

Abstract
Atherosclerosis is a chronic inflammatory disease in which both innate and adaptive immunity are involved. Although there have been major advances in the involvement of toll-like receptor 4 (TLR4) and CD36 in the initiation and development of this disease, detailed mechanisms remain unknown. Here, we show that tenascin-C (TN-C) can stimulate foam cell formation and this can be inhibited by a TLR4-blocking antibody or CD36 gene silencing. Our results identify TN-C-TLR4 activation as a common molecular mechanism in oxLDL-stimulated foam cell formation and atherosclerosis. In addition, CD36 is the major scavenger receptor responsible for the TN-C-mediated foam cell formation. Taken together, we have identified that TN-C produced by oxLDL-stimulated macrophages increases foam cell formation through TLR4 and scavenger receptor CD36.
Keywords: atherosclerosis, CD36, foam cell formation, tenascin-c, toll-like receptor 4
INTRODUCTION
Atherosclerosis-the primary cause of ischemic cardiomyopathy and ultimately a major contributing factor to death in the world today-is a chronic inflammatory disease (Lahoute et al., 2011). Atherosclerosis produces arterial plaques characterized by inflammatory infiltrates, lipid accumulation, cell death, and fibrosis (Hansson, 2005; Libby, 2006). Both innate and adaptive immunity are involved in the disease initiation and development through their response to endogenously modified structures, in particular oxidized lipoproteins (Andersson et al., 2010). Macrophages play pivotal roles in atherosclerosis development. The migration of monocytes into the intima and the conversion of monocytes/macrophages into foam cells represent the initial steps in atherosclerosis (Watanabe et al., 1985). Foam cell formation by macrophages is promoted by several extracellular factors, including LDL-especially modified LDL, such as oxidized LDL (oxLDL) or acetyl LDL (AcLDL)-and saturated free fatty acids, as well as by cellular mechanisms such as lipid uptake, metabolism, and efflux (Ito et al., 2004; Ishigaki et al., 2008). Moreover, foam cells secrete cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β, inducing cellular migration and apoptosis, which also contribute to the development of unstable plaques (Andersson et al., 2010).
The family of Toll-like receptors (TLRs) represents the major microbe-sensing system in mammals, detecting molecules derived from viruses, fungi, bacteria, and protozoa (Takeda et al., 2003). TLRs can also promote innate and adaptive immune responses, including induction of pro-inflammatory cytokines and matrix metalloproteinases (Howell et al., 2011). These make TLRs vital molecules in host defense. Given their involvement in innate immunity, it is not surprising that TLRs can affect atherosclerosis (Tedgui et al., 2011). Early studies demonstrated the expression of TLR1, TLR2, and TLR4 in both murine and human atherosclerotic plaques. Both TLR4 mutation and TLR4-neutralizing antibodies reduced foam cell formation in the presence of oxLDL, while activation of TLR4 with LPS promoted oxLDL-induced foam cell formation (Howell et al., 2011).
Although many advances have been made in characterizing the role of TLRs in atherosclerosis, additional studies aimed at elucidating the exact pathways involved in TLR signaling are warranted and may lead to the development of novel therapeutics to treat this devastating disease. In this study, oxLDL was used to stimulate macrophages differentiated from THP-1 cells. We found that in oxLDL-induced macrophages, tenascin-C (TN-C) levels were significantly elevated. We also used exogenous TN-C to stimulate macrophages, and our data suggests that the TN-C-TLRs-CD36 pathway might play a vital role in foam cell formation and atherosclerosis.
MATERIALS AND METHODS
Reagents
TNF-α ELISA kits were obtained from R&D systems (USA). TN-C ELISA kit was obtained from EIAAB USCNLIFE (China). Chol MPR2 kit was purchased from Roche Chemicals (Switzerland). TNF-α neutralizing mAb, CD36 monoclonal antibody, and TLR4 antibody were obtained from Sigma-Aldrich (USA). CD36-specific siRNAs were synthesized by Genetimes Co. (China). Lipofectamine 2000, BSA kit, TRIzol, and superscript-II MMLV-reverse transcriptase were purchased from Invitrogen (Canada). Recombinant human TN-C was obtained from R&D (USA). Antibody for western blotting was purchased from Pierce (USA). ECL Plus was obtained from Amersham (USA). Unless otherwise stated, all other substances were purchased from Sigma-Aldrich (USA).
Cell culture and treatment
The human acute monocytic leukemia cell line THP-1 (ATCC TIB-202) was cultured in RPMI 1640 medium containing 10% fetal calf serum, 2 mmol/L L-glutamine, 1.0 mmol/L sodium pyruvate, 4.5 g/liter glucose, and 100 μM/L L-ascorbic acid (Sigma) at 37°C in a humidified atmosphere containing 5% CO2. The cells were activated with 100 nM phorbol 12-myristate 13-acetate (PMA) to differentiate into macrophages. Macrophages from THP-1 cells (106/plate) were plated 24 h before treatment with a low concentration of oxLDL, or PBS as the solvent control. For further investigation, macrophages were also pre-treated with a TLR4-neutralizing antibody or CD36-specific siRNA for 60 min prior to stimulation with oxLDL.
Preparation of OxLDL
Human LDL was purified from fresh plasma of healthy donors by sequential centrifugation using a Beckman SW41 rotor as previously described (Feng et al., 2010) and dialyzed against saline with EDTA (1 mmol/L). LDL concentration (total mass) was determined by enzymatic cholesterol assay with the Chol MPR2 kit. Heavily oxidized oxLDL was prepared by incubating 200 μg/ml of the LDL with 20 μM of CuSO4 solution at 37°C for the indicated periods. The oxidative reactions were stopped with 40 μM butylhydroxytoluene in ethanol. The oxidized LDL (CuoxLDL) was concentrated by using a Vivapore membrane concentrator (Millipore, USA) before filter sterilization and was identified by changes in electrophoretic mobility (data not shown).
Real-time RT-PCR
Total RNA from macrophages (treated with oxLDL or untreated) was isolated using TRIzol with isopropanol precipitation as previously described (Khan et al., 2004). Following extraction, RNA samples were subjected to DNase treatment to degrade any contaminating DNA in the samples. Purity was assessed by measuring OD 260:280 nm.
cDNA was synthesized using 2 μg total RNA with oligo(dT) primers and superscript-II MMLV-reverse transcriptase. Real-time RT-PCR was performed in the DNA Engine Opticon 2 (Bio-rad). The reaction mixture consisted of 10 μl SYBR Green Taq ReadyMix (Sigma-Aldrich), 1.6 μl 25 mmol/L MgCl2, 1 μl of each forward and reverse 10 μmol/L primers, 4.4 μl H2O, and 2 μl cDNA template. The primer sequences for TN-C were as follows: sense: 5′-GAGACATCTGTGGAAGTGGA-3′; antisense: 5′-CGTACTCAGTGTCAGGCTTC-3′. TN-C expression was quantified in relation to β-actin expression and primers were as follows: sense: 5′-GGAGCAATGATCTTGATCTT-3′; antisense: 5′-CCTTCCTGGGCATGGAGTCCT-3′.
ELISA
Levels of TNF-α and TN-C proteins in macrophages treated with or without oxLDL were analyzed using the corresponding ELISA kits according to the manufacturer’s instructions. The results were read at 450 nm.
Lipid analysis by HPLC
Total cholesterol (TC) and cholesterol ester (CE) contents were analyzed by HPLC. Briefly, cells were washed by PBS three times and lysed by 0.1 N NaOH solution and homogenized on ice for 10 s by ultrasound. Protein concentration was measured using the BSA kit. An equal volume of trichloroacetic acid was added, and then centrifuged at 800 × g for 10 min. We used Stigmasterol to produce a standard curve and to repeat the extraction procedure. The organic phase was transferred to clear glass tubes, and dried in a vacuum-pump. The samples were dissolved in 100 μl isopropanol-acetonitrile (20:80), followed by an ultrasound water bath at room temperature for five minutes. Finally, the samples were subjected to HPLC analysis (Agilent 1100, Agilent Technology, USA).
Determination of CD36 expression by fluorescence-activated cell sorting (FACS)
CD36 expression was analyzed by FACS as described previously (Munteanu et al., 2006; Ricciarelli et al., 2000). CD36 expression was detected using fluorescein isothiocyanate-conjugated anti-human CD36 monoclonal antibody. The antibodies were diluted 1:50 in phosphate-buffered saline and 1% bovine serum albumin according to the manufacture’s protocol. A minimum of 10,000 cells/sample was assessed; data were acquired and analyzed using CellQuest software (FACScan, BD Biosciences).
CD36 gene silencing
A si-genome SMARTpool (Shanghai Genetimes) consisting of three unique 27mer siRNA duplexes and control siRNAs was used to knock down CD36 expression levels. In brief, cultured THP-1 cells were transfected with 20 μmol/L CD36 siRNA using lipofectamine 2000 1 h prior to the addition of oxLDL (5 μg/ml) and TN-C (1 μM) according to the manufacture’s protocol. CE/TC contents in macrophages were measured when cells had been treated with oxLDL and TN-C for 48h.
Western blotting
Cells transfected with CD36 specific or scrambled siRNA or cells at resting were lysed in RIPA buffer (1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS in PBS containing aprotinin and sodium orthovanadate). Lysates were cleared by centrifugation at 14,000 × g, resolved by 10% SDS-PAGE followed by electrotransfer to a nitrocellulose membrane. Blots were incubated overnight with primary Abs against CD36 at 4°C followed by horseradish peroxidase-conjugatal secondary antibodies that were detected using the ECL Plus. As internal standard, beta-Actin (β-Actin) expression was determined. CD36 expression was quantified in relation to β-Actin.
Statistical analysis
The data are expressed as means ± SEM and were analyzed by ANOVA followed by Student’s t-test. Differences were considered statistically significant at values of p < 0.05
RESULTS
TN-C expression is upregulated in oxLDL-treated macrophages
Tenascin-C is an extracellular matrix glycoprotein associated with tissue injury and repair. It is not normally expressed in most adult tissues but is specifically and transiently upregulated during acute inflammation and persistently expressed in chronic inflammation (Chiquet-Ehrismann and Chiquet, 2003). Previous studies have shown that increased TN-C levels are observed in multiple autoimmune diseases (Midwood et al., 2009), including rheumatoid arthritis, Sjogren’s syndrome (Amin et al., 2001), and diabetes (Loots et al., 1998). However, the potential function of TN-C in foam cell formation and atherosclerosis was barely understood. Our study was conducted to outline the potential role of TN-C as well as future prospects for treatment. Studies have shown that high oxLDL mediates macrophages apoptosis (Chang et al., 2006). In this study, we first used low concentrations of oxLDL (0–50 μg/ml) to stimulate macrophages. The effect of various concentrations of oxLDL on TN-C levels in macrophages was measured by real-time RT-PCR. We found that TN-C levels increased in a time- and dose-dependent manner (Fig. 1). Macrophages were treated with 0, 1, 5, 10, 20 or 50 μg/ml of oxLDL for 4 h, and TN-C expression was significantly stimulated approximately eight-fold by incubation with 5 μg/ml oxLDL(P < 0.05). However, as the oxLDL concentration increased, the TN-C expression decreased (Fig. 1A). In our previous test (results were not shown here), a high concentration of oxLDL (≥ 50 μg/ml) induced macrophage apoptosis. It may be cell apoptosis that contribute to the TN-C expression decrease when the oxLDL concentration was 50 μg/ml. We also examined the time dose effect on TN-C expression while oxLDL concentration was invariable (5 μg/ml). Incubation with 5 μg/ml of oxLDL increased the TN-C expression up to nine-fold at 6 h (Fig. 1B), and significant difference was observed between control group and 6 h or 24 h-treated group (P < 0.05). However, after this time, TN-C expression decreased; this may be associated with decreased cell viability.
Fig. 1.

OxLDL induced the expression of TN-C in macrophages in a time- and dose-dependent manner. (A) Macrophages were stimulated with different concentrations of oxLDL (0, 1, 5, 10, 20 or 50 μg/ml); (B) Macrophages were stimulated with 5 μg/ml oxLDL for different time; (C): The expression of TN-C protein in macrophages induced with 5 μg/ml oxLDL for different times were measured by ELISA. *(P < 0.05) represents significant difference to control (0 μg/ml or 0 h).
In order to further investigate the TN-C expression, an ELISA was used to measure TN-C protein levels (Fig. 1C). The significant increase of TN-C at 24 h and 48 h confirmed the time-dependent fashion of TN-C protein expression in macrophages stimulated with 5 μg/ml oxLDL(P < 0.05).
Taken together, our data suggests that oxLDL can induce TN-C expression of macrophages in a time- and dose-dependent manner.
TN-C upregulation is partly modulated by TNF-α upregulation induced by oxLDL
Previous studies have shown that oxLDL can stimulate monocyte/macrophage release of TNF-α in a dose-dependent manner, and in rheumatic aortic valve interstitial cells, TNF-α can induce the expression of TN-C (Jiang et al., 2009; Jovinge et al., 1996). To test whether oxLDL induced TNF-α expression and subsequently TNF-α induced TN-C expression in macrophages, we first analyzed TNF-α levels in oxLDL-treated macrophage using an ELISA kit as previously described (Jiang et al., 2009). Compared with control (112 ± 23 pg/ml), incubation with 5 μg/ml oxLDL for 24 h significantly increased TNF-α expression to 464 ± 12 pg/ml (P < 0.01). To further investigate the potential function of TNF-α, a blocking IgG antibody against TNF-α was adopted. Different concentration of antibody (0, 0.1, 0.5, 1, 5 μg/ml) was added to macrophages 0.5 h before or 12 h after oxLDL incubation and the TN-C variation was shown in Fig. 2. TN-C was significantly inhibited in pre-treated groups while the concentration of antibody against TNF-α was higher than 1 μg/ml. However, the inhibition of TN-C was not dose dependent and the inhibition maximized at 1 μg/ml antibody, to a level of 150 ± 14 pg/ml TN-C. Differences were also observed in the inhibition of pre-treatment and post-treatment. In the pre-treatment group, TNF-α was blocked by its antibody the moment it was secreted; in the latter group, TNF-α was induced by oxLDL for 12 h before the antibody was added, and the secreted TNF-α is available to stimulate TN-C expression prior to the antibody being added. The different expression of TNF-α may lead to the different inhibition of TN-C. However, in the pre-incubation assay of 1 μg/ml antibody against TNF-α, TN-C was still higher (150 ± 14 pg/ml) than control (112 ± 23 pg/ml) suggesting that there should be other pathways that contributed to the production of TN-C.
Fig. 2.
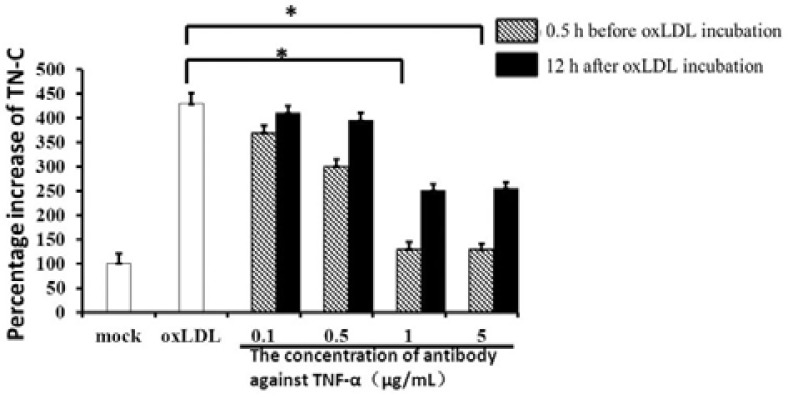
Blocking antibodies against TNF-α reduced the expression of TN-C in oxLDL-stimulated macrophages. Different concentration of blocking antibodies against TNF-α were added to macrophages 0.5 h before or 12 h after oxLDL incubation, and TN-C expression was measured by ELISA kit. *(P < 0.05), significant difference to group with oxLDLD treatment only.
In summary, TN-C expression upregulation in oxLDL-treated macrophages was primarily mediated via the upregulation of TNF-α; in other words, oxLDL stimulation could induce TN-C expression mainly through an autocrine pathway in which TNF-α was involved.
Exogenous TN-C stimulates foam cell formation in a time- and dose-dependent manner
Given the results above, we concluded that TN-C might affect foam cell formation. To exclude any interference from other factors stimulated by oxLDL, we next investigated whether exogenous TN-C could activate the foam cells formation. Exogenous TN-C was added to macrophages, and TC and CE contents-markers of foam cell formation-were measured using HPLC. As shown in Fig. 3A, 5 μg/ml oxLDL and different concentrations of exogenous TN-C were added to macrophages. TC and CE contents significantly increased to 70% compared to control (25%), and significant differences were observed between control and all other exogenous TN-C-treated groups (P < 0.05). We next analyzed the effect of time: 1 μM TN-C and 5 μg/ml oxLDL were used to stimulate macrophages, and TC and CE contents were measured at the indicated times. As shown in Fig. 3B, TC and CE contents increased as the time of treatment increased. After incubation with TN-C and oxLDL for 48 h and 72 h, TC/CE content increased to 70 and 75%, respectively, and this was significantly higher than the control or 24 h incubation group (P < 0.01). A significant difference was also observed between the control and the 24 h incubation group (P < 0.05). All these data suggested that TN-C could increase the differentiation of macrophages into foam cells.
Fig. 3.

Exogenous TN-C could increase the differentiation of macrophages into foam cells in a time- and dose-dependent manner. (A) Macrophages were induced with 5 μg/ml oxLDL and different concentration of exogenous TN-C; (B) Macrophages were stimulated with 5 μg/ml oxLDL and 1 μM TN-C for the indicated time. CE/TC contents were measured by HPLC. *(P < 0.05) and *(P < 0.01), significant difference to control (0 μM or 0 h).
Blockade of TLR4 reduces TN-C-induced macrophage foam cell formation
It is well established that TLR4 upregulation occurs on the macrophage cell surface upon exposure to oxLDL, and TLR4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plagues and is upregulated by oxLDL (Xu et al., 2001). Activation of TLR4 promotes oxLDL-induced foam cell formation (Howell et al., 2011). During inflammation of joints, TN-C is an endogenous activator of TLR4 (Midwood et al., 2009), and so we asked whether TN-C interacts with TLR4 in atherosclerosis. We analyzed the function of TLR4 in TN-C-induced foam cell formation. Macrophages were pretreated with different concentration of TLR4 monoclonal antibody 1 h prior to exogenous TN-C (1 μM) and oxLDL (5 μg/ml) treatment. After 48 h incubation, the high concentration of TLR4-blocking antibody (> 1:200 dilution) significantly decreased the differentiation of TN-C-induced macrophage into foam cells from 65% to 48% (Fig. 4; P < 0.05), while low concentration antibody (1:500 and 1:1000 dilution) only lead to a slight reduction (to a level of 58%) that have no significant difference with control. However, control antibodies did not generate any reduction on foam cell formation.
Fig. 4.

Blocking of TLR4 significantly reduced TN-C-induced foam cell formation. Different concentrations of antibody against TLR4 or control antibody were added to macrophages 1 h prior to oxLDL and TN-C stimulation, and foam cell formation was detected by measuring CE/TC contents. *(P < 0.05), significant difference to group with oxLDL and TN-C treatment only.
Increased CD36 expression associated with foam cell formation
Previous studies have shown that scavenger receptors are a receptor for oxLDL (Ashraf and Gupta, 2011). To check whether CD36 is the major scavenger receptor for TN-C-induced foam cell formation, macrophages were preincubated for 24 h with 1 μM exogenous TN-C, and the expression of CD36 was measured using flow cytometry (Fig. 5). Compared to resting cells (without TN-C treatment), a 24 h incubation with TN-C caused a rapid expression of CD36 by more than 51% in all cells (P < 0.05).
Fig. 5.

Exogenous TN-C stimulation significantly increased CD36 expression by macrophages (P < 0.05). (B) Macrophages without TN-C treatment were analyzed for CD36 expression using flow cytometry. (C) Macrophages were treated with 1 μM TN-C for 24 h, and CD36 expression was determined by flow cytometry. Significant differences were observed between resting cells and TN-C-treated cells (P < 0.05).
CD36 gene silencing reduces exogenous TN-C-stimulated foam cell formation
To further elucidate the role of CD36, CD36 gene expression was knocked down using specific siRNAs. Western blotting was performed to detect the suppression of CD36 expression. As shown in Figs. 6A and 6B, CD36 specific siRNAs transfection reduced the expression of CD36 by 67%, and significant difference was observed between cells transfected with CD36 specific siRNAs and cell at resting or cells transfected with scrambled sequence (P < 0.05). We further measuring CE/TC content to analyze the foam cell formation. As shown in Fig. 6C, control siRNA had no influence on the production of CE/TC; however, when macrophages were transfected with CD36-specific siRNA, CE/TC content significantly reduced to 39%, compared to 67% in the control group (P < 0.05).
Fig. 6.

CD36-specific siRNA transfection reduced CE/TC production in macrophages. Macrophages were transfected with scramble siRNA sequence or CD36 specific siRNA 1 h before cells were stimulated with 5 μg/ml oxLDL and 1 μM TN-C. (A, B) Cells lysates were prepared from these cells, equal amount of proteins were resolved by SDS-PAGE and CD36 expression were detected by Western blotting. Beta-actin was used as a loading control. (C) CE/TC contents were measured by HPLC 48 h after stimulation. *(P < 0.05), significant difference to group with oxLDl treatment only or cells transfected with scramble sequence.
DISCUSSION
The goal of improving therapies for atherosclerosis has been hampered by the lack of progress in identifying genes for common forms of the disease, the enormous complexity, and the important role played by environmental factors. Although many studies have supported a role of TLRs in atherosclerosis, little is known about the exact mechanism and which ligand may be present in atherosclerosis. This study has identified a novel role of TN-C in foam cell formation and has demonstrated that TN-C and TLRs are required for atherosclerosis.
Tenascin-C is a large hexameric extracellular glycoprotein. As the founding member of a family of four tenascins, TN-C is unique in its distinct pattern of expression. Little or no tenascin-C is detected in healthy adult tissues. It is transiently re-expressed upon tissue injury and down-regulated after tissue repair is complete (Midwood et al., 2011). Transient TN-C expression is also associated with acute inflammation, and persistent TN-C with chronic inflammation (Udalova et al., 2011). This study has identified a novel role for TN-C as an effector of TNF-in foam cell α formation and atherosclerosis. Here, we found that in oxLDL-induced macrophages, elevated TN-C was partly associated with increased TNF-α suggesting that TN-C is a downstream mediator of TNF-α in oxLDL-induced macrophages. Studies have shown that oxLDL can stimulate macrophage release of TNF-α (Jovinge et al., 1996). We found that the administration of TNF-α antibody could cut down the TN-C expression in a large extent suggesting that oxLDL induced TN-C release from macrophages via autocrine involving TNF-α. However, pretreatment (0.5 h before oxLDL stimulation) or post-treatment (12 h after oxLDL treatment) of TNF-α antibody neither could abolish the TN-C induction to a control level. We also found that the inhibition of TNF-α antibody on TN-C did not increase as the increasing of the TNF-α antibody concentration, and the maximum was reached at 1 μg/ml. High concentration of antibody against TNF-α could not reduce TN-C expression to a control level suggesting that there should be other signaling or cytokines that are responsible for TN-C expression in oxLDL-induced macrophages. TN-C induction was a synthetic action of different cytokines in which TNF-α play a dominating role.
Previously, studies have shown that increased TN-C levels are observed in multiple autoimmune diseases (Midwood et al., 2009), including rheumatoid arthritis, Sjogren’s syndrome (Amin et al., 2001), and diabetes (Loots et al., 1998). Our data shows that exogenous TN-C, together with oxLDL (5 μg/ml) is capable of activating foam cell formation. These results, and the tightly regulated expression pattern of TN-C, make it an attractive candidate for driving atherosclerosis. Studies from Tedgui have demonstrated an important role for TLRs in atherosclerosis (Tedgui et al., 2011). In this study, our results show that a TLR4-neutralizing antibody could reduce foam cell formation induced by exogenous TN-C, and this suggests that functional TLR4 is required for TN-C to induce foam cell formation. This is consistent with fruits of Kim et al. (2009) that TN-C is an activator of TLR4 in foam cell formation. The present study adds to the growing body of evidence that implicates Toll-like receptors in the pathogenesis of atherosclerosis.
The uptake of modified LDL leading to foam cell formation by macrophages is mediated by scavenger receptors (class A; class B, type I; and CD36). Scavenger receptors found on macrophages range from class A to G, i.e. those that share the defining feature of being able to bind the modified forms of LDL (Ashraf and Gupta, 2011). Expression of some scavenger receptors is increased in atherosclerosis, possibly as a result of a positive feedback loop mediated by oxLDL and its lipid content (Hajjar and Haberland, 1997; Han et al., 1997). Scavenger receptor-ligand interaction initiates signaling cascades that modulate macrophage activation, lipid metabolism, and inflammatory pathways that may influence the development and stability of the atherosclerotic plaque. The class B scavenger receptor CD36 is an archetypal pattern recognition receptor that binds polyanionic ligands. In addition to its function as a TLR2-TLR6 coreceptor for Staphylococcus aureus-derived lipoteichoic acid (LTA) and Mycoplasma macrophage-activating lipopeptide-2 (MALP-2) (Hoebe et al., 2005; Stuart et al., 2005), CD36 has established roles in the endocytic uptake of altered self-components, including oxidized phospholipids, apoptotic cells, and amyloid proteins. Studies from Podrez and colleagues have demonstrated the expression of scavenger receptors-especially CD36-on platelets suggesting their critical role in platelet hyper-reactivity in dyslipidemia and atheroprogression (Podrez et al., 2007). Stewart and colleagues identified CD36-TLR4-TLR6 activation as a common molecular mechanism by which atherogenic lipids and amyloid-stimulate sterile inflammation (Stewart et al., 2009). All these studies confirmed the critical role of scavenger receptor CD36 in atherosclerosis. In this study, we found that TN-C can stimulate more macrophages to express CD36. In oxLDL-induced macrophages, TN-C was upregulated due to the upregulation of TNF-α. According to our study, increased TN-C can then stimulate the expression of CD36, which is involved in the uptake of oxLDL; oxLDL uptake further induced TN-C expression and foam cell formation in which TLR4 was involved. Thus CD36 and TN-C may constitute a positive feedback loop in oxLDL-induced foam cell formation in which TLR-4 may play a critical role.
In summary, these data indicate a potential role for TN-C in foam cell formation and atherosclerosis via TLR-4. CD36 and TN-C-TLR-4 may represent a new feedback loop in oxLDL-induced foam cell formation. Further studies aims at the exact mechanism downstream of CD36 and TLR4 as well as the potential interaction of CD36 and TLR4 will shed more light on the therapy of atherosclerosis and associated diseases.
REFERENCES
- Amin K., Lúdviksdóttir D., Janson C., Nettelbladt O., Gudbjórnsson B., Valtysdottir S., Björnsson E., Roomans G., Boman G., Seveus L. Inflammation and structural changes in the airways of patients with primary Sjögren’s syndrome. Respir. Med. 2001;95:904–910. doi: 10.1053/rmed.2001.1174. [DOI] [PubMed] [Google Scholar]
- Andersson J., Libby P., Hansson G.K. Adaptive immunity and atherosclerosis. Clin. Immun. 2010;134:33–46. doi: 10.1016/j.clim.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Ashraf M.Z., Gupta N. Scavenger receptors: implications in atherothrombotic disorders. Int. J. Biochem. Cell Biol. 2011;43:697–700. doi: 10.1016/j.biocel.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Chang Y.C., Huang K.X., Huang A.C., Ho Y.C., Wang C.J. Hibiscus anthocyanins-rich extract inhibited LDL oxidation and oxLDL-mediated macrophages apoptosis. Food Chem. Toxicol. 2006;44:1015–1023. doi: 10.1016/j.fct.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R., Chiquet M. Tenascins: regulation and putative functions during pathological stress. J. Pathol. 2003;200:488–499. doi: 10.1002/path.1415. [DOI] [PubMed] [Google Scholar]
- Feng X., Zhang Y., Xu R., Xie X., Tao L., Gao H., Gao Y., He Z., Wang H. Lipopolysaccharide up-regulates the expression of Fc [alpha]/[mu] receptor and promotes the binding of oxidized low-density lipoprotein and its IgM antibody complex to activated human macrophages. Atherosclerosis. 2010;208:396–405. doi: 10.1016/j.atherosclerosis.2009.07.035. [DOI] [PubMed] [Google Scholar]
- Hajjar D.P., Haberland M.E. Lipoprotein trafficking in vascular cells. J. Biol. Chem. 1997;272:22975. doi: 10.1074/jbc.272.37.22975. [DOI] [PubMed] [Google Scholar]
- Han J., Hajjar D.P., Febbraio M., Nicholson A.C. Native and modified low density lipoproteins increase the functional expression of the macrophage class B scavenger receptor, CD36. J. Biol. Chem. 1997;272:21654. doi: 10.1074/jbc.272.34.21654. [DOI] [PubMed] [Google Scholar]
- Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Hoebe K., Georgel P., Rutschmann S., Du X., Mudd S., Crozat K., Sovath S., Shamel L., Hartung T., Zähringer U. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- Howell K.W., Meng X., Fullerton D.A., Jin C., Reece T.B., Cleveland J.C., Jr. Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. J. Surg. Res. 2011;171:27–31. doi: 10.1016/j.jss.2011.06.033. [DOI] [PubMed] [Google Scholar]
- Ishigaki Y., Katagiri H., Gao J., Yamada T., Imai J., Uno K., Hasegawa Y., Kaneko K., Ogihara T., Ishihara H. Impact of plasma oxidized low-density lipoprotein removal on atherosclerosis. Circulation. 2008;118:75–83. doi: 10.1161/CIRCULATIONAHA.107.745174. [DOI] [PubMed] [Google Scholar]
- Ito T., Yamada S., Shiomi M. Progression of coronary atherosclerosis relates to the onset of myocardial infarction in an animal model of spontaneous myocardial infarction (WHHLMI rabbits) Exp. Animals. 2004;53:339–346. doi: 10.1538/expanim.53.339. [DOI] [PubMed] [Google Scholar]
- Jiang L., Wei X., Yi D., Xu P., Liu H., Chang Q., Yang S., Li Z., Gao H., Hao G. Synergistic effects of cyclic strain and Th1-like cytokines on tenascin-C production by rheumatic aortic valve interstitial cells. Clin. Exp. Immunol. 2009;155:216–223. doi: 10.1111/j.1365-2249.2008.03747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovinge S., Ares M.P.S., Kallin B., Nilsson J. Human monocytes/macrophages release TNF-α in response to ox-LDL. Arterioscler. Thromb. Vasc. Biol. 1996;16:1573–1579. doi: 10.1161/01.atv.16.12.1573. [DOI] [PubMed] [Google Scholar]
- Khan Z.A., Cukiernik M., Gonder J.R., Chakrabarti S. Oncofetal fibronectin in diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2004;45:287. doi: 10.1167/iovs.03-0540. [DOI] [PubMed] [Google Scholar]
- Kim M., Sandra S., Anna M.P., Julia I., Annette T., Emma C., Stefan D., Nidhi S., Masahide K., Gertraud O., et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009;15:774–781. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- Lahoute C., Herbin O., Mallat Z., Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat. Rev. Cardiol. 2011;8:348–358. doi: 10.1038/nrcardio.2011.62. [DOI] [PubMed] [Google Scholar]
- Libby P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- Loots M.A.M., Lamme E.N., Zeegelaar J., Mekkes J.R., Bos J.D., Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J. Invest. Dermatol. 1998;111:850–857. doi: 10.1046/j.1523-1747.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- Midwood K., Sacre S., Piccinini A.M., Inglis J., Trebaul A., Chan E., Drexler S., Sofat N., Kashiwagi M., Orend G. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009;15:774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- Midwood K.S., Hussenet T., Langlois B., Orend G. Advances in tenascin-C biology. Cell. Mol. Life Sci. 2011;68:3175–3199. doi: 10.1007/s00018-011-0783-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munteanu A., Taddei M., Tamburini I., Bergamini E., Azzi A., Zingg J.M. Antagonistic effects of oxidized low density lipoprotein and α-Tocopherol on CD36 scavenger receptor expression in monocytes. J. Biol. Chem. 2006;281:6489–6497. doi: 10.1074/jbc.M508799200. [DOI] [PubMed] [Google Scholar]
- Podrez E.A., Byzova T.V., Febbraio M., Salomon R.G., Ma Y., Valiyaveettil M., Poliakov E., Sun M., Finton P.J., Curtis B.R. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciarelli R., Zingg J.M., Azzi A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–87. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- Stewart C.R., Stuart L.M., Wilkinson K., van Gils J.M., Deng J., Halle A., Rayner K.J., Boyer L., Zhong R., Frazier W.A. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2009;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart L.M., Deng J., Silver J.M., Takahashi K., Tseng A.A., Hennessy E.J., Ezekowitz R.A.B., Moore K.J. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J. Cell Biol. 2005;170:477–485. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Kaisho T., Akira S. Toll-like receptors. Ann. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tedgui A., Owens A.P., III, Mackman N. Nobel prize in physiology or medicine. Arterioscler. Thromb. Vasc. Biol. 2011;31:2767–2768. doi: 10.1161/ATVBAHA.111.240432. [DOI] [PubMed] [Google Scholar]
- Udalova I.A., Ruhmann M., Thomson S., Midwood K.S. Expression and immune function of tenascin-C. Crit. Rev. Immunol. 2011;31:115–145. doi: 10.1615/critrevimmunol.v31.i2.30. [DOI] [PubMed] [Google Scholar]
- Watanabe T., Hirata M., Yoshikawa Y., Nagafuchi Y., Toyoshima H. Role of macrophages in atherosclerosis. Sequential observations of cholesterol-induced rabbit aortic lesion by the immunoperoxidase technique using monoclonal anti-macrophage antibody. Lab. Invest. J. Tech. Methods Pathol. 1985;53:80. [PubMed] [Google Scholar]
- Xu X.H., Shah P.K., Faure E., Equils O., Thomas L., Fishbein M.C., Luthringer D., Xu X.P., Rajavashisth T.B., Yano J. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]