

Abstract
Multidrug resistance is the phenomenon by which, after exposure to a single chemotherapeutic agent, cancer cells evade the agent’s cytotoxic effects as well as become resistant to several classes of diverse drugs. ATP-binding cassette (ABC) transporters are a family of transporter proteins that contribute to drug resistance via an ATP-dependent drug efflux pump. P-glycoprotein (P-gp) is a prominent ABC superfamily protein encoded by the mdr gene which has the ability to mediate the cellular extrusion of xenobiotics and anticancer drugs from tumor cells. Exclusively expressed P-gp cells from the human colon cancer HCT15/DOX line showed resistance to doxorubicin while parental HCT15 cells treated with doxorubicin displayed typical signs of apoptosis. In order to verify the hypothesis that expression of MDR is controlled in part, by protein kinase C (PKC), expression patterns of different PKC isoforms were examined in both cell lines. Of the PKC isoforms evaluated, the membrane translocation and expression levels of PKCα were strikingly increased in HCT15/DOX cells. PKCα reversed doxorubicin-induced apoptosis through the scavenging of ROS as well as inhibition of PARP cleavage. In addition, inhibition of PKCα with Go6976, a specific inhibitor of classical PKC, led to reduced MDR expression and increased doxorubicin-induced apoptosis. Knockdown of PKCα by siRNA diminished the protective effects of PKCα for doxorubicin-induced apoptosis. These results suggested that over-expression and activity of PKCα is closely associated with the regulation of the MDR phenotype in human colon cancer HCT15 cells and provided insight into a new strategy for inhibiting doxorubicin resistance in human cancers.
Keywords: apoptosis, doxorubicin, multidrug resistance, PKCα
INTRODUCTION
Membrane transporter families play a pivotal role in the circulation and excretion of many clinically essential therapeutic drugs. P-glycoprotein (P-gp) is a 170-kDa ATP-dependent transmembrane glycoprotein which is the best characterized drug efflux pump and known to confer resistance to a variety of structurally and functionally unrelated anti-cancer drugs. This phenomenon is known as multidrug resistance (MDR) and represents a serious problem in the chemotherapeutic management of cancers. Manifestation of the MDR phenotype is most often due to the over-expression of drug efflux pumps in the plasma membrane of cancer cells (Endicott and Ling, 1989; Gottesman et al., 2002). It has been reported that P-gp drug efflux activity can be modulated through phosphorylation by different families of protein kinases (Gottesman and Pastan, 1993). The protein kinase responsible for the phosphorylation of P-gp is believed to be protein kinase C (PKC) (Ling, 1997). PKC is a serine/threonine kinase involved in the signal transduction required for cellular proliferation and differentiation (Clemens et al., 1992; Nishizuka and Kikkawa, 2003). Classical and novel PKC groups respond to activation by phorbol esters (Gill et al., 2001). Conventional PKCs are, like the originally described kinase activity, dependent on Ca2+ and phospholipids for their activation. This group includes PKCα, PKCβI, PKCβII and PKCγ. A second group, including the novel PKCs, does not require Ca2+ but are phospholipid dependent and known PKCδ, PKCɛ, PKCη and PKCθ. A third, more recently identified group includes PKCξ and PKCλ/τ. These are known as atypical PKCs, which require neither phospholipids nor Ca2+ for their activation (Mellor and Parker, 1998).
A wide array of research has postulated that specific isoforms of PKCs may be either pro-apoptotic or anti-apoptotic depending on the condition and stimulus of the cell type (Leszczynski, 1995; Lucas and Sánchez-Margalet, 1995). Of the PKC isoforms, PKCα has been reported to be of particular determinant in the development and prolongation of the MDR phenotype. Studies have shown that PKCα is expressed abundantly in surgical specimens of human colon cancer and contributes to drug resistance as observed in colon cancer (Gravitt et al., 1994). Breast carcinoma cells also develop resistance against doxorubicin through elevated levels of P-gp and PKCα, and display the MDR phenotype (Budworth et al., 1997). Additionally, doxorubicin was screened against a panel of human tumor cell lines derived from cancer patients. This study revealed that increased PKCα levels were associated with reduced drug efficacy, which means that one or more substrates of this kinase may be related to the MDR phenotype (Gariboldi et al., 2003).
Doxorubicin is commonly used for the treatment of different types of cancer. However, its efficacy is often reduced because it is a substrate for P-gp. Therefore, in order to investigate the individual roles of various PKC isoforms in MDR, we examined the expression and activity of the major isoforms of PKC in the parent HCT15 cell line and its drug-resistant subline, HCT15/DOX. It was observed that the expression level and membrane translocation of PKCα alone markedly increased in HCT15/DOX cells and functioned as an antagonist to doxorubicin-induced apoptosis. Additionally, inhibition of PKCα by a specific inhibitor, Go6967, and by using PKCα SiRNA, both lead to reduced MDR activity and enhanced doxorubicin-induced apoptosis. The results of this study demonstrated that over-expression of an endogenous PKCα conferred increased MDR through the modulation of reactive oxygen species (ROS) generation in human colon cancer HCT15 cells.
MATERIALS AND METHODS
Materials
Doxorubicin (adriamycin), 4,6-Diamidino-2-phenylindole (DAPI) and propidium iodide were purchased from Sigma-Aldrich (USA). Dichlorofluorescein diacetate (DCFHDA), dihydrohodamine 123 (DHR 123), and rhodamine 123 were obtained from Molecular Probes (USA). The electrophoresis reagents and Bio-Rad protein assay kit were purchased from Bio-Rad Laboratories (USA). The anti-PKC isotypes antibodies were obtained from Cell Signaling Technology Inc. (USA). These chemicals were used according to the manufacturer’s instructions.
Cell culture
The human colorectal carcinoma (HCT15) cells were obtained from ATCC, CCL-225. An doxorubicin-resistant subline of HCT15 cells, named HCT15/doxorubicin (HCT15/DOX), were established by continuous exposure to doxorubicin as described previously (Choi et al., 1996). Cells were cultured at a density of 5 × 105 in RPMI-1640 medium containing 10% heat inactivated fetal bovine serum (FBS), L-glutamine and supplemented with 1% (v/v) antibiotic-penicillin streptomycin (Gibco, Invitrogen Corporation) at 37°C in a 5% CO2-humidified atmosphere. Exponentially growing cells were seeded at 1 × 106 cells per dish and then the cells were exposed to various agents.
Preparation of the cytosolic and membrane protein fractions
Cytosolic and particulate membrane fractions of cells were prepared as described previously but with slight modification (Tentori et al., 2001). Briefly, the cells were sonicated in buffer A (20 mM Tris, pH 7.5, 250 mM sucrose, 10 mM ethyleneglycol tetra-acetic acid (EGTA), 2 mM ethylenediamine tetra-acetic acid (EDTA), 1 mM sodium fluoride (NaF), 1 mM sodium orthovanadate (Na3VO4), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin) and centrifuged for 10 min at 1,000 × g to remove cell debris. The supernatants were then centrifuged at 100,000 × g for 30 min and the resulting supernatants were saved as the cytosolic fractions. Proteins in the pellets were extracted with buffer B [20 mM Tris, pH 7.5, 1% sodium dodecyl sulfate (SDS), 150 mM NaCl, 1 mM EGTA, 1 mM NaF, 1 mM Na3VO4 and various protease inhibitors]. Following centrifugation at 100,000 × g for 30 min, the supernatants were referred to as the particulate membrane fractions.
Determination of apoptosis
Cell viability was determined using a trypan blue exclusion test. Morphological analysis of apoptosis was performed after staining using Hoechst 33342 dye. The cells were fixed in 4% para-formaldehyde and permeabilized with PBS/0.5% Triton X-100, and the nuclei were stained for 20 min using Hoechst dye. The coverslips were then washed, mounted onto slides and viewed with a fluorescence microscope. Apoptotic DNA fragments were isolated according to a previously described method (Ma et al., 2000). The DNA was separated by electrophoresis on 1% agarose gels, stained with ethidium bromide and visualized under ultraviolet light.
Cell cycle analysis
The cells were trypsinized and collected by centrifugation at 1,000 × g for 10 min. For fixation, 70% ethanol was added and the cell suspension was kept overnight at 4°C. The cells were then stained with propidium iodide solution (50 μg/ml propidium iodide, 0.1% Triton X-100, 0.1 mM EDTA and 50 μg/ml RNase) for 20 min at 4°C. The stained DNA was analyzed by flow cytometer (Becton Dickinson).
Measurement of ROS
Intracellular ROS concentration was measured using the oxidant-sensitive fluorescent probe, DCFHDA, with inverted microscope. Cells were grown at a density of 1 × 106 cells per 35 mm culture dish and maintained in the growth medium for 24 h. Cells were exposed to 5 μM DCFHDA for 20 min and were then washed with PBS. DCF fluorescence (excitation, 480 nm; emission, 520 nm) was imaged using an inverted microscope (Zeiss Axiovert 200). For FACS analyses, cellular DCF fluorescence measurement included at least 10,000 events/test using a flow cytometer with a fluorescein isothiocyanate filter.
Dihydrorhodamine 123 accumulation assay
The HCT15 cells were incubated with 10 μM of DHR123 in the culture medium for 20 min and then washed three times with ice-cold PBS. The cells were imaged on an inverted microscope using FITC fluorescence intensity.
Protein kinase assay
The PKC activity was determined by measuring the transfer of 32P-ATP to a peptide substrate that was captured on P81 phosphocellulose papers. Aliquots of cell lysates were incubated with 50 mM Tris at pH 7.5 with 200 μM PKCα substrate AAKIQASFRGHMARKK, 40 mM of MgCl2 and 400 μM of ATP. After 5 min at 30°C, the reactions were terminated by adding EDTA to a final concentration of 20 mM. Relative PKC activity was then quantified with a liquid scintillation counter.
Western blotting
In brief, aliquots of protein extracts (30 μg) from cells of different treatment groups were suspended in 0.1 M Tris-HCl buffer (pH 7.4) containing 1% SDS, 0.05% β-mercaptoethanol, 2.5% glycerol and 0.001% bromophenol blue, and subsequently fractionated by 10% SDS-polyacrylamide gel electrophoresis. The proteins were transferred electrophoretically onto nitrocellulose membranes (0.2 μm, Schleicher and Schuell). The membranes were blocked using 5% non-fat dry milk and 0.1% Tween 20 in Tris buffered saline (TBS). The membranes were subsequently probed with primary antibody in TBS containing 3% non-fat dry milk and 0.1% Tween 20. The antibody-antigen complexes were detected using goat anti-mouse IgG or goat anti-rabbit IgG peroxidase conjugates followed by the use of an enhanced chemiluminescence (ECL) detection kit (Amersham Corp.).
Reverse-transcription polymerase chain reaction (RT-PCR)
In order to determine if HCT15 cells expressed transcriptional levels of mdr, RT-PCR was performed. Total RNA was isolated from the doxorubicin-resistant HCT15/DOX and parental HCT15 cells using TRI reagent (Molecular Research Center, Inc.). RT-PCR was performed using an access RT-PCR kit (Promega). The following PCR conditions were employed: 1 cycle at 95°C for 5 min, 30 cycles at 95°C for 1 min, 55°C for 1 min, 72°C for 1 min, and an additional extension step of 5 min at 72°C. The amplified PCR products were analyzed by 1 % agarose gel electrophoresis and ethidium bromide staining. The following primer sequences for mdr1 gene sequence were utilized as follows, 5′-GCCTGGCAGCTGGAAGACAAATACACAAAATT-3′ (sense) and 5′-CAGACAGCAGCTGACAGTCCAAGAACA GGACT-3′ (antisense), corresponding to the previously published cDNA sequence (Limthrakul et al., 2004), and 5′-GTCDDAGTCAACGGATTTGG-3′ (sense) and 5′-GGGTGGA ATCATATTTGGAACTTG-3′ (antisense) for GAPDH as an internal loading control (Oligo, Macrogen).
Generation of the siRNA-PKCα HCT15 clones
The siRNA-PKCα pool was obtained from Santa Cruz Biotechnology (SC-36243) and utilized according to the manufacturer’s instruction. HCT15 cells were transfected using Lipofectamine RNAiMAX (Invitrogen, USA) according to the manufacturer’s recommendations. Cells were cultured for 24 h at densities of 1 × 106 cells per dish to 40–50% confluence. The cells were then transfected with 1 nmol of siRNA. At the indicated times after transfection, the cells were treated with 5 μM doxorubicin for 24 h. The effects of scrambled siRNA were also evaluated according to the modified protocol as described previously (Nakura et al., 2011).
Statistical analysis
All data are presented as the mean values ± SD (standard deviation) and originate from 3 separate experiments. The data was evaluated using SPSS for the student t-test and subjected to one-way analysis of variance (ANOVA). The significance of the difference between the means was determined by the Tukey range test and considered significant at P ≤ 0.05.
RESULTS
Doxorubicin induced oxidative-stress apoptosis
The quinone moiety of the anthracyclines are known to act as catalysts in the formation of ROS including superoxide anion and hydrogen peroxide (Doroshow et al., 1986). At relevant physiological concentrations, a consequence of treatment with doxorubicin is the induction of apoptosis associated with DNA fragmentation and cell shrinkage (Bose et al., 1995; Skladanowski and Konopa, 1993). HCT15 and HCT15/DOX cells were treated with 2 and 4 μM of doxorubicin for 12, 24 and 36 h, and the cell lysates from both cell lines were analyzed by Western blot. Doxorubicin induced apoptosis through poly (ADP-ribose) polymerase (PARP) and lamin B cleavage in the HCT15 cells only, and failed to induce apoptosis in the HCT15/DOX cells (Fig. 1A). Additionally, doxorubicin induced DNA fragmentation with rapid kinetics in the parental cells (Fig. 1B). Lamin B and PARP were cleaved with similar kinetics as the DNA fragmentation, yielding cleavage bands (Fig. 1B). The use of doxorubicin in cancer therapy has been hampered by the ability of cancer cells to develop doxorubicin resistance (Dolfini et al., 1997). In order to evaluate the degree of apoptosis induced by doxorubicin, both cell types were treated with doxorubicin for 12 and 24 h. As shown in Fig. 1C, doxorubicin induced 13% apoptosis in parental cells, but no apoptosis was observed in the resistant cell line. These results indicated that doxorubicin is hindered in inducing apoptosis in HCT15/DOX cell lines.
Fig. 1.

Doxorubicin induced oxidative-stress apoptosis in HCT15 and HCT15/DOX cells. HCT15 and HCT15/DOX cells were incubated in the absence (con) or presence of doxorubicin for 24 h. (A) Cleavages of PARP and lamin B were analyzed by Western blot. (B) Analysis of doxorubicin-induced DNA fragmentation of cells by agarose gel electrophoresis. (C) At the indicated time, the cells were harvested and stained with propidium iodide and their DNA content was analyzed by flow cytometry. (D) Doxorubicin induces ROS generation. HCT15 and HCT15/DOX cells were treated with doxorubicin for the indicated time and further incubated with DCFHDA for 20 min. Accumulation of the probe in the cells was measured by an increased emission at 520 nm when the sample was excited at 480 nm. Fluorescence images of DCF-loaded cells were obtained under a microscope.
In order to investigate the role of oxidative stress in doxorubicin-induced apoptosis, we used a cell-permeable fluorescent dye, DCFHDA, to examine ROS generation in both types of cells in response to doxorubicin stimulation. A considerable increase in oxidant-induced 2′,7′-dichlorofluorescein fluorescence was observed in the HCT15 cells but not in the HCT15/DOX cells (Fig. 1D). It is assumed that doxorubicin-induced apoptosis occurs through increase in ROS generation, but only in parental cells. Under resistance conditions, it does not induce intracellular oxidative stress.
MDR expression in HCT15 and HCT15/DOX cells
Expression of the mdr1 gene was evaluated in the parental and HCT15/DOX cells using RT-PCR. After RNA isolation, the expression of mdr1 was determined using mdr1-specific primers producing a 243 bp RT-PCR product. Control RT-PCR was carried out in parallel with GAPDH. Little mdr expression was revealed in parental cells, while robust expression was observed in the HCT15/DOX cells (Fig. 2A). Gel electrophoresis was performed in order to detect the expression of MDR1 in both cell lines. The cell lysates were analyzed by Western blot with anti-MDR1. The expression of MDR was very high in the HCT15/DOX cells while remaining very little or undetected in the parental cell lines (Fig. 2B).
Fig. 2.

Over-expression of MDR in doxorubicin resistant HCT15/DOX cells. (A) RT-PCR analysis of P-glycoprotein (MDR). Total RNA was isolated from HCT15 parental and resistant cells, and RT-PCR was performed. PCR products were separated on 1.2% agarose gel and photographed. GAPDH was also examined as a reference. (B) Western blot identification of MDR expression in HCT15 and HCT15/DOX cells. Whole-cell lysates were extracted from HCT15 cells and separated by SDS-PAGE and the resulting proteins were detected by immunoblotting. (C) Mitochondria and membrane potential were revealed by rhodamine 123. Rhodamine 123 retention was evaluated after accumulation for 30 min and 30 min of efflux in dye-free medium.
In order to gain further insight into the multiple resistance mediated by HCT15/DOX cells, rhodamine 123 was used as a molecular probe and functional assay because this fluorescent dye is considered to be a relatively specific substrate for P-gp, reduced retention of rhodamine 123 in the cells indicates the functioning of P-gp (Altenberg et al., 1994; Gravitt et al., 1994). Rhodamine 123 accumulation was evaluated for 30 min along with 30 min efflux in dye-free medium. As shown in Fig. 2C, doxorubicin-resistant subline HCT15/DOX significantly reduced the retention of rhodamine 123 as compared to the parental cells.
Differential expressions of PKC isoforms
It was realized very early that the mechanism of action for PKC induced reversal of MDR would require a specific substrate. Therefore, the anti-apoptotic activity of PKC and the effects of doxorubicin on PKC activity were determined. PKC was measured in the parental and resistant cells with a radioisotope assay that utilizes the phosphorylation of a synthetic substrate AAKIQASFRGHMARPKK. The PKC activity increased almost six-fold after treatment with doxorubicin in the HCT15/DOX cell lines (Fig. 3A). This result suggested that the activity of PKC is implicated in the doxorubicin resistance observed in the HCT15/DOX cells.
Fig. 3.

PKCα is specifically activated in doxorubicin resistant cell lines. (A) Measurement of PKC activity in HCT15 and HCT15/DOX cells. Lysates from HCT15 and HCT15/DOX cells were used for the kinase assay using the specific PKC substrate peptide (AAKIQA SFRGHMARKK) and the 32P-incorporation was measured by scintillation counter. Notably, specific up-regulation of PKCα was detected in the doxorubicin-resistant HCT15/DOX cells. (B) HCT15 and HCT15/DOX cells were treated with doxorubicin. The cells were fractionated into cytosolic and membrane fractions and equal amounts of lysates were used for immunoblotting with PKC isoform (α, βI and ξ)-specific antibodies.
PKC isotypes α, βI and ξ were evaluated by Western blot in the investigation into their role in drug resistance. It was noted that the PKC α, βI and ξ-isoforms were detectable in the cytosol and in membrane portion of both cell lines, although some differences in their expressions were apparent. βI- and ξ-PKC protein levels were similar in the parental and resistant cell lines, but an obvious difference was observed in PKCα expression. As shown in Fig. 3B, a higher level of PKCα was found in the HCT15/DOX cell membranes than in the cytosol, as compared to the results with the HCT15 cells. Furthermore, total expression of PKC was marsedly decreased in α the parental cell line and highly expressed in the HCT15/DOX cell line.
Protein level of PKCα was reduced by doxorubicin in HCT15 cells
The conventional PKC family contains many isoforms including PKCα and PKCβI. In order to scrutinize the effects of doxorubicin on the expression of PKCα and PKCβI in parental and resistant cell lines, we performed immunoblot experiments on their separated cytosolic and membrane fractions. As shown in Fig. 4, doxorubicin treatment inhibited PKCα expression in the membrane fractions whereas the PKCβI isoform remained the same at both fractions in the HCT15 cells. Furthermore, the transient expression of PKCα in the cytosol suggests the translocation from cytosol to the membrane. In contrast, PKCα in the HCT15/DOX cells seemed to predominate in regards to doxorubicin treatment in the cytosol and membrane fractions (Fig. 4). The relative expression of PKCβI in the cytosolic and membrane fractions was not significantly altered by doxorubicin. This data suggests that PKCβI has no major role, whereas PKCα mediates, at least in part, multidrug resistance.
Fig. 4.
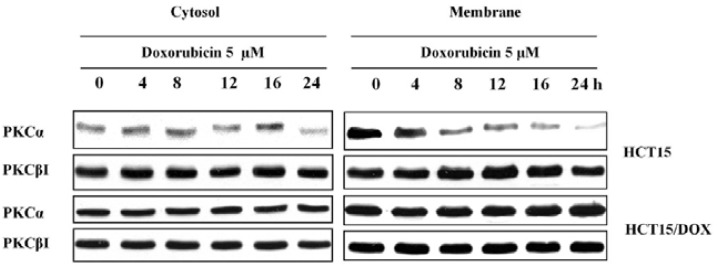
Western blot analysis of PKC isoforms in HCT15 and HCT15/DOX cells. HCT15 and HCT15/DOX cells were treated with doxorubicin for the indicated time. The cells were fractionated into cytosolic and membrane fractions and equal amounts of lysates were used for immunoblotting with PKC isoform-specific antibodies.
PKCα activity blocks ROS production in HCT15 and HCT15/DOX cells
Increasing evidence suggests that the mitochondria are the main site of ROS generation and high levels of ROS may mediate the induction of apoptosis (Fleury et al., 2002). In order to investigate if doxorubicin silled cancer cells through ROS generation, we evaluated the redox status in the HCT15 and HCT15/DOX cells using oxidation-sensitive fluorescent dye, DCFHDA. Doxorubicin treatment increased oxidant-induced 2′,7′-dichlorofluorescein fluorescence, only in the parental cells, but with treatment of the cells with Go6976, a rapid accumulation of fluorescence was observed in both cell types (Fig. 5). The occurrence of ROS generation were displayed by the mitochondrial-specific redox-sensitive fluorophore dihydrorhodamine (DHR) 123 (Fig. 5) when compared to both types of cell lines, either with or without inhibiting PKCα.
Fig. 5.

Inhibition of PKCα by Go6976 induces ROS generation in doxorubicin treated HCT15/DOX cells. Both cell types were challenged with Go6976 and then incubated with doxorubicin. ROS generation was exhibited by DCFHDA. Dihydrorhodamine 123 results indicated mitochondrial ROS generation.
Go6976 reversed the PKCα inhibition of doxorubicin - induced apoptosis in HCT15/DOX cells
Much compelling evidence has illuminated the anti-apoptotic roles of Bcl2 and Bad proteins. In fact, PKCα can phosphorylate Bcl2 at Ser70, a site which is critical for its anti-apoptotic effects. In order to determine the relative contribution of the PKCα isoform to decreased activity of doxorubicin in HCT15/DOX cells, we treated both cell types with classical PKC inhibitor, Go6976. After treatment, a significant decrease was observed in Bcl2 and p-Bad in the parental fraction as well as in HCT15/DOX cells lines (Fig. 6A). Upon the inhibition of PKCα with inhibitor Go6976, doxorubicin-induced apoptosis increased 45% in the parental cells and 17% in the HCT15/DOX cells relative to the untreated cells (Fig. 6B). These results are in agreement with previously published work (La Porta et al., 1998) that indicated PKCα plays a pivotal role in drug resistance and mediation of anti-apoptotic events in doxorubicin treated cells. Furthermore, inhibition of PKCα led to increased cleavage of PARP in both types of cells (Fig. 6A).
Fig. 6.

Go6976 reversed the PKCα inhibition of doxorubicin-induced apoptosis in HCT15/DOX cells. Cells were pretreated, either with or without 5 μM Go6976, for 2 h and then with 5 μM of doxorubicin. (A) Lysates of HCT15/DOX cells were used for Western blotting to detect PARP, Bcl2 and p-Bad, and actin was used as a control. (B) The cells were harvested and stained with propidium iodide and their DNA content was analyzed by flow cytometry.
Knockdown of PKCα by SiRNA induced apoptosis in HCT15/DOX cells
To gain further insight into the anti-apoptotic effects of PKCα, targeted siRNA was used against PKCα mRNA in order to knockdown PKCα in the resistant cell line. The result was analyzed by FACS and Western blot. As shown in Fig. 7A, apoptotic cell death increased in transfected PKCα siRNA cells as compared to control siRNA transfected cells. Additionally, knockdown of PKCα down-regulated Bcl2 and p-Bad as well as enhanced PARP cleavage and induced apoptosis in the resistant cell line (Fig. 7B). We also examined the dose dependent effects of siRNA by exposing HCT15/DOX cells to 5 μM doxorubicin along with different concentrations of siRNA (Fig. 7B). These results clearly indicated that depletion of PKCα was associated with doxorubicin-induced apoptosis in the resistant cell line.
Fig. 7.

RNA interference-mediated inhibition of PKCα increased doxorubicin-induced apoptosis. (A) FACS analysis of the cell cycle. Cells were pre-incubated with either siRNA, scrambled siRNA, or vehicle alone for 24 h followed by stimulation with doxorubicin. Cell cycle analysis was done by FACS. (B) PKCα protein levels of siRNA-PKCα cells. HCT15/DOX cells were transfected with various concentrations of siRNA and then incubated with either doxorubicin or carrier for 24 h. Total cell lysates were prepared and used for Western blotting. (C) PKCα inhibition induces ROS generation. HCT15/DOX cells were transfected with siRNA and then incubated with doxorubicin or carrier for 24 h. PKCα siRNA diminished the protective effect of PKCα and induced ROS generation as shown through DCFHDA fluorescence and translocation of DHR.
Conventional evidence has indicated that doxorubicin-induced apoptosis is a complex multifunctional process in which ROS generation production plays a vital role in the induction of apoptosis (Konorev et al., 1999). In order to investigate whether or not doxorubicin-induced apoptosis in HCT15/DOX cells is associated with the generation of cellular oxidative stress, the levels of intracellular peroxides were evaluated with DCFHDA. It was assumed that PKCα reversed doxorubicin-induced apoptosis in resistant cells through decrease in DCF-detectable ROS. As shown in Fig. 7C, transfection of cells with PKCα siRNA, followed by doxorubicin treatment, diminished the protective effect of PKCα and induced ROS generation. Therefore, it is likely that ROS production is the agent of doxorubicin-induced apoptosis in HCT15/DOX cells. Additionally, the HCT15/DOX cells transfected with scramble siRNA mediated or induced the same effect as the control cells without transfection (Fig. 7C). These results suggested that PKCα provides protection from the apoptotic actions of doxorubicin and enhanced MDR by decreasing the steady-state level of intracellular oxidants.
DISCUSSION
The mechanisms of MDR relative to the treatment failure of chemotherapeutic drugs has been studied extensively for more than two decades, but the exact molecular mechanism mediating the chemotherapy resistance and the role of drug resistance genes involved in the transport of anticancer agents remains unclear. There are a number of different mechanisms by which cancer cells develop MDR (Takara et al., 2006). The most often reported mechanism with known clinical significance is activation of ATP-dependent efflux pumps resulting in reduced intra-cellular drug concentrations, in which P-gp is the best known efflux pump gene alteration. MDR can also be caused by proteins involved in the control of apoptosis, such as Bcl2 (Han et al., 2002). The current study postulated a role for PKC in the regulation of the MDR phenotype. Other studies have shown that PKC produces prominent activity in MDR cell lines and that PKC inhibitors could be of therapeutic value in reversing MDR and inhibiting P-gp phosphorylation (Fine et al., 1996; O’Brian, 2001). It has been shown that P-gp is phosphorylated by PKC and that this phosphorylation entirely modulates P-gp efflux function (Ling, 1997). At present, the PKC family consists of at least 12 isoforms whose expression and regulation varies between cell types. In particular, the over-expression and activity of the PKCα isoform is closely associated with the development of the MDR phenotype, but in some cancers the expression patterns of PKCβI, PKCγ and PKCη mediate MDR. Similarly, the PKCθ isoform is also activated by diverse stimuli and participates in MDR, but this contribution is minor and occurs only in concert with PKCα (Aquino et al., 1999; Gill et al., 2001). In addition, the pro- and anti-apoptotic roles of PKCs isoforms has been well documented (Leszczynski, 1995; Lucas and Sánchez-Margalet, 1995). Previously, it has been shown that over-expression of PKCα resulted in increased mitochondrial PKCα localization and Bcl2 phosphorylation, which are required for their anti-apoptotic function (Ruvolo et al., 1998). Therefore, it is pertinent to understand the mechanism by which PKCα is activated in the apoptotic process.
HCT15/DOX is an established doxorubicin resistant cell line selected by the stepwise exposure of parental HCT15 cells to increasing concentrations of doxorubicin. Studies have shown that HCT15/DOX cells possess characteristics of classical MDR including over-expression of P-gp as well as high resistance to the cytotoxicity of doxorubicin (Choi et al., 1996). The above mentioned results imply that doxorubicin is hindered by some protein residues which may alter the resistant cell efflux rate. The over-expression of PKCα has been radically elevated yet no significant difference was observed in the expression of PKCβI, PKCβII and PKCγ in colon cancer cells (Gravitt et al., 1994). Furthermore, the expression patterns of PKCα and PKCβI in the resistant cell line remained constant upon the treatment of doxorubicin, but in HCT15 cells the membrane translocation of PKCα was reduced. After treatment with doxorubicin, similar expression levels of PKCβI in the cytosol and membrane revealed that it shared no major contribution to the effect, suggesting that only PKCα has a role in MDR. This finding means that PKCα is capable of enhancing mdr gene expression and inducing MDR because the total protein expression of PKCα was also increased in the HCT15/DOX cells (Kuranami et al., 1995). The over-expression of PKCα in HCT15/DOX cells, not only induced MDR, but also reversed apoptosis through the down-regulation of ROS generation in doxorubicin treated cells. Doxorubicin has been reported to induce apoptosis in various cancer cell lines through ROS generation (Gouazé et al., 2001), but the exact role of ROS in the resistance of chemotherapy remains to be elucidated. Early evidence has shown that ROS production is responsible for Gαqmediated activation of phospholipase C (PLC)-β, which is followed by the activation of a complex network of signaling proteins governing cell-cycle transition and redox balance (Bae et al., 2011). Although PKC stimulated the induction of ROS generation, this effect seemed to be cell and tissue dependent (Inoguchi et al., 2003).
The current study proved that PKCα was over-expressed in resistant cells as compared to parental cells and attenuated doxorubicin induced oxidative-stress apoptosis. In fact, PKCα phosphorylated Bcl2, p-Bad, decreased PARP cleavage and prevented apoptosis in doxorubicin treated resistant cell lines. But incubation with PKCα inhibitor Go6976, doxorubicin induced apoptosis in the parental fraction and the HCT15/DOX cells line. Inhibition of PKCα activity has been shown to efficiently restore the drug-sensitive phenotype of cancer cells (Wang et al., 2010). To gain further insight into the possible effects of PKCα on MDR in human colon cancer cells, we transfected the cells with PKCα-siRNA because it has been shown that transfection of exogenous PKCα into cells over-expressing the MDR gene conferred an increased MDR phenotype into these cells (Yu et al., 1991). The knockdown of PKCα, through a PKCα-siRNA dose, dependently down-regulated Bcl2 and p-Bad, enhanced PARP cleavage and induced apoptosis in the resistant cell line. These results were in agreement with a previously published report that suggested that transfection of PKCα antisense into cells reversed the MDR phenotype (Osborn et al., 1999).
In conclusion, the results presented here support the notion that PKCα activation phosphorylates P-gp and over-expression of MDR, and ultimately leads to drug resistance. PKCα reversed doxorubicin induced apoptosis through the scavenging of ROS and expression of anti-apoptotic proteins such as Bcl2 as well as inhibited PARP cleavage. Using PKCα inhibitor Go6976 and PKCα siRNA knockdown revealed that PKCα provides a major contribution to drug resistance. We hope that the outcome of this study will make further insights into doxorubicin-induced apoptosis as well as therapeutic strategies directed against PKCα as a tool for restructuring the resistance of tumors against cytotoxic agents.
Acknowledgments
This work was supported by the Nuclear Research and Development Program of the National Research Foundation of Korea funded by the Korean Ministry of Education, Science and Technology (grant code: 2011-0006331).
REFERENCES
- Altenberg G.A., Vanoye C.G., Han E.S., Deitmer J.W., Reuss L. Relationships between rhodamine 123 transport, cell volume, and ion-channel function of P-glycoprotein. J. Biol. Chem. 1994;269:7145–7149. [PubMed] [Google Scholar]
- Aquino A., Warren B.S., Omichinski J., Hartman K.D., Glazer R.I. Protein kinase C-gamma is present in doxorubicin resistant HL-60 leukemia cells. Biochem. Biophys. Res. Commun. 1990;166:723–728. doi: 10.1016/0006-291x(90)90869-o. [DOI] [PubMed] [Google Scholar]
- Bae Y.S., Oh H., Rhee S.G., Yoo Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R., Verheij M., Haimovitz-Friedman A., Scotto A., Fuks Z., Kolesnick R.N. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- Budworth J., Gant T.W., Gescher A. Co-ordinate loss of protein kinase C and multidrug resistance gene expression in revertant MCF-7/ADR breast carcinoma cells. Br. J. Cancer. 1997;75:1330–1335. doi: 10.1038/bjc.1997.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens M.J., Trayner I., Menaya J. The role of protein kinase C isoenzymes in the regulation of cell proliferation and differentiation. J. Cell Sci. 1992;103:881–888. doi: 10.1242/jcs.103.4.881. [DOI] [PubMed] [Google Scholar]
- Choi S.U., Kim N.Y., Choi E.J., Kim K.H., Lee C.O. Establishment of doxorubicin-resistant subline derived from HCT15 human colorectal cancer cells. Arch. Pharm. Res. 1996;19:342–347. [Google Scholar]
- Dolfini E., Dasdial T., Arancia G., Molinari A., Calcabrini A., Scheper R.J., Flens M.J., Gariboldi M.B., Monti E. Characterization of a clonal human colon adenocarcinoma line intrinsically resistant to doxorubicin. Br. J. Cancer. 1997;76:67–76. doi: 10.1038/bjc.1997.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroshow J.H., Locker G.Y., Myers C.E. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alter alterations produced by doxorubicin. J. Clin. Invest. 1980;65:128–135. doi: 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J.A., Ling V. The biochemistry of P-glycoprotein mediated multidrug resistance. Ann. Rev. Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- Fine R.L., Chambers T.C., Sachs C.W. P-glycoprotein, multidrug resistance and protein kinase. C. Stem Cells. 1996;14:47–55. doi: 10.1002/stem.140047. [DOI] [PubMed] [Google Scholar]
- Fleury C., Mignotte B., Vayssiere J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- Gariboldi M.B., Ravizza R., Riganti L., Meschini S., Calcabrini A., Marra M., Arancia G., Dolfini E., Monti E. Molecular determinants of intrinsic resistance to doxorubicin in human cancer cell lines. Int. J. Oncol. 2003;22:1057–1064. [PubMed] [Google Scholar]
- Gill P.K., Gescher A., Gant T.W. Regulation of MDR1 promoter activity in human breast carcinoma cells by protein kinase C isozymes alpha and theta. Eur. J. Biochem. 2001;268:4151–4157. doi: 10.1046/j.1432-1327.2001.02326.x. [DOI] [PubMed] [Google Scholar]
- Gottesman M.M., Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- Gottesman M., Fojo T., Bates S. Multidrug resistance in cancer: role of ATP-depen dent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Gouazé V., Mirault M.E., Carpentier S., Salvayre R., Levade T., Andrieu-Abadie N. Glutathione peroxidase-1 over-expression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol. Pharmacol. 2001;60:488–496. [PubMed] [Google Scholar]
- Gravitt K.R., Ward N.E., Fan D., Skibber J.M., Levin B., O’Brian C.A. Evidence that protein kinase C-alpha activation is a critical event in phorbol ester-induced multiple drug resistance in human colon cancer cells. Biochem. Pharmacol. 1994;48:375–381. doi: 10.1016/0006-2952(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Han Y., Han Z.Y., Zhou X.M., Shi R., Zheng Y., Shi Y.Q., Miao J.Y., Pan B.R., Fan D.M. Expression and function of classical protein kinase C isoenzymes in gastric cancer cell line and its drug-resistant sublines. World J. Gastroenterol. 2002;8:441–445. doi: 10.3748/wjg.v8.i3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoguchi T., Sonta T., Tsubouchi H., Etoh T., Kakimoto M., Sonoda N., Sato N., Sekiguchi N., Kobayashi K., Sumimoto H., et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J. Am. Soc. Nephrol. 2003;14:S227–232. doi: 10.1097/01.asn.0000077407.90309.65. [DOI] [PubMed] [Google Scholar]
- Konorev E.A., Kennedy M.C., Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: The role of reactive oxygen and nitrogen intermediates. Arch. Biochem. Biophys. 1999;368:421–428. doi: 10.1006/abbi.1999.1337. [DOI] [PubMed] [Google Scholar]
- Kuranami M., Powell C.T., Hug H., Zeng Z., Cohen A.M., Guillem J.G. Differential expression of protein kinase C isoforms in human colorectal cancers. J. Surg. Res. 1995;58:233–239. doi: 10.1006/jsre.1995.1036. [DOI] [PubMed] [Google Scholar]
- La Porta C.A., Dolfini E., Comolli R. Inhibition of protein kinase C-alpha isoform enhances the P-glycoprotein expression and the survival of LoVo human colon adenocarcinoma cells to doxorubicin exposure. Br. J. Cancer. 1998;78:1283–1287. doi: 10.1038/bjc.1998.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leszczynski D. Regulation of cell cycle and apoptosis by protein kinase C in rat myeloid leukemia cell line. Oncol. Res. 1995;7:471–480. [PubMed] [Google Scholar]
- Limtrakul P., Anuchapreeda S., Buddhasukh D. Modulation of human multidrug-resistance MDR-1 gene by natural curcuminoids. BMC Cancer. 2004;4:13. doi: 10.1186/1471-2407-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling V. Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother. Pharmacol. 1997;40:S3–S8. doi: 10.1007/s002800051053. [DOI] [PubMed] [Google Scholar]
- Lucas M., Sanchez-Margalet V. Protein kinase C involvement in apoptosis. Gen. Pharmacol. 1995;26:886–887. doi: 10.1016/0306-3623(94)00295-x. [DOI] [PubMed] [Google Scholar]
- Ma Y.Y., Wei S.J., Lin Y.C., Lung J.C., Chang T.C., Whang-Peng J., Liu J.M., Yan D.M., Yang W.K., Shen C.Y. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19:2739–2744. doi: 10.1038/sj.onc.1203597. [DOI] [PubMed] [Google Scholar]
- Mellor H., Parker P.J. The extended protein kinase C superfamily. Biochem. J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakura A., Higuchi C., Yoshida K., Yoshikawa H. PKCα suppresses osteoblastic differentiation. Bone. 2011;48:476–484. doi: 10.1016/j.bone.2010.09.238. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y., Kikkawa U. Early studies of protein kinase C: a historical perspective. Methods Mol. Biol. 2003;233:9–18. doi: 10.1385/1-59259-397-6:9. [DOI] [PubMed] [Google Scholar]
- O’Brian C.A., Ward N.E., Stewart J.R., Chu F. Prospects for targeting protein kinase C isozymes in the therapy of drug resistant cancer--an evolving story. Cancer Met. Rev. 2001;20:95–100. doi: 10.1023/a:1013186430906. [DOI] [PubMed] [Google Scholar]
- Osborn M.T., Berry A., Ruberu M.S., Ning B.T., Bell L.M., Chambers T.C. Phorbol ester induced MDR1 expression in K562 cells occurs independently of mitogen-activated protein kinase signaling pathways. Oncogene. 1999;18:5756–5764. doi: 10.1038/sj.onc.1202943. [DOI] [PubMed] [Google Scholar]
- Ruvolo P.P., Deng X., Carr B.K., May W.S. A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J. Biol. Chem. 1998;273:25436–25442. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- Skladanowski A., Konopa J. Doxorubicin and daunomycin induce programmed cell death (apoptosis) in tumour cells. Biochem. Pharmacol. 1993;46:375–382. doi: 10.1016/0006-2952(93)90512-u. [DOI] [PubMed] [Google Scholar]
- Takara K., Sakaeda T., Okumura K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr. Pharm. 2006;12:273–228. doi: 10.2174/138161206775201965. [DOI] [PubMed] [Google Scholar]
- Tentori L., Portarena I., Bonmassar E., Graziani G. Combined effects of adenovirus-mediated wild-type p53 transduction, temozolomide and poly (ADP-ribose) polymerase inhibitor in mismatch repair deficient and non-proliferating tumor cells. Cell Death Differ. 2001;8:457–469. doi: 10.1038/sj.cdd.4400832. [DOI] [PubMed] [Google Scholar]
- Wang N., Li Z., Tian F., Feng Y., Huang J., Li C., Xie F. PKCα inhibited apoptosis by decreasing the activity of JNK in MCF-7/ADR cells. Exp. Toxicol. Pathol. 2010;64:459–464. doi: 10.1016/j.etp.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Yu G., Ahmad S., Aquino A., Fairchild C.R., Tepel J.B., Ohno S., Suzuki K., Tsuro T., Cowan K.H., Glazer R.I. Transfection with protein kinase Cα confers increased multidrug resistance to MCF–7 cells expressing P-glycoprotein. Cancer Commun. 1991;3:181–189. doi: 10.3727/095535491820873263. [DOI] [PubMed] [Google Scholar]