc cAGCT-3′) and SP1CAS (5′-gggtgggggc ggggcgcccg ccccgcccca aggggcggga agggcggg-3′). The 2 subcloned fragments were mixed and directionally ligated to the KpnI/SacI-digested promoter-less pGL2 basic plasmid (Promega, USA). The culture supernatant of hybridoma S12 cells was used to for the LMP1 monoclonal antibody. Anti-Flag M2 antibody was purchased from Sigma, while monoclonal antip52 (C-5), polyclonal anti-Sp1 (Sc-59), anti-p50, anti-RelA (p65) (A-G), anti-RelB, and anti-c-Rel antibodies were purchased from Santa Cruz Biotechnology.
Transfection and reporter assay
Human embryonic kidney 293T cells and mouse embryonic fibroblasts (MEFs) were used in this study. One day before transfection, 3 × 105 293T cells were seeded in each well of 6- well plates, and then the cells were transfected the following day with 1 μg of CD99 promoter luciferase reporters, 0.25 μg pGKβ-galactosidase, and various amounts of effector plasmids, as indicated in the appropriate figures. Twenty hours after transfection, cells were harvested, washed with PBS, and lysed in the 1× lysis buffer for luciferase. They were incubated on ice for 30 min followed by centrifugation for 15 min at 4℃, and then subjected to reporter assays and western blotting, using 20 μg of protein extract per well.
Analysis of p100 processing and immunoprecipitation
293T cells or mouse embryonic fibroblasts were transfected with LMP1. Two or two-and-a-half days after transfection, cells were lysed in 2× SDS sample buffer. The lysates were separated on a 10% denaturing gel, transferred to a nitrocellulose membrane, and then probed with anti-p52 monoclonal antibody (C-5) purchased from Santa Cruz Biotechnology.
RESULTS
Heterologous NF-κB activators, but not JNK activity, repress the Sp1 site-rich CD99 promoter
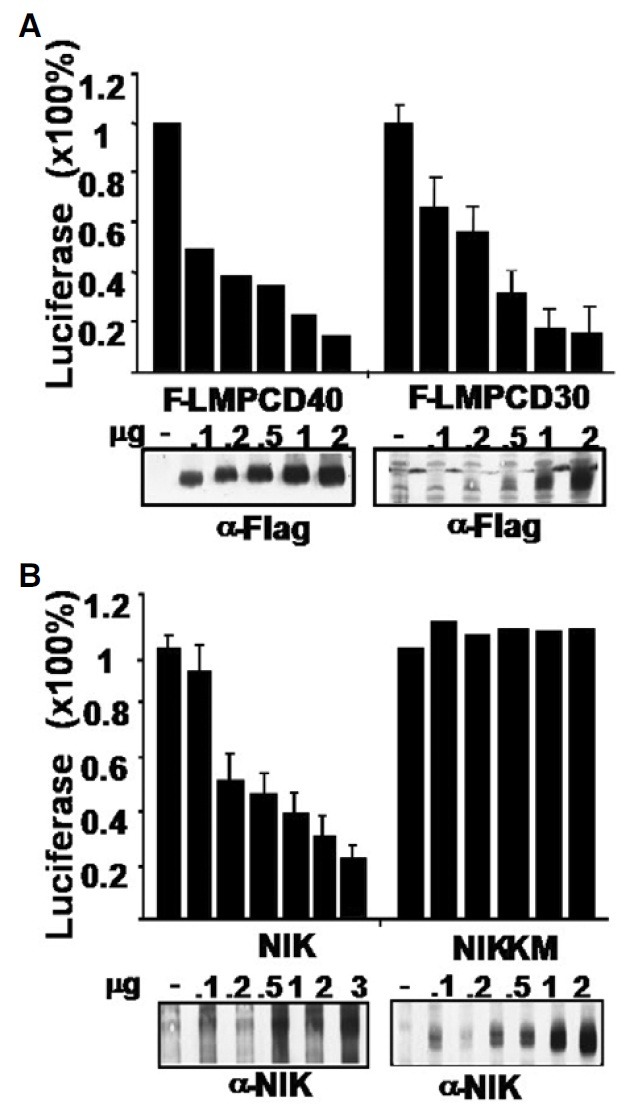
Non-EBV-infected HL cells constitutively express CD30 or CD40, strong activators of NF-κB. Instead of using the native form of CD40 or CD30 that requires a ligand, we used chimeric proteins consisting of the LMP1 transmembrane domain (aa 1- 187) fused to the cytoplasmic tail of CD40 or CD30, so as to convey a ligand-independent, constitutive NF-κB signals. Both LMP1-CD40 and LMP1-CD30 efficiently repressed the CD99 -1643~+123-Luc reporter in a dose-dependent manner (Fig. 1A). We also tested NF-κB-Inducing Kinase (NIK) for repression of the CD99 promoter. Transfection of NIK inhibited the CD99 promoter, whereas the NF-κB-defective NIK kinase domain mutant (NIK-KM) did not (Fig. 1B).
Fig. 1. NF-κB inhibits the Sp1 binding site-rich CD99 promoter (-1643~+123-Luc). (A) Heterologous NF-κB activators, LMP1-CD40 and LMP1-CD30, inhibit the CD99 promoter. (B) Transfection of NIK but not NIK-KM, a kinase mutant defective in kinase activity, represses the CD99 promoter. In (A, B), 293T cells were transfected with the indicated amount of effector, CD99 promoter (-1643 ~+123-Luciferase), and pGKβ-galactosidase, and subjected to reporter assays and Western blotting 24 h after transfection, using equal amounts of protein extracts.
As previously known (Lee et al., 2001), LMP1 inhibited the -1643~+123-Luc CD99 promoter in a dose-dependent manner (Supplementary Figs. 1A and 2A). We investigated which of the LMP1-propagated signals, NF-κB or JNK activity is responsible for the repression. Transfection of the LMP1 TES1 domain (denoted as 1-231), the mutated TES1 domain (AA) or the TES2 domain (ID) cripple NF-κB activity by approximately 70%, 40% and 70%, respectively, compared to transfection of wild type LMP1. While transfection of the wild-type LMP1 represses CD99, transfection of each of the TES constructs significantly rescued CD99 repression, and the repression of the CD99 promoter inversely correlated with the NF-κB activity that each LMP1 mutant retained (Supplementary Fig. 1B). A constitutive NF-κB-specific inhibitor was tested, IκBαAA, in which the serine residues at positions 32 and 36 were mutated to alanine residues. As a result, IκBαAA alone activated CD99 promoter activity in a dose-dependent manner (Supplementary Fig. 1C). Co-transfection of IκBαAA relieved the LMP1-mediated represssion of the CD99 promoter, further demonstrating that NF-κB activity is the central player in repression of the CD99 promoter (Kim et al., 2000; Lee et al., 2001) (Supplementary Fig. 1D).
Jun kinase (JNK) is one of the signaling molecules activated by LMP1. We next investigated whether JNK is the inhibitory signaling molecule in LMP1-mediated CD99 promoter repression. The JNK dominant negative inhibitor (JNKDN) alone did not affect activity of the CD99 promoter (Supplementary Fig. 2A). In addition, neither JNKDN nor JNK interacting inhibitory protein (JIP) relieved LMP1-mediated repression (Supplementary Figs. 2B and 2C). These results indicate that LMP1- mediated activation of JNK does not likely affect the activity of the CD99 promoter.
NF-κB exerts its inhibitory role through Sp1 sites in the CD99 promoter
The nucleotide sequence between bases -1643 and +123 of the CD99 promoter has no NF-κB binding sites. Instead, it has 10 copies of a Sp1-binding site. A series of CD99 promoter deletion mutants were used to locate a cis DNA sequence(s) on the CD99 promoter that mediates LMP1-dependent repression. Neither deletion of the 1st through 5th Sp1 sites from the 5′ end (del1-5sp) nor internal deletions in the 5th (del5sp) nor 8th Sp1 site (del8sp) abolished LMP1 and NF-κB2 (p52)-mediated promoter repression. These results suggest that the repressive effect of LMP1 or p52 on the promoter is likely mediated through redundant Sp1-binding sequences. In other words, LMP1 and NF-κB2 (p52) might be able to repress any promoter that has Sp1 site(s), such as the CD99 promoter (Fig. 2A).
Fig. 2. LMP1 represses the CD99 promoter via Sp1 sites. (A) 293T cells were co-transfected with LMP1 and a CD99 promoter that has a deletion of Sp1 sites (denoted as black squares). LMP1 and NF- κB2 (p52) repressed any promoter that contained one or more Sp1 site(s). (B) LMP1 represses a synthetic Sp1 reporter (CD99SP 1 × 10Luc), which includes the 10 Sp1 sites collected from the CD99 promoter. (C) In the control, pCDNA3-Sp1 activates the CD99SP 1 × 10Luc reporter, whereas p52 contransfection with the Sp1 represses the same reporter to the basal level 72 h after transient transfection in 293T cells. (D) In B lymphocytes-derived Burkitt’s lymphoma BJAB cells, pcDNA3-Sp1 activates the same CD99SP 1 × 10Luc and -1643~+123-Luc reporter (not shown), while p52 transfection represses the same reporter activity significantly to lower than that of the Sp1 or vector control 24 h after transfection.
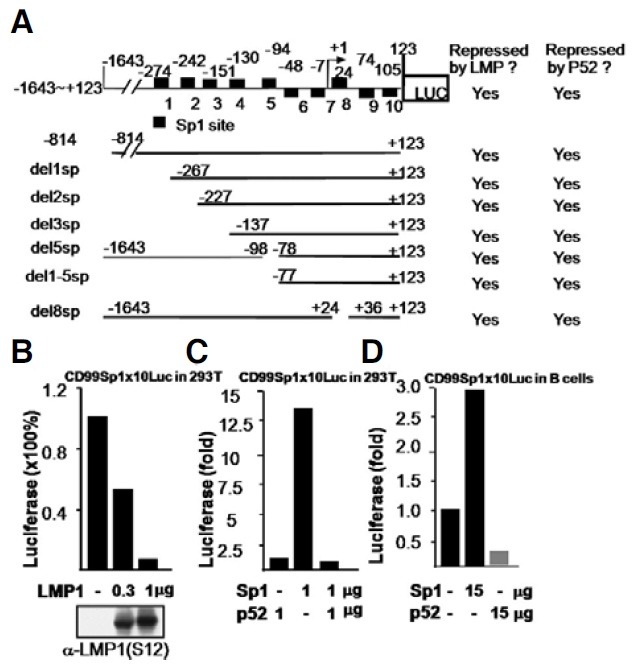
Since Sp1 is a constitutively active transcription factor, the effect of Sp1 on promoter activity was tested. As expected, Sp1 strongly augmented activity of the -1643~+123-Luc promoter in a dose-dependent manner (Supplementary Fig. 1E). Next, to determine if the Sp1 sites are directly used by LMP1 for the repression, a synthetic Sp1 reporter consisting of all 10 Sp1 sites from the CD99 promoter was tested. LMP1 strongly repressed activity of the synthetic Sp1 reporter (Fig. 2B). In a control experiment, pCDNA3-Sp1 transfection activated the cotransfected synthetic Sp1 reporter in dose-dependent manner 72 h after transfection. This Sp1-activated luciferase was repressed to the level of p52 alone by co-transfection of p52 (Fig. 2C). Whether or not p52 represses the same CD99 promoter in B lineage cells was addressed using B lymphocytes-derived Burkitt’s lymphoma BJAB cells (Fig. 2D). In a control experiment, pcDNA3-Sp1 also activated the same CD99SP1x10Luc reporter, while p52 transfection significantly repressed the same reporter to the level of lower than that of the Sp1 alone or vector control 24 h after transfection (Fig. 2D). These results suggest that Sp1 sites are used by LMP1 and P52 for repression of the CD99 promoter.
NF-κB2 (p52) inhibits the CD99 promoter
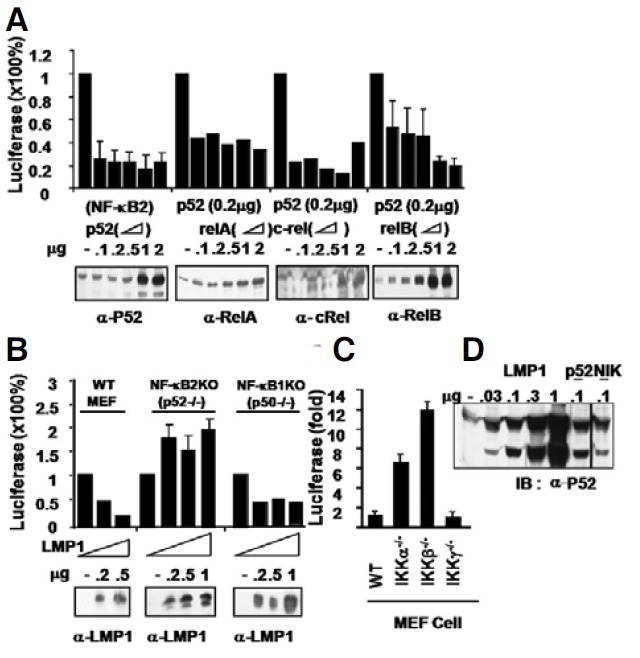
We further investigated which NF-κB subunit(s) is primarily responsible for the repression, among NF-κB1 (p50), NF-κB2 (p52), RelA (P65), RelB and c-Rel. These subunits were transiently transfected alone or in various combinations into 293T cells. Transfection of NF-κB1 (p50) or c-Rel had only a marginally inhibitory effect on the CD99 promoter, while RelA or RelB slightly induced the promoter (Supplementary Fig. 3A). Interestingly, NF-κB2 (p52) alone or in combination with RelA, c-Rel or RelB strongly inhibited the CD99 promoter (Fig. 3A). In contrast, NF-κB1 (p50)/RelA did not repress the promoter, and in fact even slightly augmented it, while p50/c-Rel or p50/RelB and RelA/c-Rel repressed it at a low dose but activated it at a high dose, leaving the role of other NF-κB subunits ambiguous (Supplementary Fig. 3B).
Fig. 3. NF-κB2 (P52) exhibits major inhibitory activity on the CD99 promoter. (A) p52 alone or in combination with RelA, c-Rel, RelB, and p65/c-Rel was transfected into 293T cells for the repression assay. (B) Mouse embryonic fibroblasts from wild-type control mice, and NF-κB2 precursor (p100/p52) - or NF-κB1 precursor (p105/ p50)-deficient mice were transfected with LMP1. (C) MEFs from wild-type control, IKKα-/-, IKKβ-/-, and IKKγ-/- mice were transiently transfected. In (A-C), cells were transiently co-transfected with the -1643~+123-Luc CD99 promoter and pGKβ-galactosidase control reporter, and subjected to reporter assays and Western blotting 20 h (A) or 48 h after transfection, using equal amounts of protein extracts (B). The p52 heteromeric complexes with all other subunits exhibited significant repressive activity in a dose-dependent manner, whereas the p50 heteromeric complexes had no or a much lesser inhibitory effect (Supplementary Fig. 3). (D) LMP1 stimulates p100 processing to p52. Approximately 105 293T cells were transfected with increasing amounts (0, 0.03, 0.1, 0.3, and 1 μg) of pCDNALMP1, RSV-p52 (0.1 μg), or NIK (0.1 μg) as controls. At posttransfection day 2, cell lysates were separated and transferred to nitrocellulose before being probed with the anti-p52 mouse monoclonal antibody (C-5, Santa Cruz). The processing of p100 by LMP1 results in 3 major bands, likely terminating at aa 445 (not fully processed, upper band) or aa 405 (lower band, p52).
NF-κB2 (p52) is required for LMP1-mediated CD99 promoter repression
To further investigate the roles of the p50 hetero complex or the p52 heterocomplex on NF-κB activity, we next transfected LMP1 into mouse embryonic fibroblast cells derived from wildtype, NF-κB1 (p105/p50)-/-, or NF-κB2 (p100/p52)-/- mouse embryos. Consistent with the results above, the CD99 promoter was repressed in wild-type and NF-κB1 (p105/p50)-/- MEFs, but not in NF-κB2 (p100/p52)-/- MEFs, indicating that LMP1 represses the CD99 promoter primarily through activation of NF-κB2 (Fig. 3B). We next compared the basal level of uninduced CD99 promoter activity in MEFs derived from wild-type control, IKKα-/-, IKKβ-/-, and IKKγ-/- embryos. These cells were transiently transfected with the -1643~+123-Luc CD99 promoter and the pGKβ galactosidase control reporter without LMP1. While the transfection efficiency did vary by no more than 2-fold, deficiency in IKKα or IKKβ strongly augmented CD99 promoter activity by 6-fold and 11-fold, respectively, whereas deficiency in IKKα did not affect promoter activity, indicating that IKKγ or IKKβ- mediates LMP1-dependent repression of the CD99 promoter (Fig. 3C). These data indicate that the NF-κB2 (p52) homodimer or heterodimer is the major inhibitory molecule, regardless of the NF-κB binding partner, whereas NF-κB1 (p50) likely only has a minor inhibitory effect.
To link LMP1 to p52 (NF-κB2) in the cell, we confirmed that LMP1 induces the processing of p100 to form p52. LMP1 transfection in 293T cells consistently induced the appearance of p52 in a dose-dependent manner (Fig. 3D). The appearance of p52 upon LMP1 introduction was verified by transfection of control p52 and NIK, a positive control effector that is known to induce processing of p100 to form p52 in the same 293T cells (Fig. 3D). The induction of P52 from P100 by LMP1 is consistent with those results that LMP1-mediated P52 is the major inhibitory player in the repression of the CD99 promoter (shown in Figs. 3A and 3B).
DISCUSSION
Given that (1) HLRSCs express very low or undetectable levels of CD99 (Kim et al., 1998), (2) HLRSCs constitutively express NF-κB-activating TNFR-1 family proteins (CD30, CD40 or LMP1) (Gruss et al., 1997; Pallesen et al., 1991; Schwab et al., 1982; Young et al., 1991), and (3) biochemical or genetic alterations that affect NF-κB activation frequently occur in HLRSCs (Cabannes et al., 1999; Emmerich et al., 1999; Wood et al., 1998), the results in this study demonstrating that NF-κB2 represses CD99 expression are relevant, and it is very likely to play an important role in the pathogenesis of HL. On average, over 50% of HL cases are associated with latent infections with the Epstein Barr virus, and that those EBV-infected HL cells invariably express EBV LMP1.
Ligand engagement of CD99 results in the upregulation of MHC class I and MHC class II, apoptosis, and cell-cell adhesion. Decreased surface expression of CD99 in HLRSCs has been associated with the downregulation of MHC-I or MHC-II, which is possibly why these cells are better able to avoid cytotoxic T lymphocytes (CTL) attack or resist apoptosis. Therefore, constitutive lack of CD99 surface expression in HLRSCs may provide the cells with a favorable environment for survival.
Compared to mature germinal center B cells, which express high-affinity B cell receptor (BCR), HL cells usually express lowaffinity B cell receptor, making them highly vulnerable to cell death unless survival signals are provided. Despite having lowaffinity BCRs, HL cells can be resistant to signal-induced apoptosis due to constitutively active NF-κB survival signals (Bours et al., 1992).
In this study, NF-κB appeared to inhibit the CD99 promoter through Sp1 sites, because (1) the CD99 promoter contains no NF-κB sites but multiple Sp1 sites, (2) LMP1 sufficiently repressed a synthetic Sp1 reporter (Fig. 3B), (3) LMP1 induces p52 production (Fig. 3D), and (4) the p52 subunit efficiently represses the promoter in vivo (Fig. 3B). All these findings suggest that LMP1-induced p52 plays the inhibitory roles in the repression of the promoter.
It is known that Sp1 interacts with many cellular factors in vitro or in vivo, including RelA, v-Rel, NF-κB1, and NF-κB2 (Perkins et al., 1994; Sif and Gilmore, 1994). Interaction of Sp1 with p52 is likely to inhibit Sp1 protein binding to Sp1 sites in the promoter, in a similar manner to what has been reported (Lin et al., 2009). Similar to the current study, the physical interaction of Sp1 with the NF-κB subunit RelA did activate an HIV promoter, in which Sp1 sites are adjacent to NF-κB sites (Perkins et al., 1994). On the other hand, it was recently shown that Sp1 interaction with NF-κB represses the Hepatitis B Virus (HBV) CP promoter, in which multiple Sp1 sites but no NF-κB sites are present (Lin et al., 2009). A zinc finger DNA-binding domain of Sp1 interacts with the N-terminal 43 amino acids of the DNAbinding domain of RelA (p65) in cells (Lin et al., 2009). The Sp1 co-transfection with p52 may decrease Sp1 binding to Sp1-binding sites in vivo, although this was not the case in an in vitro gel shift assay (data not shown). It is equally possible that Sp1 may interact with p52, resulting in the recruitment of transcriptional repressors, without affecting Sp1 DNA-binding activities, which ultimately leads to the repression. Whether or not Sp1 interacts physically with NF-κB2 subunit needs to be further studied.
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
Acknowledgments
We thank Drs. S.H. Park at Seoul National University and I.S. Lee at Konkuk University for providing reporter constructs and CD99 antibodies. This study was supported in part by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare Affairs, Republic of Korea (grant to MSK, A092255). We have no competing financial interests. This work was in part supported by 5R01CA085180-10 granted to Elliott Kieff, Channing Laboratory, Brigham and Women’s Hospital.
References
- 1.Bernard G., Zoccola D., Deckert M., Breittmayer J.P., Aussel C., Bernard A. The E2 molecule (CD99) specifically triggers homotypic aggregation of CD4+ CD8+ thymocytes. J. Immunol. (1995);154:26–32. [PubMed] [Google Scholar]
- 2.Bernard G., Breittmayer J.P., de Matteis M., Trampont P., Hofman P., Senik A., Bernard A. Apoptosis of immature thymocytes mediated by E2/CD99. J. Immunol. (1997);158:2543–2550. [PubMed] [Google Scholar]
- 3.Boiocchi M., De Re V., Dolcetti R., Carbone A., Scarpa A., Menestrina F. Association of Epstein-Barr virus genome with mixed cellularity and cellular phase nodular sclerosis Hodgkin’s disease subtypes. Ann. Oncol. (1992);3:307–310. doi: 10.1093/oxfordjournals.annonc.a058187. [DOI] [PubMed] [Google Scholar]
- 4.Bours V., Burd P.R., Brown K., Villalobos J., Park S., Ryseck R.P., Bravo R., Kelly K., Siebenlist U. A novel mitogen-inducible gene product related to p50/p105-NF-kappa B participates in transactivation through a kappa B site. Mol. Cell. Biol. (1992);12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braeuninger A., Kuppers R., Strickler J.G., Wacker H.H., Rajewsky K., Hansmann M.L. Hodgkin and Reed-Sternberg cells in lymphocyte predominant Hodgkin disease represent clonal populations of germinal center-derived tumor B cells. Proc. Natl. Acad. Sci. USA. (1997);94:9337–9342. doi: 10.1073/pnas.94.17.9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brauninger A., Hansmann M.L., Strickler J.G., Dummer R., Burg G., Rajewsky K., Kuppers R. Identification of common germinal-center B-cell precursors in two patients with both Hodgkin’s disease and non-Hodgkin’s lymphoma. N. Engl. J. Med. (1999);340:1239–1247. doi: 10.1056/NEJM199904223401604. [DOI] [PubMed] [Google Scholar]
- 7.Cabannes E., Khan G., Aillet F., Jarrett R.F., Hay R.T. Mutations in the IκBα gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. (1999);18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 8.Emmerich F., Meiser M., Hummel M., Demel G., Foss H.D., Jundt F., Mathas S., Krappmann D., Scheidereit C., Stein H., et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood. (1999);94:3129–3134. [PubMed] [Google Scholar]
- 9.Gelin C., Aubrit F., Phalipon A., Raynal B., Cole S., Kaczorek M., Bernard A. The E2 antigen, a 32 kd glycoprotein involved in T-cell adhesion processes, is the MIC2 gene product. EMBO J. (1989);8:3253–3259. doi: 10.1002/j.1460-2075.1989.tb08485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S., May M.J., Kopp E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. (1998);16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 11.Gruss H.J., Herrmann F., Gattei V., Gloghini A., Pinto A., Carbone A. CD40/CD40 ligand interactions in normal, reactive and malignant lympho-hematopoietic tissues. Leuk. Lymphoma. (1997);24:393–422. doi: 10.3109/10428199709055580. [DOI] [PubMed] [Google Scholar]
- 12.Hahn J.H., Kim M.K., Choi E.Y., Kim S.H., Sohn H.W., Ham D.I., Chung D.H., Kim T.J., Lee W.J., Park C.K., et al. CD99 (MIC2) regulates the LFA-1/ICAM-1-mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J. Immunol. (1997);159:2250–2258. [PubMed] [Google Scholar]
- 13.Hatzivassiliou E., Miller W.E., Raab-Traub N., Kieff E., Mosialos G. A fusion of the EBV latent membrane protein-1 (LMP1) transmembrane domains to the CD40 cytoplasmic domain is similar to LMP1 in constitutive activation of epidermal growth factor receptor expression, nuclear factor-kappa B, and stress-activated protein kinase. J. Immunol. (1998);160:1116–1121. [PubMed] [Google Scholar]
- 14.Herbst H., Niedobitek G., Kneba M., Hummel M., Finn T., Anagnostopoulos I., Bergholz M., Krieger G., Stein H. High incidence of Epstein-Barr virus genomes in Hodgkin’s disease. Am. J. Pathol. (1990);137:13–18. [PMC free article] [PubMed] [Google Scholar]
- 15.Herbst H., Pallesen G., Weiss L.M., Delsol G., Jarrett R.F., Steinbrecher E., Stein H., Hamilton-Dutoit S., Brousset P. Hodgkin’s disease and Epstein-Barr virus. Ann. Oncol. 3 Suppl. (1992a);4:27–30. doi: 10.1093/annonc/3.suppl_4.s27. [DOI] [PubMed] [Google Scholar]
- 16.Herbst H., Steinbrecher E., Niedobitek G., Young L.S., Brooks L., Muller-Lantzsch N., Stein H. Distribution and phenotype of Epstein-Barr virus-harboring cells in Hodgkin’s disease. Blood. (1992b);80:484–491. [PubMed] [Google Scholar]
- 17.Hodgkin T. On some morbid appearances of the absorbant glands and spleen. Med.-Chir. Trans. (1832);17:68. doi: 10.1177/095952873201700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jundt F., Anagnostopoulos I., Bommert K., Emmerich F., Muller G., Foss H.D., Royer H.D., Stein H., Dorken B. Hodgkin/ Reed-Sternberg cells induce fibroblasts to secrete eotaxin, a potent chemoattractant for T cells and eosinophils. Blood. (1999);94:2065–2071. [PubMed] [Google Scholar]
- 19.Kieff E., Rickinson A.B., Knipe D.M., Howley P.M. Epstein-Barr virus and its replication. In Fields Virology. Lippincott Williams and WIlkins; Philadelphia: (2001). pp. 2511–2574. [Google Scholar]
- 20.Kim S.H., Choi E.Y., Shin Y.K., Kim T.J., Chung D.H., Chang S.I., Kim N.K., Park S.H. Generation of cells with Hodgkin’s and Reed-Sternberg phenotype through downregulation of CD99 (Mic2). Blood. (1998);92:4287–4295. [PubMed] [Google Scholar]
- 21.Kim S.H., Shin Y.K., Lee I.S., Bae Y.M., Sohn H.W., Suh Y.H., Ree H.J., Rowe M., Park S.H. Viral latent membrane protein 1 (LMP-1)-induced CD99 down-regulation in B cells leads to the generation of cells with Hodgkin's and Reed- Sternberg phenotype. Blood. (2000);95:294–300. [PubMed] [Google Scholar]
- 22.Krappmann D., Emmerich F., Kordes U., Scharschmidt E., Dorken B., Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. (1999);18:943–953. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- 23.Kuppers R., Hansmann M.L., Rajewsky K. Clonality and germinal centre B-cell derivation of Hodgkin/Reed-Sternberg cells in Hodgkin’s disease. Ann. Oncol. (1998);9(Suppl 5):S17–20. doi: 10.1093/annonc/9.suppl_5.s17. [DOI] [PubMed] [Google Scholar]
- 24.Kuppers R., Rajewsky K. The origin of Hodgkin and Reed/Sternberg cells in Hodgkin’s disease. Annu. Rev. Immunol. (1998);16:471–493. doi: 10.1146/annurev.immunol.16.1.471. [DOI] [PubMed] [Google Scholar]
- 25.Lee I.S., Shin Y.K., Chung D.H., Park S.H. LMP1- induced downregulation of CD99 molecules in Hodgkin and Reed-Sternberg cells. Leuk. Lymphoma. (2001);42:587–594. doi: 10.3109/10428190109099318. [DOI] [PubMed] [Google Scholar]
- 26.Levy R., Dilley J., Fox R.I., Warnke R. A human thymus-leukemia antigen defined by hybridoma monoclonal antibodies. Proc. Natl. Acad. Sci. USA. (1979);76:6552–6556. doi: 10.1073/pnas.76.12.6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Y.C., Hsu E.C., Ting L.P. Repression of hepatitis B viral gene expression by transcription factor nuclear factorkappaB. Cell. Microbiol. (2009);11:645–660. doi: 10.1111/j.1462-5822.2008.01280.x. [DOI] [PubMed] [Google Scholar]
- 28.Luftig M., Yasui T., Soni V., Kang M.S., Jacobson N., Cahir- McFarland E., Seed B., Kieff E. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc. Natl. Acad. Sci. USA. (2004);101:141–146. doi: 10.1073/pnas.2237183100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marafioti T., Hummel M., Anagnostopoulos I., Foss H.D., Huhn D., Stein H. Classical Hodgkin’s disease and follicular lymphoma originating from the same germinal center B cell. J. Clin. Oncol. (1999);17:3804–3809. doi: 10.1200/JCO.1999.17.12.3804. [DOI] [PubMed] [Google Scholar]
- 30.Ohno T., Stribley J.A., Wu G., Hinrichs S.H., Weisenburger D.D., Chan W.C. Clonality in nodular lymphocyte-predominant Hodgkin’s disease. N. Engl. J. Med. (1997);337:459–465. doi: 10.1056/NEJM199708143370704. [DOI] [PubMed] [Google Scholar]
- 31.Pallesen G., Hamilton-Dutoit S.J., Rowe M., Young L.S. pression of Epstein-Barr virus latent gene products in tumour cells of Hodgkin’s disease. Lancet. (1991);337:320–322. doi: 10.1016/0140-6736(91)90943-j. [DOI] [PubMed] [Google Scholar]
- 32.Park C.K., Shin Y.K., Kim T.J., Park S.H., Ahn G.H. High CD99 expression in memory T and B cells in reactive lymph nodes. J. Korean Med. Sci. (1999);14:600–606. doi: 10.3346/jkms.1999.14.6.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins N.D., Agranoff A.B., Pascal E., Nabel G.J. An interaction between the DNA-binding domains of RelA(p65) and Sp1 mediates human immunodeficiency virus gene activation. Mol. Cell. Biol. (1994);14:6570–6583. doi: 10.1128/mcb.14.10.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettersen R.D., Bernard G., Olafsen M.K., Pourtein M., Lie S.O. CD99 signals caspase-independent T cell death. J. Immunol. (2001);166:4931–4942. doi: 10.4049/jimmunol.166.8.4931. [DOI] [PubMed] [Google Scholar]
- 35.Rickinson A.B., Kieff E., Howely P.M. Epstein-Barr virus. In Fields Virology. Lippincott Williams and Wilkins; Philadelphia: (2001). pp. 2575–2628. [Google Scholar]
- 36.Schwab U., Stein H., Gerdes J., Lemke H., Kirchner H., Schaadt M., Diehl V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. (1982);299:65–67. doi: 10.1038/299065a0. [DOI] [PubMed] [Google Scholar]
- 37.Shin Y.K., Kim Do N., Lee S.K. Association between Epstein-Barr virus infection and chemoresistance to docetaxel in gastric carcinoma. Mol. Cells. (2011);32:173–179. doi: 10.1007/s10059-011-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sif S., Gilmore T.D. Interaction of the v-Rel oncoprotein with cellular transcription factor Sp1. J. Virol. (1994);68:7131–7138. doi: 10.1128/jvi.68.11.7131-7138.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sohn H.W., Choi E.Y., Kim S.H., Lee I.S., Chung D.H., Sung U.A., Hwang D.H., Cho S.S., Jun B.H., Jang J.J., et al. Engagement of CD99 induces apoptosis through a calcineurinindependent pathway in Ewing's sarcoma cells. Am. J. Pathol. (1998);153:1937–1945. doi: 10.1016/S0002-9440(10)65707-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staudt L.M. The molecular and cellular origins of Hodgkin’s disease. J. Exp. Med. (2000);191:207–212. doi: 10.1084/jem.191.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sylla B.S., Hung S.C., Davidson D.M., Hatzivassiliou E., Malinin N.L., Wallach D., Gilmore T.D., Kieff E., Mosialos G. Epstein-Barr virus-transforming protein latent infection membrane protein 1 activates transcription factor NF-kappaB through a pathway that includes the NF-kappaB-inducing kinase and the IkappaB kinases IKKalpha and IKKbeta. Proc. Natl. Acad. Sci. USA. (1998);95:10106–10111. doi: 10.1073/pnas.95.17.10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe K., Yamashita Y., Nakayama A., Hasegawa Y., Kojima H., Nagasawa T., Mori N. Varied B-cell immunophenotypes of Hodgkin/Reed-Sternberg cells in classic Hodgkin’s disease. Histopathology. (2000);36:353–361. doi: 10.1046/j.1365-2559.2000.00830.x. [DOI] [PubMed] [Google Scholar]
- 43.Wood K.M., Roff M., Hay R.T. Defective Ikappa Balpha in Hodgkin cell lines with constitutively active NF-kappaB. Oncogene. (1998);16:2131–2139. doi: 10.1038/sj.onc.1201735. [DOI] [PubMed] [Google Scholar]
- 44.Young L.S., Deacon E.M., Rowe M., Crocker J., Herbst H., Niedobitek G., Hamilton-Dutoit S.J., Pallesen G. Epstein-Barr virus latent genes in tumour cells of Hodgkin’s disease. Lancet. (1991);337:1617. doi: 10.1016/0140-6736(91)93322-z. [DOI] [PubMed] [Google Scholar]